

Abstract
Objective
We compared the response to antipsychotic treatment between patients with and without tardive dyskinesia (TD) and examined the course of TD.
Method
This analysis compared 200 patients with DSM-IV–defined schizophrenia and TD and 997 patients without TD, all of whom were randomly assigned to receive one of 4 second-generation antipsychotics. The primary clinical outcome measure was time to all-cause treatment discontinuation, and the primary measure for evaluating the course of TD was change from baseline in Abnormal Involuntary Movement Scale (AIMS) score. Kaplan-Meier survival analysis and Cox proportional hazards regression models were used to compare treatment discontinuation between groups. Changes in Positive and Negative Syndrome Scale (PANSS) and neurocognitive scores were compared using mixed models and analysis of variance. Treatment differences between drugs in AIMS scores and all-cause discontinuation were examined for those with TD at baseline. Percentages of patients meeting criteria for TD postbase-line or showing changes in AIMS scores were evaluated with χ2 tests. Data were collected from January 2001 to December 2004.
Results
Time to treatment discontinuation for any cause was not significantly different between the TD and non-TD groups (χ21 =0.11, P = .743). Changes in PANSS scores were not significantly different (F1,974 = 0.82, P = .366), but patients with TD showed less improvement in neurocognitive scores (F1,359=6.53, P =.011). Among patients with TD, there were no significant differences between drugs in the decline in AIMS scores (F3,151 = 0.32, P = .811); 55% met criteria for TD at 2 consecutive visits postbaseline, 76% met criteria for TD at some or all postbaseline visits, 24% did not meet criteria for TD at any subsequent visit, 32% showed a ≥ 50% decrease in AIMS score, and 7% showed a ≥ 50% increase in AIMS score.
Conclusions
Schizophrenia patients with and without TD were similar in time to discontinuation of treatment for any cause and improvement in psychopathology, but differed in neurocognitive response. There were no significant differences between treatments in the course of TD, with most patients showing either persistence of or fluctuation in observable symptoms.
Trial Registration
clinicaltrials.gov Identifier: NCT00014001
The likelihood of tardive dyskinesia (TD) being irreversible served as a major impetus in antipsychotic drug development. Although controversy arose over the severity and reversibility of TD, a consensus emerged that TD followed a persistent or fluctuating course, but gradually stabilized or diminished, with few patients showing progression.1,2
Because medication withdrawal risks relapse in schizophrenia, it became important to examine the course of TD while maintaining antipsychotic treatment. Data on the change in prevalence of TD during treatment have been inconclusive, with some studies showing an increase3–6 and others, a decrease7–9 or no change at all.10,11 However, prevalence rates as well as mean dyskinesia scores obscure the dynamics of TD in individual patients. Although estimates have varied,1,12–15 roughly 50% of patients have persistent TD symptoms, 10% to 30% have a reduction in symptoms, and 10% to 30% show increased symptoms during treatment.16 Long-term studies estimated that from 2% to 23% of patients show loss of observable TD symptoms during treatment with first-generation drugs.5,17–20 Similarly, studies of second-generation agents have shown reduction of TD ratings,21–23 with some showing greater reductions,15,19,22,24–26 lesser reductions,13,27 or no difference6,8 compared with first-generation agents.
Studying the course of TD in parallel with clinical symptoms is also important because of evidence suggesting an association between TD, symptom severity, treatment response, and prognosis of schizophrenia.2,28 For example, several cross-sectional studies have found an association between TD and severity of cognitive deficits, as well as positive and negative symptoms of schizophrenia.2,28 In addition, several prospective studies have shown that the presence of TD in schizophrenia correlates with poor response to treatment, lower rates of symptom remission, longer hospital stays, greater risk of relapse, lower quality of life and functioning, a progressive course, and higher mortality.29–33 This evidence suggests the possibility that TD may be a phenotypic manifestation of a more severe, chronic, and refractory subtype of schizophrenia.29,34
The Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Schizophrenia Trial offered the opportunity to examine treatment response in a large sample of schizophrenia patients with TD at baseline randomly assigned to receive second-generation antipsychotics compared to patients without TD in the setting of a controlled trial. In addition, we sought to further delineate the course of TD during treatment with second-generation antipsychotics, thereby enabling the development of treatment recommendations for management of symptoms of both schizophrenia and dyskinesias in patients with existing TD. We hypothesized that patients with TD at baseline are more likely to discontinue treatment for any cause compared with non-TD patients; the course of TD varies among individuals; and there are no differences between second-generation antipsychotics in the outcome of dyskinesias or symptoms of schizophrenia in patients with TD.
METHOD
Study Setting and Design
This was a post hoc study analysis based on the CATIE Schizophrenia Trial, which was conducted between January 2001 and December 2004 at 57 US sites and included a series of treatment phases, which has been described previously.35 Patients were initially randomly assigned to receive olanzapine, perphenazine, quetiapine, risperidone, or ziprasidone under double-blind conditions and studied for up to 18 months or until treatment was discontinued for any reason (Phase 1). Patients with TD were excluded from randomization to perphenazine and were assigned to one of the 4 second-generation antipsychotics (Phase 1a). Ziprasidone was approved for use by the US Food and Drug Administration during the trial and was added after 40% of the patients had been enrolled. Patients who discontinued their first assigned treatment were invited to participate in subsequent phases of the trial. The data presented in the current report deal only with the time from initial randomization until the first medication was discontinued for patients with TD (Phase 1a) or without TD (Phase 1).
Participants
The study was approved by an institutional review board at each site, and written informed consent was obtained from all participants or their legally authorized representatives. Eligible patients were 18 to 65 years of age with a diagnosis of schizophrenia, as determined on the basis of the Structured Clinical Interview of the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. Patients were excluded if they had a diagnosis of schizoaffective disorder, mental retardation, or other cognitive disorders; an unstable and serious medical condition; past adverse reactions to the proposed treatments; or treatment-resistant schizophrenia or if they were in their first episode of schizophrenia, pregnant, or breast-feeding. Data were collected from January 2001 to December 2004.
Although 1,493 patients were enrolled in the trial, all data from 1 site (33 patients) were excluded prior to analysis due to concerns about data integrity, and 17 patients were randomized but did not start taking study medications (for CONSORT diagram, see Lieberman et al35).
The primary patient population for this analysis consisted of 200 patients randomly assigned to receive second-generation antipsychotics who met modified Schooler-Kane criteria for TD at baseline36 having at least 1 Abnormal Involuntary Movement Scale (AIMS)37 item with a value of 3, or at least 2 items with a value of 2 at the baseline visit. A history of at least 3 months of total antipsychotic exposure was not required. Although there were 212 patients in the CATIE study with TD at baseline as determined by modified Schooler-Kane criteria, 12 of these were randomly assigned inadvertently to perphenazine and were excluded from the present analyses. A comparison (non-TD) group consisted of 997 patients assigned to receive second-generation antipsychotics who did not meet modified Schooler-Kane criteria for TD at baseline. However, 67 of these patients had a history of TD, and 68 patients had 1 AIMS item rated 2; these patients were considered indeterminate and excluded from secondary supportive analyses, yielding a more restricted comparison group (n = 862).
Interventions
Identical-appearing capsules contained olanzapine (7.5 mg), quetiapine (200 mg), risperidone (1.5 mg), perphenazine (8 mg), or ziprasidone (40 mg). Medications were flexibly dosed with 1 to 4 capsules daily, as judged by the study doctor. Overlap in administration of the antipsychotic drugs that patients received before study entry was permitted for the first 4 weeks after randomization to allow a gradual transition to study medication. Concomitant medications were permitted, except for additional antipsychotic agents. Further details about blinding, later phases of treatment, and modal dosing have been presented previously.33
Measures
Basic sociodemographic data were recorded at baseline. Patients were screened for alcohol and drug abuse, treatment with antipsychotics or anticholinergics, duration of illness, and measures of symptom severity (Positive and Negative Syndrome Scale [PANSS]),38 TD (AIMS),37 parkinsonism (Simpson-Angus Abbreviated scale [SAS]),39 and akathisia (Barnes Akathisia Scale [BAS]).40 Clinical measures were collected at baseline, month 1, and month 3 and quarterly thereafter until 18 months or time of treatment discontinuation. Cognitive functioning was measured at baseline and months 2, 6, and 18 by completing 11 neurocognitive tests, as described in detail previously41,42; 5 neurocognitive domain scores were calculated from 9 neurocognitive test summary scores and standardized to create Z-scores (mean [SD] = 0 [1]) for each domain. A neurocognitive composite score was calculated by creating a Z-score of the average of the 5 standardized domain scores.
Statistical Methods
The primary clinical outcome measure was time to all-cause treatment discontinuation. Secondary outcomes included discontinuations for intolerability, inefficacy, and patient decision; rates of discontinuations; mean modal dose; and change from baseline in the PANSS and neurocognitive composite scores. For all clinical measures, supportive analyses were also conducted in which 135 patients with indeterminate evidence of TD were excluded from the non-TD group.
Kaplan-Meier survival curves were used to estimate the time to discontinuation of treatment in patients with and without TD at baseline. The TD groups were compared using Cox proportional hazards regression models, stratifying according to site, with adjustment for treatment group, age, duration of illness, baseline PANSS score, and antipsychotic drugs taken before study entry. For all Cox proportional hazards regression models, the proportional hazards assumption was evaluated, and no substantial violations were noted. Sites with 15 or fewer patients were grouped according to the sites’ health care systems. The mean modal doses of antipsychotics by treatment group were compared for patients with and without TD at baseline using an analysis of covariance (ANCOVA) adjusting for factors shown to be related to dosing assignment, including age, gender, body mass index, baseline PANSS score, service use, akathisia, and baseline quality of life.
Change from baseline in PANSS scores was compared for patients with and without TD using mixed models, adjusting for the same covariates as for time to discontinuation as well as terms representing time (treated as a classification variable) and baseline-by-time and TD-by-time interactions. A random subject effect and a spatial power covariance structure were used to adjust standard errors for the correlation of observations within subject.
Change from baseline in the neurocognitive composite scores at 6 months was compared for the TD groups with an ANCOVA, adjusting for demographic and other baseline measures found to be associated with the overall composite score.41,42 A large pool of potential predictors was identified a priori, and from these, the following covariates were selected for inclusion in the adjusted analysis: baseline neurocognitive score, treatment, investigator site, race, duration of illness, education, depression, and acute EPS at baseline. This same group of covariates was used in each of the domain score models for consistency.
In all clinical comparisons, the interaction between TD and treatment group was explored in order to assess whether the effect of TD differed among the second-generation antipsychotics. If an interaction was found (P <.10), exploratory analyses of the relationship between TD and the outcome were further conducted for each of the 4 treatments individually using a Bonferroni correction for multiple comparisons. Type I error was maintained at 5% by applying a threshold of significance for α = .05/4 = .013 for these pair-wise comparisons.
The primary outcome measure used to evaluate the course of TD was change from baseline in total AIMS score. Secondary outcome measures included change in global, distress, and impairment of function items on the AIMS; percentage of patients meeting Schooler-Kane criteria for at least 2 consecutive visits postbaseline; percentage of visits at which patients met modified Schooler-Kane criteria; and percentage of patients with at least a 50% change in AIMS score (excluding month 1). In addition, treatment differences with respect to all-cause discontinuation are described for patients with TD at baseline.
Changes in AIMS total score and global items were compared among the treatment groups for those patients with TD at baseline, using mixed models as described above. The AIMS analyses were conducted with adjustment for significant predictors that were identified from a large a priori set likely to have a relationship with the outcome, including a CATIE design variable for study entrance after the ziprasidone treatment option became available. The covariates selected for the adjusted models were baseline AIMS score, duration of illness, and baseline PANSS score. Potential interactions between covariates and treatment group were also explored. For comparison, we also analyzed changes in mean SAS scores and the global BAS score during the study for patients with TD at baseline using mixed models and the same covariates used in the AIMS analysis, plus baseline mean SAS score and BAS global score, respectively, and use of first-generation antipsychotics at baseline.
Among patients with TD at baseline, the percentage of patients who met Schooler-Kane criteria at 2 consecutive visits, the percentage of follow-up visits at which patients met Schooler-Kane criteria, and the percentage of patients whose average AIMS score changed by at least 50% were compared across treatment groups with χ2 tests. Additionally, the percentage of patients taking antipsychotics prior to randomization was compared for those who did and did not meet Schooler-Kane criteria at 2 consecutive visits in follow-up using a χ2 analysis. Only patients with at least 2 postbaseline visits were included in these analyses (n=143).
The original CATIE trial was designed to have a statistical power of 85% to identify an absolute difference of 12% in the rates of discontinuation between any 2 atypical agents. The current analyses represent a tertiary post hoc examination of outcomes specifically in patients with TD, which was not factored into the original design of the trial. However, if the rate of discontinuation due to any cause (primary outcome) was 10% higher in patients with TD (84%) compared to those without TD (74%), then our analysis was calculated to have a statistical power of 85% to detect that degree of difference given the sample sizes of 200 in the TD group and 977 in the non-TD group, using a 2-sided test with α = .05 (using nQuery Advisor; Statistical Solutions; Saugus, Massachusetts).
RESULTS
Patient Characteristics at Baseline
Compared with non-TD patients at baseline, patients with TD were significantly older, with longer antipsychotic treatment but fewer recent exacerbations; had higher ratings of symptom severity and acute extrapyramidal side effects, but lower neurocognitive scores; and were more likely to have received anticholinergics, antipsychotics in general, and first-generation antipsychotics (Table 1).
Table 1.
Demographic and Clinical Characteristics of Patients With and Without Tardive Dyskinesia (TD) at Baselinea
| Characteristic | TD (n = 200) | Non-TD (n = 997) | Non-TD, Restricted (n = 862)b | TD vs Non-TD
|
TD vs Non-TD (restricted)b
|
||||
|---|---|---|---|---|---|---|---|---|---|
| Test | df | P Value | Test | df | P Value | ||||
| Age, mean (SD), y | 47.2 (9.4) | 39.4 (11.0) | 38.7 (10.9) | T = 9.37 | 1195 | <.001 | T = 10.17 | 1060 | <.001 |
| Male, % | 79 | 72 | 73 | χ2 = 3.70 | 1 | .054 | χ2 = 2.85 | 1 | .091 |
| White, % | 65 | 60 | 60 | χ2 = 1.74 | 1 | .187 | χ2 = 1.60 | 1 | .207 |
| Hispanic, % | 13 | 12 | 13 | χ2 = 0.14 | 1 | .704 | χ2 = 0.00 | 1 | .967 |
| Years since first prescribed antipsychotic, mean (SD) | 21.6 (10.7) | 13.1 (10.1) | 12.6 (9.7) | T = 10.35 | 1125 | <.001 | T = 11.20 | 999 | <.001 |
| Exacerbation in previous 3 mo, % | 20 | 29 | 30 | χ2 = 7.16 | 1 | .007 | χ2 = 8.09 | 1 | .004 |
| Education, mean (SD), y | 12.1 (2.3) | 12.1 (2.3) | 12.1 (2,3) | T = 0.21 | 1189 | .835 | T = 0.25 | 1054 | .805 |
| PANSS score, mean (SD) | 78.8 (17.7) | 75.4 (17.3) | 75.3 (17.3) | T = 2.54 | 1185 | .011 | T = 2.58 | 1053 | .010 |
| Cognitive function, mean (SD) | −0.31 (0.96) | 0.05 (1.00) | 0.09 (0.98) | T = 4.40 | 1089 | <.001 | T = 4.86 | 965 | <.001 |
| Acute EPS, % | 12 | 3 | 3 | χ2 = 26.36 | 1 | <.001 | χ2 = 33.21 | 1 | <.001 |
| Anticholinergic, % | 27 | 15 | 14 | χ2 = 15.48 | 1 | <.001 | χ2 = 19.36 | 1 | <.001 |
| First-generation antipsychotic, % | 29 | 17 | 15 | χ2 = 13.83 | 1 | <.001 | χ2 = 17.13 | 1 | <.001 |
| Antipsychotic use prior to baseline, % | χ2 = 11.80 | 2 | .003 | χ2 = 14.19 | 2 | .001 | |||
| No antipsychotic | 25 | 28 | 29 | ||||||
| Atypical only | 45 | 52 | 53 | ||||||
| Typical (and possibly atypical) | 31 | 20 | 19 | ||||||
| Alcohol abuse, % | 34 | 30 | 30 | χ2 = 1.14 | 1 | .285 | χ2 = 1.18 | 1 | .278 |
| Drug abuse, % | 29 | 26 | 26 | χ2 = 0.84 | 1 | .361 | χ2 = 0.59 | 1 | .444 |
- For whites: non-TD, n = 995; non-TD (restricted), n = 860.
- For years since first prescribed antipsychotic: TD, n = 187; non-TD, n = 940, non-TD (restricted), n = 814.
- For education: TD, n = 199; non-TD, n = 992; non-TD (restricted), n = 857.
- For PANSS: non-TD, n = 987; non-TD (restricted), n = 855.
- For cognitive function: TD, n = 177; non-TD, n = 914; non-TD (restricted), n = 790.
- For acute EPS: non-TD, n = 991; non-TD (restricted), n = 859.
- For first-generation antipsychotic: TD, n = 185; non-TD, n = 839; non-TD (restricted), n = 723.
- For antipsychotic use prior to CATIE: TD, n = 195; non-TD, n = 980; non-TD (restricted), n = 849.
Excludes patients who did not meet modified Schooler-Kane criteria36 at baseline but had a history of TD or 1 item on the Abnormal Involuntary Movement Scale rated as mild.
Abbreviations: CATIE = Clinical Antipsychotic Trials of Intervention Effectiveness, EPS = extrapyramidal symptoms, PANSS = Positive and Negative Syndrome Scale.
Treatment Discontinuation
There was no significant difference between TD and non-TD groups in time to discontinuation for any cause adjusted for covariates (χ21= 0.11, P=.743) (Figure 1). There were also no significant differences between groups in time to discontinuation due to inefficacy (χ21= 1.87, P= .171), intolerability (χ21 = 0.01, P = .913), or patient decision (χ21 = 0.13, P = .722). The rates of discontinuation (74%) were identical between the groups (Table 2). The median time to discontinuation was 4.7 months for the TD group versus 5.4 months for the non-TD group.
Figure 1.
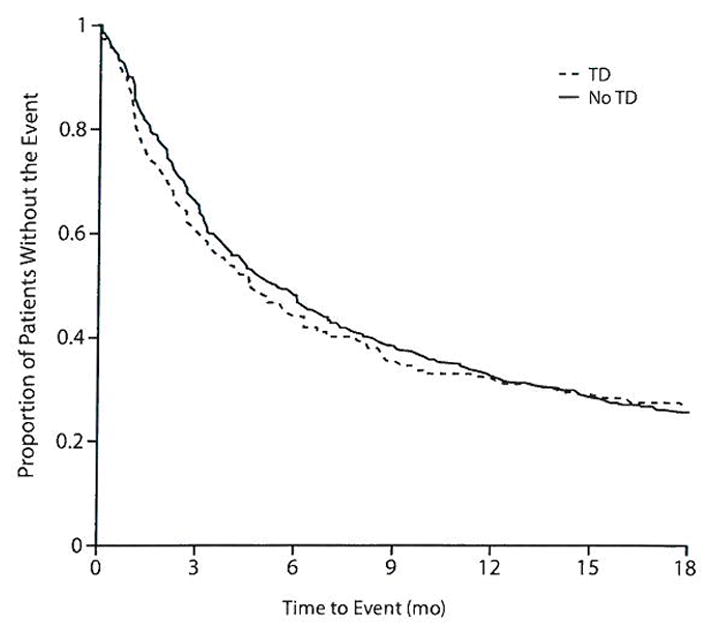
All-Cause Discontinuation Between Patients With and Without Tardive Dyskinesia (TD) at Baselinea
aP = .743 for difference between TD groups in model adjusting for treatment, age, baseline Positive and Negative Syndrome Scale score, investigator site, duration of illness, and antipsychotic medications taken prior to study entry.
Table 2.
Discontinuations by Baseline Tardive Dyskinesia (TD) Status and Treatment Group, n (%)
| Reason for Discontinuation | Olanzapine | Quetiapine | Risperidone | Ziprasidone | Total |
|---|---|---|---|---|---|
| TD | n = 54 | n = 62 | n = 56 | n = 28 | n = 200 |
| All | 31 (57) | 51 (82) | 44 (79) | 21 (75) | 147 (74) |
| Efficacy | 9 (17) | 17 (27) | 17 (30) | 5 (18) | 48 (24) |
| Tolerance | 8 (15) | 7 (11) | 8 (14) | 8 (29) | 31 (16) |
| Patient decision | 13 (24) | 25 (40) | 18 (32) | 8 (29) | 64 (32) |
| Non-TD | n = 281 | n = 274 | n = 285 | n = 157 | n = 997 |
| All | 184 (65) | 224 (82) | 208 (73) | 126 (80) | 742 (74) |
| Efficacy | 39 (14) | 75 (27) | 74 (26) | 39 (25) | 227 (23) |
| Tolerance | 54 (19) | 42 (15) | 26 (9) | 20 (13) | 142 (14) |
| Patient decision | 69 (25) | 88 (32) | 89 (31) | 55 (35) | 301 (30) |
| Non-TD (restricted)a | n = 233 | n = 241 | n = 250 | n = 138 | n = 862 |
| All | 151 (65) | 193 (80) | 183 (73) | 110 (80) | 637 (74) |
| Efficacy | 33 (14) | 64 (27) | 66 (26) | 35 (25) | 198 (23) |
| Tolerance | 43 (18) | 35 (15) | 23 (9) | 15 (11) | 116 (13) |
| Patient decision | 58 (25) | 76 (32) | 77 (31) | 48 (35) | 259 (30) |
Excludes patients who did not meet modified Schooler-Kane criteria36 at baseline but had a history of TD or 1 item on the Abnormal Involuntary Movement Scale rated as mild.
Tests of the interaction between TD and drug treatment group were not significant for time to discontinuation due to any cause (χ23 = 4.65, P =. 199), inefficacy (χ23 = 1.36, P = .716), or patient decision (χ23 = 2.30, P = .512), but did reveal a differential impact of treatment on the relationship between TD and time to discontinuation due to intolerability (χ23 =10.21, P=.017). Exploring this relationship for each treatment revealed shorter times to discontinuation for ziprasidone and risperidone than for olanzapine and quetiapine among patients with TD. However, after controlling for multiple comparisons, the hazard ratio (HR) for discontinuation due to intolerability in the TD group versus the non-TD group was significant only for ziprasidone (HR = 4.65, χ21= 7.21, P = .007). All of the above treatment discontinuation results between TD groups, and TD group–treatment interactions, were unchanged when the indeterminate patients were removed from the non-TD group in supportive analyses.
Although we did not find a statistically significant TD group–by-treatment interaction for all-cause discontinuation, we found that patients with TD at baseline showed a longer time to discontinuation for any cause on olanzapine compared to the other treatments (HR vs quetiapine [0.43; χ21 = 8.03, P = .005], risperidone [0.36; χ21= 12.47, P < .001], and ziprasidone [0.37; χ21= 8.28, P = .004]) (Figure 2). The median time to all-cause discontinuation for TD patients receiving olanzapine was 11.8 months compared with quetiapine (4.1 months), risperidone (3.2 months), and ziprasidone (2.6 months).
Figure 2.
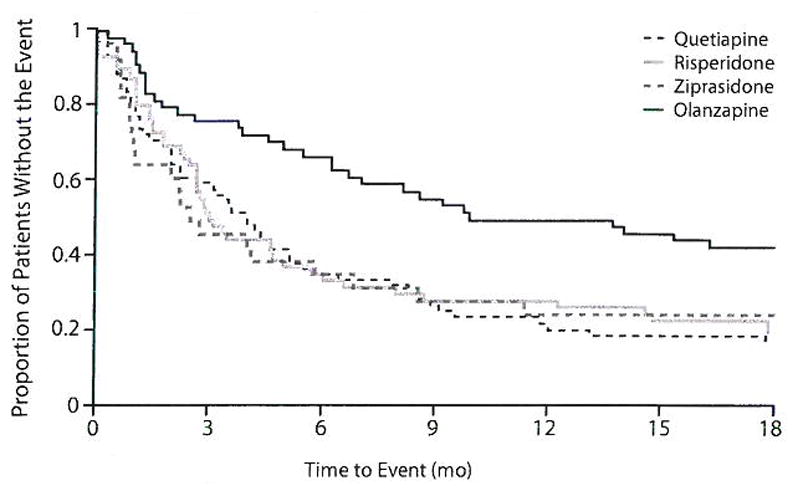
All-Cause Discontinuation by Treatment for Patients With Tardive Dyskinesia (TD) at Baselinea
aBelween-group differences significant for olanzapine vs quetiapine (P = .005), risperidone (P < .001), and ziprasidone (P = .004) in model adjusting for age, baseline Positive and Negative Syndrome Scale score, investigator site, duration of illness, and other antipsychotic medications taken prior to study entry.
Efficacy Measures
Analysis of change in the PANSS total score adjusted for covariates revealed no significant differences between TD and non-TD patient groups (F1,974 = 0.82, P =.366; Figure 3). Similarly, there were no differences for changes in positive (F1,914 = 0.03, P = .871) or negative (F1,988 = 0.27, P = .606) symptoms or general psychopathology (F1,923 = 0.10, P = .746). There were no significant TD group-by-treatment interactions for PANSS outcomes, and the results were consistent when patients with indeterminate TD were excluded.
Figure 3.
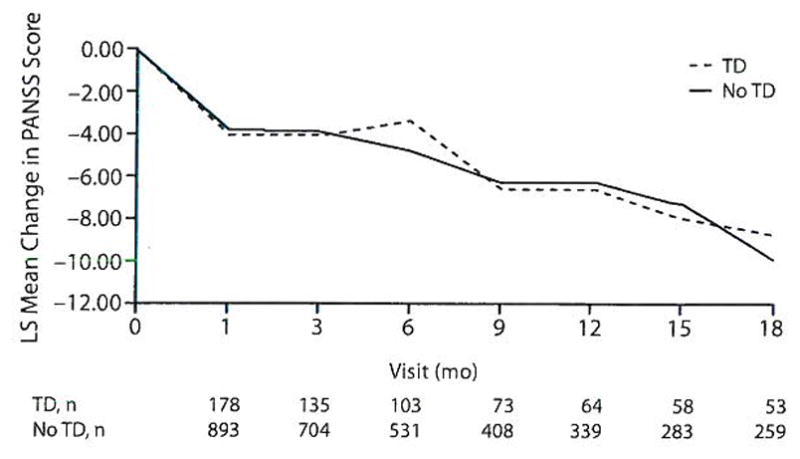
PANSS: Adjusted Repeated-Measures Model Between Patients With and Without Tardive Dyskinesia (TD) at Baselinea
aAdjusted P = .366 for overall TD effect. Model adjusted for baseline PANSS score, investigator site, treatment, age, duration of illness, and other antipsychotics taken prior to study entry.
Abbreviations: LS = least squares, PANSS = Positive and Negative Syndrome Scale.
We found a small but statistically significant difference in the adjusted mean change of the neurocognitive composite Z-score at 6 months when we compared TD (0.06) with non-TD (0.27) patients (F1,359 = 6.53, P = .011; Table 3). On subscores, non-TD patients showed significantly greater improvement in verbal memory (F1,359 = 4.17, P = .042) and a trend for greater improvement in processing speed (F1,359 = 3.69, P = .056). Results using the restricted non-TD group were similar, and there were no significant group-by-treatment interactions.
Table 3.
Model Adjusteda Mean Change in Cognitive Function at 6 Months for Patients With and Without Tardive Dyskinesia (TD) at Baselineb
| Measure | TD (n = 64), Adjusted Mean (SE) | Non-TD (n = 353), Adjusted Mean (SE) | TD vs Non-TD
|
||
|---|---|---|---|---|---|
| F | df | P Value | |||
| Cognitive composite (Z-score) change from baseline | 0.06 (0.08) | 0.27 (0.04) | 6.53 | 1,359 | .011 |
| Verbal memory | −0.17 (0.12) | 0.10 (0.06) | 4.17 | 1,359 | .042 |
| Working memory | 0.14 (0.10) | 0.18 (0.05) | 0.14 | 1,358 | .713 |
| Processing speed | 0.10 (0.08) | 0.26 (0.04) | 3.69 | 1,359 | .056 |
| Vigilance | 0.10 (0.12) | 0.31 (0.06) | 2.47 | 1,295 | .117 |
| Reasoning | 0.09 (0.10) | 0.22 (0.05) | 1.41 | 1,359 | .236 |
Model adjusted for baseline cognitive score, treatment, investigator site, race, duration of illness, education, depression at baseline, and acute extrapyramidal symptoms at baseline.
Means are based on number of patients with data available. Slight changes in sample sizes from those given in the column headings are as follows: working memory: TD, n = 63; vigilance: TD, n = 56; Non-TD, n = 297.
Dose Effects
Mean modal dose was not significantly different between TD groups after adjusting for covariates (F1,1054 = 0.33, P = .568); however, there was a significant TD group–by-treatment interaction (F3,1051 = 2.99, P = .030). We found a significantly higher adjusted mean modal dose of olanzapine for patients with TD (23.65 mg) versus those without TD (19.65 mg). This difference was noteworthy even when adjusted for covariates although not statistically significant when Bonferroni correction was applied (F1,296 = 6.12, P = .014). The overall percentage of patients who received the maximum daily dose of all treatments was nearly the same for those with and without TD (43% and 42%, respectively).
Course of TD
There was a significant reduction in adjusted total AIMS scores for all patients with TD at baseline (mean change at 18 months in total AIMS score = −2.38 ± 0.44, T52 = −5.44, P < .001), but treatment differences between second-generation antipsychotics were not statistically significant (F3,151 = 0.32, P = .811) (Figure 4). Similarly, the change in the AIMS global (F3,162 = 0.72, P=.544), distress (F3,146 = 0.81, P= .490), and impairment (F3,152 = 1.12, P = .343) item scores were not significantly different between treatments. The mean SAS score decreased from baseline to 18 months in the TD patients (−0.10), but this was not statistically significant (T52= 1.68, P = .099). Similarly, the BAS global score declined during the study (−0.32), and this change was statistically significant (T52 = −2.26, P = .028).
Figure 4.
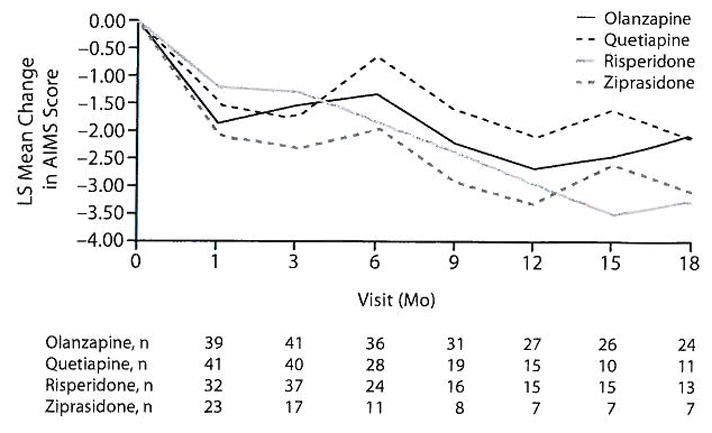
AIMS: Adjusted Repeated-Measures Model for Patients With Tardive Dyskinesia (TD) at Baselinea
aAdjusted P = .811 for overall treatment difference. Model adjusted for baseline AIMS score, baseline PANSS score, and duration of illness.
Abbreviations: AIMS = Abnormal Involuntary Movement Scale, LS= least squares, PANSS = Positive and Negative Syndrome Scale.
Analyzing patients with TD at baseline who had at least 2 follow-up visits (n = 143), we found that 78 (55%) met Schooler-Kane criteria at 2 consecutive visits postbaseline. Forty-nine (34%) met criteria for TD at all visits, while 35 (24%) did not meet criteria at any subsequent visit. The percentage of TD patients meeting criteria for TD postbaseline was not significantly different between the treatment groups (χ23 = 1.60, P =.659). Only 10 (7%) patients showed an increase in AIMS score of at least 50%, compared to 46 (32%) patients who showed a decrease of at least 50%.
The effects of prior antipsychotic use on persistence of TD were also explored for this subgroup of patients. We found no significant association between use of first-generation antipsychotics prior to randomization and TD during follow-up (χ21 = 0.00, P = .946). This lack of association was also noted for any prior antipsychotic use (χ21=1.41, P = .236).
DISCUSSION
We found no significant differences between schizophrenia patients with and without TD who were treated with one of 4 second-generation antipsychotics in the time to treatment discontinuation for all causes, patient decision, inefficacy, or intolerability. Although we found significantly greater symptom severity among patients with TD at baseline, we observed a similar degree of improvement in psychopathology compared with non-TD patients. However, we did find that patients with TD had lower neurocognitive scores at baseline and showed less improvement in cognition at 6 months. This suggests that TD may be associated with severe psychopathology, but that patients with TD are equally likely to respond to treatment with any one of these 4 second-generation agents as those without TD, except for cognition.
Thus, the findings from CATIE provide only partial support for the notion that TD in schizophrenia is associated with severe psychopathology, impaired cognition, and poor response to treatment.29,30,34,43 There are several reasons for discrepant findings between studies on this point. Measures of outcome differ; here, we chose treatment discontinuations in addition to symptom reduction as a practical measure that integrates both patient and clinician judgment. Patients in CATIE were older and chronically ill, with a long duration of prior exposure to antipsychotic drugs, compared with patients in some studies.29,31 However, the effects of age and duration of illness were adjusted for in the clinical analyses.
In addition, the association between TD, psychopathology, and poor response could be a treatment artifact reflecting the fact that patients with severe symptoms may receive higher drug doses for longer periods, with poor adherence, resulting secondarily in a greater incidence of TD. Indeed, patients with TD in our sample were older, had more severe symptoms at baseline, and had been treated for longer periods of time than non-TD patients, although these differences were also adjusted for in the analyses. Alternatively, the association between TD and acute extrapyramidal side effects could adversely affect treatment adherence, resulting in earlier discontinuations, poor symptom control, and relapse. In fact, TD patients had higher ratings of acute extrapyramidal side effects and were more likely to be receiving anticholinergics at baseline, the latter possibly contributing to impaired cognition as well.44 We reported previously that among all patients in the trial, 45 patients discontinued treatment for acute extrapyramidal side effects, whereas only 3 patients discontinued because of TD.45
It is of particular interest that we found no significant group-by-treatment interaction for all-cause time to discontinuation, indicating that the treatment differences previously published35 were not altered by TD status at baseline. Thus, the relatively longer duration of treatment among patients receiving olanzapine was replicated in the TD group. It is relevant to note that patients with TD received higher doses of olanzapine than patients without TD, although this was not significant when corrected for multiple comparisons. The reasons for this are unclear; patients with TD in general had more severe psychopathology at baseline, perhaps requiring higher doses, but this should apply to all treatment groups.28 Conversely, anticholinergic effects of higher doses of olanzapine could have unmasked TD in some patients, adding them to the TD group, but quetiapine was associated with the highest rate of anticholinergic symptoms and did not show a similar dose effect.35
Moreover, quetiapine was similar to risperidone and ziprasidone in overall effectiveness in both TD and non-TD groups. In Phase 1b of CATIE, Stroup et al46 reported that quetiapine and olanzapine were significantly more effective than risperidone in patients who discontinued perphenazine for intolerability and acute extrapyramidal symptoms. It was therefore surprising that patients with TD did not show a more robust response to quetiapine. This suggests that susceptible patients may be less tolerant of drugs likely to cause acute extrapyramidal symptoms, but, in comparison, TD by itself is unlikely to affect treatment discontinuation regardless of drug chosen, as found in our earlier analysis.45
Regarding the course of TD, we found that the mean total AIMS score decreased from baseline in patients with TD, although there were no significant differences in change of AIMS scores between the 4 second-generation agents at the dosages used in the trial (Figure 4). Similarly, there were no differences between treatments in changes in global ratings of TD, distress, or impairment of functioning.
The present findings are consistent with previous evidence on the variable course of TD. For the majority of our patients, TD symptoms were either persistent (34%) or fluctuating (42%) between visits, with few patients (7%) showing a significant increase in severity. Although difficult to compare, the changes in TD were within the range of previous studies of first-1,13,16 and second-generation drugs,19,23,25 with similar percentages of patients showing increases, reduction, or lack of diagnosable TD symptoms.
Although 45% of patients no longer met criteria for persistent TD, and 24% fell below criteria on all visits postbaseline, we cannot say whether these reductions represent reversal or simply suppression of TD. In many antipsychotic trials, patients with TD for whom rating scores decreased or fell below a threshold were termed “remitted,” implying reversibility of TD, but these cases are properly considered only as masked due to drug suppression.36 To distinguish remission from suppression, a few studies discontinued second-generation drugs, with some reporting continued absence of TD23,25 but others reporting unmasking and reappearance of TD.21,47 Thus, whether TD can resolve during maintenance treatment with any antipsychotic remains an open question. It would be difficult to design studies to test this question in schizophrenia patients, given the ethical quandaries raised by stopping medication treatment purposely and risking psychotic relapse. Although some investigators have proposed that suppression of TD is the result of increases in drug-induced parkinsonism,48 we found that ratings of parkinsonism also declined during the study, consistent with previous evidence that the antidyskinetic effect of antipsychotics is independent of their acute parkinsonian effects.13,21,22,49–51
There are a number of limitations to acknowledge. First, this analysis is post hoc because CATIE was not designed to study the course and correlates of TD. For example, modified diagnostic criteria for TD were applied at baseline, and a 3-month history of exposure was not required. We did not have comprehensive measures of previous treatment history or onset of drug-induced movement disorders. The response of TD could have been affected by switching from a prior antipsychotic medication to the randomized CATIE treatment assignment, resulting in withdrawal dyskinesias; however, cross-titration was allowed for up to 1 month, few cases of TD occurred in the first month of the trial,45 the 1 -month visit was excluded from postbaseline AIMS analyses, and treatment with antipsychotics prior to enrollment was found to have no significant effect on persistence of TD. As with other trials of antipsychotics, training to rate movement disorders was not as rigorous as for ratings of psychopathology, although the double-blind design should have resulted in all groups being affected equally. We did not have interrater reliability data across sites on the AIMS. We also did not control for or include changes in antipsychotic doses during the trial, which could affect severity of TD; however, correlations between dose and long-term course of TD have not been firmly established.1,13,52 In addition, the results of the study are specific for the drugs used at the doses in the trial, which may not apply to higher doses used in practice. Another limitation was the high rate of discontinuations, which resulted in relatively short duration of treatment and follow-up; length of follow-up has been associated with increasing rates of stabilization and improvement of TD.1,10 Nevertheless, the duration of treatment was comparable to those in prior studies of second-generation antipsychotics; for example, the average duration of the trials referenced in the review by Correll et al53 was only 8.8 months. Finally, there were no attempts at dose reduction or withdrawal in the trial, so we cannot say whether decreases in AIMS scores represented reversal or suppression of TD symptoms. Instead, we adopted a conservative view, describing only changes in scores.
We chose to assess only the 6-month time point for cognition, protecting against short-term effects of treatment switching at the 2-month visit and avoiding dilution of effects and statistical power from attrition at the 18-month visit. In addition, differences in cognition could have been affected by the greater use of anticholinergics at baseline among patients with TD.44 However, Keefe et al42 previously reported no significant difference in mean neurocognitive score changes during treatment between patients in the CATIE trial who did and did not receive anticholinergic medications at baseline and found no interaction between anticholinergics and treatment group for the composite score. We also found that a similar percentage of patients with (5.0%) and without (5.7%) TD first received anticholinergics within 6 months after the start of the trial. Longer duration of illness or greater age among TD patients could also affect neurocognitive results, but after covariate adjustment that considered these factors, TD patients still had significantly lower neurocognitive scores than non-TD patients.
Finally, the patient sample included primarily older patients with moderate symptoms and a chronic course with long-term exposure to antipsychotics, such that the findings may not apply to other ages or stages of schizophrenia. Furthermore, we cannot address relative effects of first- and second-generation drugs, because the study design did not allow for patients with TD to be assigned to perphenazine. The small numbers of patients in some treatment groups, especially for ziprasidone, limit conclusions. Lastly, clozapine may be particularly advantageous in treatment and dyskinesia outcomes for schizophrenia patients with TD but was not included in this phase of the study.
In conclusion, our findings suggest that while TD may be associated with more severe psychopathology and cognitive impairment in chronic schizophrenia, patients with existing TD are equally likely to respond to treatment with any of these 4 second-generation drugs compared to patients without TD, except for cognition. The prognosis of TD was variable between individuals, with the majority of patients showing persistence or fluctuations between visits. There were no significant differences between drug treatments on the course of TD at the dosages used in the trial. Few patients showed significant increases in TD ratings during treatment.
Acknowledgments
Funding/support: Supported by the Clinical Antipsychotic Trials of Intervention Effectiveness project, National Institute of Mental Health (NO1 MH90001). This article was based on results from the Clinical Antipsychotic Trials of Intervention Effectiveness project, supported by the National Institute of Mental Health (NO1 MH90001). The aim of this project is to examine the comparative effectiveness of antipsychotic drugs in conditions for which their use is clinically indicated including schizophrenia and Alzheimer’s disease. The project was carried out by principal investigators from Columbia University, the University of North Carolina, Duke University, the University of Southern California, the University of Rochester, and Yale University in association with Quintiles, Inc.; the program staff of the Division of Interventions and Services Research of the National Institute of Mental Health; and investigators from 57 sites in the United States (CATIE Investigators Group). AstraZeneca Pharmaceuticals LP, Bristol-Myers Squibb Company, Forest Pharmaceuticals, Inc., Janssen Pharmaceutica Products, L.P., Eli Lilly and Company, Otsuka Pharmaceutical Co., Ltd., Pfizer Inc., and Zenith Goldline Pharmaceuticals, Inc., provided medications for the studies. This material is based upon work also supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research Development, with resources and the use of facilities at the Philadelphia Veterans Affairs Medical Center.
Footnotes
Drug names: clozapine (Clozaril, FazaClo, and others), olanzapine (Zyprexa), quetiapine (Seroquel), risperidone (Risperdal and others), ziprasidone (Geodon).
Disclaimer: The content of this work does not represent the views of the Department of Veterans Affairs or the United States Government.
Potential conflicts of interest: Dr Caroff has been a consultant for Eli Lilly and has received grant/research support from Pfizer, Bristol-Myers Squibb, and Ortho-McNeil Neurologies. Dr V. G. Davis is an employee of the Collaborative Studies Coordinating Center in the Department of Biostatistics, University of North Carolina. Dr Miller has been a consultant for Otsuka, Hoffmann-La Roche, and Schering Plough and has received research funds from the National Institute of Mental Health and the Foundation for the National Institute of Health. Dr S. M. Davis is an employee of Quintiles. Dr Rosenheck has been a consultant for Eli Lilly, GlaxoSmithKline, Bristol-Myers Squibb, Organon, and Janssen; has received research support from Bristol-Myers Squibb, Eli Lilly, Janssen, AstraZeneca, and Wyeth; and provided expert testimony for the plaintiffs in UFCW Local 1776 and Participating Employers Health and Welfare Fund, et al vs Eli Lilly and Company; for the respondent in Eli Lilly Canada Inc vs Novapharm Ltd and Minister of Health, respondent; and for the Patent Medicines Prices Review Board, Canada, in the matter of Janssen Ortho Inc. and “Risperdal Consta.” Dr McEvoy has received research funding from AstraZeneca, Forest, Eli Lilly, Janssen, Pfizer, Sanofi-Aventis, and Dainippon Sumitomo; consulting or advisory board fees from Pfizer, Bristol-Myers Squibb, Indevus, and Eli Lilly; lecture fees from Janssen and Bristol-Myers Squibb; and honoraria from Eli Lilly. Dr Saltz has received research funds from Cephalon, AstraZeneca, and Novartis and lecture fees from Janssen and Novartis. Dr Chakos has been a consultant for Roche and has received grant/research support from Pfizer, Solvay, AstraZeneca, Janssen, Roche, and Organon. Dr Swartz has received consulting and education fees from AstraZeneca, Bristol-Myers Squibb, Pfizer, Eli Lilly, and Comprehensive Neuroscience and has received grant/research support and honoraria from Eli Lilly. Dr Keefe is currently receiving or in the past 12 months has received investigator-initiated research funding support from the National Institute of Mental Health, Allon, Novartis, and the Singapore National Medical Research Council and an unrestricted educational grant from AstraZeneca; and has received honoraria from or served as a consultant or advisory board member for Abbott, AstraZeneca, BiolineRx, Bristol-Myers Squibb, Cephalon, Dainippon Sumitomo, Eli Lilly, Johnson & Johnson, Lundbeck, Memory, Merck, Neurosearch, Orion, Orexigen, Otsuka, Pfizer, Roche, Targacept, Sanofi-Aventis, Shire, Wyeth, and Xenoport. In the past, he has received honoraria from or served as a consultant or advisory board member for Acadia, Cortex, Cyberonics, Forest, Gabriel, GlaxoSmithKline, Repligen, Saegis, and Schering Plough and has received research funding from AstraZeneca, Eli Lilly, Janssen, and Pfizer. He receives royalties from the Brief Assessment of Cognition in Schizophrenia (BACS) testing battery and the MATRICS Battery (BACS Symbol Coding). Dr Stroup has received research funding from Eli Lilly and consulting fees from Janssen, Eli Lilly, Lundbeck, Solvay, GlaxoSmithKline, and Bristol-Myers Squibb. Dr Lieberman has received research funding from AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Janssen, and Pfizer and consulting and educational fees from AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Forest, GlaxoSmithKline, Janssen, Novartis, Pfizer, and Solvay. Drs Campbell and Riggio report no financial or other relationship relevant to the subject of this article.
References
- 1.Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE, Gardos G, editors. Tardive Dyskinesia and Neuroleptics: From Dogma to Reason. Washington, DC: American Psychiatric Press, Inc; 1986. pp. 76–97. [Google Scholar]
- 2.American Psychiatric Association. Tardive Dyskinesia: A Task Force Report of the American Psychiatric Association. Washington, DC: American Psychiatric Association; 1992. [Google Scholar]
- 3.Barnes TR, Kidger T, Gore SM. Tardive dyskinesia: a 3-year follow-up study. Psychol Med. 1983;13(1):71–81. doi: 10.1017/s003329170005008x. [DOI] [PubMed] [Google Scholar]
- 4.Cavallaro R, Regazzetti MG, Mundo E, et al. Tardive dyskinesia outcomes: clinical and pharmacologic correlates of remission and persistence. Neuropsychopharmacology. 1993;8(3):233–239. doi: 10.1038/npp.1993.26. [DOI] [PubMed] [Google Scholar]
- 5.Chouinard G, Annable L, Ross-Chouinard A, et al. A 5-year prospective longitudinal study of tardive dyskinesia: factors predicting appearance of new cases. J Clin Psychopharmacol. 1988;8(suppl):21S–26S. [PubMed] [Google Scholar]
- 6.Halliday J, Farrington S, Macdonald S, et al. Nithsdale Schizophrenia Surveys 23: movement disorders. 20-year review. Br J Psychiatry. 2002;181(5):422–427. doi: 10.1192/bjp.181.5.422. [DOI] [PubMed] [Google Scholar]
- 7.Eberhard J, Lindström E, Levander S. Tardive dyskinesia and antipsychotics: a 5-year longitudinal study of frequency, correlates and course. Int Clin Psychopharmacol. 2006;21(1):35–42. doi: 10.1097/01.yic.0000182120.51672.7d. [DOI] [PubMed] [Google Scholar]
- 8.Modestin J, Wehrli MV, Stephan PL, et al. Evolution of neuroleptic-induced extrapyramidal syndromes under long-term neuroleptic treatment. Schizophr Res. 2008;100(1–3):97–107. doi: 10.1016/j.schres.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 9.Barron ET, McCreadie RG. One year follow-up of tardive dyskinesia. Br J Psychiatry. 1983;143:423–424. [PubMed] [Google Scholar]
- 10.Gardos G, Casey DE, Cole JO, et al. Ten-year outcome of tardive dyskinesia. Am J Psychiatry. 1994;151(6):836–841. doi: 10.1176/ajp.151.6.836. [DOI] [PubMed] [Google Scholar]
- 11.Richardson MA, Casey DE. Tardive dyskinesia status: stability or change. Psychopharmacol Bull. 1988;24(3):471–475. [PubMed] [Google Scholar]
- 12.Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: The Fourth Generation of Progress. New York, NY: Raven Press, Ltd; 1995. pp. 1485–1495. [Google Scholar]
- 13.Jeste DV, Wyatt RJ. Therapeutic strategies against tardive dyskinesia: two decades of experience. Arch Gen Psychiatry. 1982;39(7):803–816. doi: 10.1001/archpsyc.1982.04290070037008. [DOI] [PubMed] [Google Scholar]
- 14.Glazer WM, Morgenstern H, Doucette JT. The prediction of chronic persistent versus intermittent tardive dyskinesia: a retrospective follow-up study. Br J Psychiatry. 1991;158(6):822–828. doi: 10.1192/bjp.158.6.822. [DOI] [PubMed] [Google Scholar]
- 15.Rosenheck RA. Evaluating the cost-effectiveness of reduced tardive dyskinesia with second-generation antipsychotics. Br J Psychiatry. 2007;191(3):238–245. doi: 10.1192/bjp.bp.106.035063. [DOI] [PubMed] [Google Scholar]
- 16.Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583–609. doi: 10.1093/schbul/23.4.583. [DOI] [PubMed] [Google Scholar]
- 17.Kane JM, Woerner M, Borenstein M, et al. Integrating incidence and prevalence of tardive dyskinesia. Psychopharmacol Bull. 1986;22(1):254–258. [PubMed] [Google Scholar]
- 18.Glazer WM, Morgenstern H, Doucette JT. Predicting the long-term risk of tardive dyskinesia in outpatients maintained on neuroleptic medications. J Clin Psychiatry. 1993;54(4):133–139. [PubMed] [Google Scholar]
- 19.Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158(4):503–510. doi: 10.1192/bjp.158.4.503. [DOI] [PubMed] [Google Scholar]
- 20.Tarsy D, Baldessarini RJ. Epidemiology of tardive dyskinesia: is risk declining with modern antipsychotics? Mov Disord. 2006;21(5):589–598. doi: 10.1002/mds.20823. [DOI] [PubMed] [Google Scholar]
- 21.Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl) 1978;56(1):75–80. doi: 10.1007/BF00571412. [DOI] [PubMed] [Google Scholar]
- 22.Chouinard G. Effects of risperidone in tardive dyskinesia: an analysis of the Canadian multicenter risperidone study. J Clin Psychopharmacol. 1995;15(suppl 1):36S–44S. doi: 10.1097/00004714-199502001-00007. [DOI] [PubMed] [Google Scholar]
- 23.Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985–996. doi: 10.1016/j.pnpbp.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 24.Rosenheck R, Perlick D, Bingham S, et al. Department of Veterans Affairs Cooperative Study Group on the Cost-Effectiveness of Olanzapine. Effectiveness and cost of olanzapine and haloperidol in the treatment of schizophrenia: a randomized controlled trial. JAMA. 2003;290(20):2693–2702. doi: 10.1001/jama.290.20.2693. [DOI] [PubMed] [Google Scholar]
- 25.Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102–106. [PubMed] [Google Scholar]
- 26.Tenback DE, van Harten PN, Slooff CJ, et al. SOHO Study Group. Effects of antipsychotic treatment on tardive dyskinesia: a 6-month evaluation of patients from the European Schizophrenia Outpatient Health Outcomes (SOHO) Study. J Clin Psychiatry. 2005;66(9):1130–1133. [PubMed] [Google Scholar]
- 27.Claus A, Bollen J, De Cuyper H, et al. Risperidone versus haloperidol in the treatment of chronic schizophrenic inpatients: a multicentre double-blind comparative study. Acta Psychiatr Scand. 1992;85(4):295–305. doi: 10.1111/j.1600-0447.1992.tb01473.x. [DOI] [PubMed] [Google Scholar]
- 28.Miller DD, McEvoy JP, Davis SM, et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophr Res. 2005;80(1):33–43. doi: 10.1016/j.schres.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 29.Lieberman JA, Alvir J, Geisler S, et al. Methylphenidate response, psycho-pathology and tardive dyskinesia as predictors of relapse in schizophrenia. Neuropsychopharmacology. 1994;11 (2):107–118. doi: 10.1038/npp.1994.40. [DOI] [PubMed] [Google Scholar]
- 30.Ascher-Svanum H, Zhu B, Faries D, et al. Tardive dyskinesia and the 3-year course of schizophrenia: results from a large, prospective, naturalistic study. J Clin Psychiatry. 2008;69(10):1580–1588. doi: 10.4088/jcp.v69n1008. [DOI] [PubMed] [Google Scholar]
- 31.Yarden PE, Discipio WJ. Abnormal movements and prognosis in schizophrenia. Am J Psychiatry. 1971;128(3):317–323. doi: 10.1176/ajp.128.3.317. [DOI] [PubMed] [Google Scholar]
- 32.Chong SA, Tay JA, Subramaniam M, et al. Mortality rates among patients with schizophrenia and tardive dyskinesia. J Clin Psychopharmacol. 2009;29(1):5–8. doi: 10.1097/JCP.0b013e3181929f94. [DOI] [PubMed] [Google Scholar]
- 33.Kelly DL, Weiner E, Ball MP, et al. Remission in schizophrenia: the relationship to baseline symptoms and changes in symptom domains during a one-year study. J Psychopharmacol. 2009;23(4):436–441. doi: 10.1177/0269881108093883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crow TJ. The two-syndrome concept: origins and current status. Schizophr Bull. 1985;11(3):471–486. doi: 10.1093/schbul/11.3.471. [DOI] [PubMed] [Google Scholar]
- 35.Lieberman JA, Stroup TS, McEvoy JP, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 36.Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486–487. doi: 10.1001/archpsyc.1982.04290040080014. [DOI] [PubMed] [Google Scholar]
- 37.Guy W. Abnormal Involuntary Movement Scale (AIMS). ECDEU Assessment Manual for Psychopharmacology. US Department of Health, Education and Welfare publication (ADM) 76–338. Alcohol, Drug Abuse, and Mental Health Administration. Rockville, MD: National Institute of Mental Health Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
- 38.Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261–276. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- 39.Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl. 1970;45(S212):11–19. doi: 10.1111/j.1600-0447.1970.tb02066.x. [DOI] [PubMed] [Google Scholar]
- 40.Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672–676. doi: 10.1192/bjp.154.5.672. [DOI] [PubMed] [Google Scholar]
- 41.Keefe RS, Bilder RM, Harvey PD, et al. Baseline neurocognitive deficits in the CATIE schizophrenia trial. Neuropsychopharmacology. 2006;31(9):2033–2046. doi: 10.1038/sj.npp.1301072. [DOI] [PubMed] [Google Scholar]
- 42.Keefe RS, Bilder RM, Davis SM, et al. CATIE Investigators; Neurocognitive Working Group. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE trial. Arch Gen Psychiatry. 2007;64(6):633–647. doi: 10.1001/archpsyc.64.6.633. [DOI] [PubMed] [Google Scholar]
- 43.Tenback DE, van Harten PN, Slooff CJ, et al. SOHO Study Group. Worsening of psychosis in schizophrenia is longitudinally associated with tardive dyskinesia in the European Schizophrenia Outpatient Health Outcomes study. Compr Psychiatry. 2007;48(5):436–440. doi: 10.1016/j.comppsych.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 44.Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055–1062. doi: 10.1176/appi.ajp.2009.09010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller DD, Caroff SN, Davis SM, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279–288. doi: 10.1192/bjp.bp.108.050088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stroup TS, Lieberman JA, McEvoy JP, et al. CATIE Investigators. Effectiveness of olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia after discontinuing perphenazine: a CATIE study. Am J Psychiatry. 2007;164(3):415–427. doi: 10.1176/ajp.2007.164.3.415. [DOI] [PubMed] [Google Scholar]
- 47.Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472–477. doi: 10.4088/jcp.v59n0906. [DOI] [PubMed] [Google Scholar]
- 48.Fernandez HH, Krupp B, Friedman JH. The course of tardive dyskinesia and parkinsonism in psychiatric inpatients: 14-year follow-up. Neurology. 2001;56(6):805–807. doi: 10.1212/wnl.56.6.805. [DOI] [PubMed] [Google Scholar]
- 49.Beasley CM, Jr, Tollefson G, Tran P, et al. Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology. 1996;14(2):111–123. doi: 10.1016/0893-133X(95)00069-P. [DOI] [PubMed] [Google Scholar]
- 50.Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696–701. doi: 10.4088/jcp.v65n0516. [DOI] [PubMed] [Google Scholar]
- 51.Hirsch SR, Kissling W, Bauml J, et al. A 28-week comparison of ziprasidone and haloperidol in outpatients with stable schizophrenia. J Clin Psychiatry. 2002;63(6):516–523. doi: 10.4088/jcp.v63n0609. [DOI] [PubMed] [Google Scholar]
- 52.Rosenheck RA, Davis VG, Davis SM, et al. Can a nonequivalent choice of dosing regimen bias the results of flexible dose double blind trials? the CATIE schizophrenia trial. Schizophr Res. 2009;113(1):12–18. doi: 10.1016/j.schres.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414–425. doi: 10.1176/appi.ajp.161.3.414. [DOI] [PubMed] [Google Scholar]