

Abstract
Since dendritic cells operate as professional antigen-presenting cells (APCs) and hence are capable of jumpstarting the immune system, they have been exploited to develop a variety of immunotherapeutic regimens against cancer. In the few past years, myeloid-derived suppressor cells (MDSCs) have been shown to mediate robust immunosuppressive functions, thereby inhibiting tumor-targeting immune responses. Thus, we propose that the immunomodulatory activity of MDSCs should be carefully considered for the development of efficient anticancer immunotherapies.
Keywords: antigen presentation, cancer, dendritic cells, myeloid-derived suppressor cell, T cells
Introduction
Cancer is caused by the uncontrolled growth of transformed cells, initially forming a localized (primary) lesion and then colonizing distant organs (a process that is known as metastatic dissemination). Both these manifestations of cancer can significantly interfere with the physiological functions of the organism. Malignant cells exhibit consistent alterations in protein expression, survival and proliferation, mostly originating from the accumulation of mutations (reflecting a high degree of genetic instability) and epigenetic changes. Thus, neoplastic cells usually express an array of mutated proteins that provides them with some degree of immunogenicity (quasi antigens). At least theoretically, such an acquired immunogenicity allows the immune system to identify and destroy cancer cells.
However, the immune system must overcome 2 major obstacles to effectively fight cancer. The first of such barriers is represented by the standard immunological tolerance toward self antigens, which impedes the activation of quasi-antigen-specific T cells. The second barrier stems from various immunosuppressive mechanisms set in place by neoplastic cells, which are largely responsible for the failure of conventional immunotherapeutic anticancer regimens. Such a systemic state of immunosuppression is caused by the expansion of potent immunomodulatory cells, including (but not limited to) myeloid-derived suppressor cells (MDSCs). Anticancer immunotherapy thus attempts at overcoming these obstacles by stimulating the immune system through a variety of procedures and interventions. In this regard, dendritic cell (DC)-based approaches deserve special attention. The refinement of ex vivo DC expansion protocols has boosted the development of several immunotherapeutic strategies against cancer, as DCs operate as central controllers of innate and adaptive responses. However, the administration of DCs loaded with tumor-associated antigens (TAAs) or previously treated with potent immunostimulants to cancer patients results in limited therapeutic responses, especially as compared with the expectations raised by preclinical data. Here, we review the systems that are currently available for the ex vivo evaluation of immunotherapeutic anticancer regimens, as well as the reasons for the limited predictive value of preclinical results obtained with this approach. We propose that testing immunomodulatory strategies ex vivo should focus not only on DCs but also on the immunosuppressive cells that are found in the tumor microenvironment, hence faithfully mimicking physiological conditions. This might allow for the identification (and further development) of immunotherapeutic regiments that are truly capable of overcoming the immunosuppressive effects of the tumor microenvironment.
Recognition of Tumor-Associated Antigens by the Immune System
Cancer immunotherapy relies on activating or boosting pre-existing tumor-specific cytotoxic T lymphocyte (CTL) responses. Generally, antigenic peptides are recognized by specific T-cell receptors (TCRs) once complexed with MHC molecules and exposed on the surface of antigen-presenting cells (APCs) (Fig. 1). The specific recognition of MHC-peptide complexes by TCRs results in the delivery of a first activating signal to T lymphocytes. However, such a signal is insufficient for the activation of T cells, implying that APCs must trigger additional signaling events to elicit antigen-specific immune responses.1 These signals are delivered by the binding of a wide range of ligands to specific receptors, which can be stimulatory or inhibitory, expressed on the surface of T cells. The integration of these signal transduction cascades regulate the degree of T-cell activation.2 A classical co-stimulatory interaction is represented by the binding of CD80, which is expressed on DCs, to CD28, which is found at the surface of T cells. Conversely, a strong inhibitory signal is delivered upon the interaction of CD80 with cytotoxic T lymphocyte-associated protein 4 (CTLA4) as well as upon the binding of programmed cell death 1 (PDCD1) ligand 1 (PD-L1, official name CD274) with PDCD1 (best known as PD-1) (Reviewed in Refs. 1, 3). A variety of additional ligand-receptor interactions can regulate T-cell co-stimulation (Fig. 1). Finally, cytokines that are present in the microenvironment where antigen presentation occurs confer T cells with specific effector functions, mainly through the differentiation of specific CD4+ helper T-cell subtypes (Reviewed in ref. 1).

Figure 1. Activation of T cells by antigen-presenting cells. Antigenic peptides (rhomboids) complexed with MHC molecules on the surface of dendritic cells (DCs) are recognized by cognate T-cell receptors (TCRs), delivering a first activatory signal (signal 1) to T cells (top). A second signal (signal 2) is delivered to T cells upon the integration of positive (activatory) and negative (inhibitory) co-stimulation, originating from the interaction of specific receptors expressed on the T-cell surface and their ligands. A third signal (signal 3) is delivered by cytokines found in the microenvironment where antigen presentation occurs, which are often secreted by antigen-presenting DCs. Such a cytokine priming generally directs the polarization of T-cell responses. CD40L, CD40 ligand; CTLA4, cytotoxic T lymphocyte-associated protein 4; PD-1, programmed cell death 1; PD-L1, PD-1 ligand 1; PD-L1, PD-1 ligand 2.
The immune responses against infectious agents rely on the recognition of microbial molecules via pattern recognition receptors (PRRs) expressed by professional APCs, including DCs. The binding of such microbe-associated molecular patterns (MAMPs) to PRRs favors the maturation of DCs, resulting in the upregulation of co-stimulatory molecules, enhanced cytokine secretion and increased expression of MHC molecules (Reviewed in ref. 4). As they mature, DCs migrate to secondary lymphoid tissues where they present antigenic peptides to antigen-specific T cells. However, most TAAs are either aberrantly overexpressed self proteins or quasi-antigens. Thus, the frequency of circulating TAA-specific T cells is low, mostly because of their efficient removal by clonal deletion in the thymus (central tolerance). The autoreactive T cells that survive thymic selections either bear TCRs that display a low affinity for cognate antigens or differentiate into regulatory T cells (Tregs).5 Nonetheless, the immune system is capable of recognizing, controlling, and eliminating cancer cells.6,7 Thus, the efficient activation of autoreactive TAA-specific T cells as well as the inhibition of the immunosuppressive activity of Tregs stand out as key goals for anticancer immunotherapy. Another significant challenge in this context is to neutralize the profound state of systemic immunosuppression that characterizes cancer patients with a large tumor burden. Tumors actively secrete a variety of molecules that act in the bone marrow to divert myeloid differentiation, resulting in the accumulation of MDSCs.8 MDSCs are able to exit the bone marrow, distribute to peripheral organs, and actively infiltrate neoplastic lesions, hence inhibiting antitumor immune responses via both antigen-specific and non-specific mechanisms.9-11 The expansion of MDSCs in cancer patients is responsible (at least for a large part) for the inefficacy of standard immunotherapeutic regimens. Thus, MDSCs have attracted great interest from the fields of experimental and clinical tumor immunology.12 In summary, an ideal immunotherapeutic intervention against cancer would have to (1) stimulate the presentation of TAAs to T cells, while (2) counteracting the immunosuppressive activity of Tregs and MDSCs.
The Discovery of Dendritic Cells and their Impact in Biomedical Research
After the recent award of the Nobel Prize to Ralph Steinman (for the first time in history, posthumously), it is worthy to briefly comment on the impact that his work had on immunology. Indeed, although Langerhans cells (a particular type of DCs) had previously been described by Langerhans, the discovery of conventional DCs can be attributed to Steinman,13 who in 1973 demonstrated the capacity of these cells to either strongly activate14 or inhibit T cell-mediated immune responses.15,16 The characteristics of each DC lineage described since have extensively been described elsewhere.17
The line of investigation focusing on DC biology has significantly been stimulated in the mid-1990s, when systems for the differentiation of myeloid DCs ex vivo were first developed. These protocols relied on the murine bone marrow or purified monocytes as a starting material, and on granulocyte macrophage colony-stimulating factor (GM-CSF) as a central differentiation stimulus.18,19 Within 7 y from the publication of ex vivo DC differentiation protocols, the number of papers on DCs had increased from about 80 to approximately 500 per y, and this figure climbed further to 1000 per y 10 ys afterwards (Fig. 2). Because of its simplicity and reproducibility, the production of DCs ex vivo has de facto revolutionized the study of DC biology.
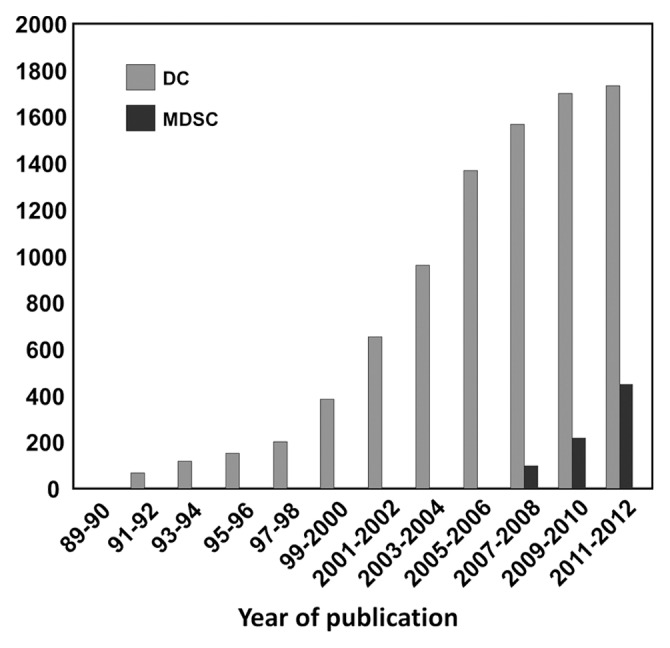
Figure 2. Number of publications dealing with conventional dendritic cells and myeloid-derived suppressor cells. The approximate number of publications dealing with conventional dendritic (DCs) or myeloid-derived suppressor cells (MDSCs), as retrieved by searching PubMed (http://www.ncbi.nlm.nih.gov/pubmed) for entries whose title contains the term “dendritic cell” or “myeloid-derived suppressor cells,” is represented as a function of publication biennium.
The differentiation of myeloid DCs ex vivo presents many advantages for biomedical research. First, cell numbers are not a limiting factor in this context. Indeed, close to 50 x 106 DCs can be easily obtained from the material collected from a single mouse within 1 wk. Although the human system is comparatively less efficient, mostly owing to a limited initial supply of peripheral blood mononuclear cells (PBMCs), it is still an amenable protocol of key clinical relevance. During differentiation, DCs tend to mature, implying that experimental assessments can easily be performed at different DC maturation stages. The large numbers of DCs that can be obtained ex vivo are compatible with their use for the systematic monitoring of immunomodulatory agents. DC cultures also provided a means to thoroughly study DC responses to large collections of maturation stimuli, and in particular how these agents influence intracellular signal transduction and antigen presentation by DCs (reviewed in ref. 4). These studies have generated a significant amount of experimental evidence in support of the so-called “danger signal” model, originally put forward by Charles Janeway and Polly Matzinger to explain the general regulation of immune responses.20,21 This model de facto establishes a link between innate and adaptive immunity. In brief, it postulates that APCs possess receptors for the recognition of a wide range of molecules that can be found in microbial products (MAMPs) as well as in cellular components that are released in the course of inflammation or trauma (the so-called damage-associated molecular patterns, DAMPs). The recognition of MAMPs and DAMPs triggers the phenotypic and functional maturation of DCs, resulting in the upregulation of both MHC and co-stimulatory molecules. Thus, only when professional APCs encounter danger signals of this type, their antigen-presenting capability is strongly improved. Although the danger model has weak points, it is both elegant and simple. The development of protocols for the differentiation of DCs ex vivo has allowed for the systematic study of the effects of MAMPs and DAMPs on DCs, in terms of functional responses and intracellular signaling (Reviewed in ref. 22). Along similar lines, the refinement of procedures for the co-culture of DCs and T cells has allowed for the dissection of the cellular/molecular mechanisms of antigen-presentation and T-cell polarization, as recently reviewed by Liechtenstein and colleagues.1
Dendritic Cell-Based Vaccines in Anticancer Immunotherapy
The possibility to differentiate DCs ex vivo in large-scale had an immediate therapeutic application: the development of anticancer vaccines based on these professional APCs. DCs are key regulators of immune responses and are possibly the immune cells with the most prominent adjuvant effects. Immature DCs exhibit an intense phagocytic activity, which provides them with a consistent amount of antigenic peptides, including TAAs, for loading on MHC molecules.17 Furthermore, DCs are highly susceptible to genetic engineering via viral vectors, including adenoviral, retroviral, lentiviral, and poxviral particles, as well as non-viral systems.17,23 Finally, the maturation state of DCs can be manipulated to boost or suppress immune responses.22 These APCs are therefore ideal vaccines for anticancer immunotherapy. DCs generated ex vivo have indeed been extensively employed to assess T-cell responses to infectious agents in vitro24-27 and harnessed for the development of anticancer immunotherapeutic regimens.28-34 Moreover, DCs have been successfully used to suppress immune responses, in both animal models and humans.15,16,35-40
Undoubtedly, the refinement of DC production systems has promoted the use of DCs in various clinical settings, including anticancer immunotherapy.41 Since these methods have become part of the research routine, scientists generally follow the same pipeline for developing new antineoplastic interventions, which involves the following steps: (1) treatment of DC cultures with a specific agent, to assess its ability to stimulate DC maturation or alter cytokine secretion; (2) assessment of how this agent influence DC-mediated antigen presentation to T cells, ex vivo; (3) testing of DCs in vivo, in experimental models that allow for the assessment of T-cell responses, and (4) testing of DCs activated according to optimal protocols in therapeutic/preventive experimental tumor models, in vivo.
Undoubtedly, a sizeable amount of preclinical research based on DCs amplified ex vivo has generated interesting insights into the immunobiology of DCs, often highlighting promising therapeutic activity.17,42-45 Surprisingly, however, these encouraging preclinical results have not always been translated into a clinical success in cancer patients.46,47
Why are there such significant discrepancies between preclinical and clinical data on the immunotherapeutic anticancer profile of DCs? At least in part, this reflects the conditions employed to assess the antigen-presenting capacities of DCs in vitro, which are highly controlled. While this is certainly advantageous from an experimental point of view, this experimental setting may not appropriately reproduce the conditions in which tumor-specific immune responses occur. For instance, these assays often rely on well-defined, immunogenic TAAs or even model xenoantigens, such as ovalbumin (OVA).33,48,49 The use of tumor lysates to load DCs for clinical applications may also be disadvantageous, as tumor lysates (1) contain both defined and undefined TAAs, and (2) are generally tolerogenic, hence inhibiting the therapeutic activity of DCs unless additional immunostimulatory agents are also employed.50-56 Finally, upon infusion, DCs must counteract the strongly immunosuppressive state of cancer patients. This is perhaps the most prominent factor accounting for the discrepancy between the preclinical and clinical immunotherapeutic activity of DCs against cancer.
From an experimental point of view, much effort and resources would be saved if candidate treatments were appropriately assessed early in the course of preclinical development. To this aim, it would be ideal to either establish DC-T cell co-culture assays in (immunosuppressive) conditions that closely resemble the tumor microenvironment, or utilize the immunomodulatory myeloid counterpart of DCs that robustly infiltrate neoplastic lesions: MDSCs.
MDSCS: Discovery and Definition
The dissemination of malignant cells from the primary lesion is in itself a key pathological feature of cancer. In addition, a major complication for cancer patients, especially individuals in the late stages of disease (when tumor load is generally consistent), is their state of systemic immunosuppression, which prevents the immune system from eliminating transformed cells. Growing neoplasms produce indeed a wide range of cytokines and metabolites that alter the differentiation of myeloid cells, facilitating the accumulation of cell populations exerting strong immunosuppressive effects.57 Myeloid cells including tolerogenic DCs, tumor-infiltrating macrophages and granulocytes have been known for a long time to play an important role in various aspects of tumor progression, including neoangiogenesis. However, only recently specific subsets of myeloid cells have been identified as specialized immunosuppressive cells that accumulate in cancer patients.58,59 The recognition of MDSCs as a specific cell lineage remains rather controversial. Still, recent experimental evidence indicates that MDSCs differ from other myeloid cells in many aspects. MDSCs comprise indeed a heterogeneous population of cells exhibiting cancer-specific phenotypic and functional characteristics. In mice, MDSCs were originally described as myeloid CD11b+ cells that express high levels of the granulocyte-specific epitope GR1. GR1 is shared by 2 surface markers, namely, lymphocyte antigen 6 complex, locus C1 (Ly6C1) and lymphocyte antigen 6 complex, locus G (Ly6G). Thus, based on the relative expression levels of these markers, murine MDSCs can be classified into a monocytic (Ly6C1highLy6Gneg/low) and granulocytic (Ly6C1highLy6Ghigh) subsets. As such, monocytic and granulocytic MDSCs can also be identified in cancer patients.60 These cells, when isolated from tumor-bearing hosts, exhibit a pronounced capacity to functionally inhibit T cells, via both antigen-specific and non-specific mechanisms.61 The immunosuppressive functions of MDSCs mainly originate from the expression of enzymes that deplete the extracellular microenvironment of essential amino acids, such as inducible nitric oxide synthase (iNOS), indoleamine 2,3-deoxygenase (IDO) and arginase 1, as well as from the secretion of immunosuppressive cytokines such as interleukin (IL)-10 and transforming growth factor β (TGFβ).61 The growing importance of MDSCs in biomedical research is well represented by the fact that, following their “official” definition, the number of papers dealing with these cells is steadily increasing (Fig. 2).
Differentiation of MDSCs Ex Vivo
As mentioned above, the establishment of a protocol for the amplification of conventional DCs ex vivo provided a significant boost to the corresponding area of biomedical research. The development of a similar system for the differentiation and amplification of MDSCs could hence entail another significant step forward. However, the current systems for the ex vivo differentiation of MSDCs are inefficient and fail to achieve significant levels of amplification. In addition, MDSCs isolated from tumor-bearing mice generally do not proliferate ex vivo in the presence of GM-CSF, and their survival is thus severely compromised.62 Considering the reports published so far, GM-CSF stands out as a key factor for MDSC differentiation, in vitro and in vivo. This is particularly interesting in view of the fact that GM-CSF-based therapies are currently used in patients affected by some types of neoplasms as an immunostimulatory regimen.63-66 Indeed, taking into consideration the key role of GM-CSF in the tumor-associated expansion of MDSCs, the administration of this cytokine as a standalone intervention might not be an ideal choice for anticancer immunotherapy, at least in some circumstances.67-69
One of the first methods to obtain murine MDSCs from the bone marrow relied on high concentrations of GM-CSF coupled to lipopolysaccharide (LPS), a protocol allowing for the expansion of highly immature myeloid cells with robust immunosuppressive activities.57 Possibly, these cells represented activated MDSCs, although nomenclature guidelines had not yet been formulated at that time. Bone marrow-derived “immature” myeloid cells obtained with GM-CSF and LPS exerted strong immunosuppressive effects upon cell-to-cell contact (antigen-specific immunosuppression) and by expressing high levels of iNOS (non-specific immunosuppression). In line with these observations, the production of GM-CSF by breast carcinoma cells has soon been identified as one of the key drivers of MDSC expansion.70 In this context, MDSCs could be generated upon the addition of GM-CSF to the bone marrow, and similar results could be obtained by using culture medium conditioned by breast carcinoma cells. Still, the authors of this paper only obtained 28% of differentiated cells exhibiting a CD11b+GR1+ phenotype, of which Ly6Ghigh cells did not exert immunosuppressive effects.70 Moreover, the addition of GM-CSF-neutralizing antibodies did not completely abrogate the differentiation of MDSCs, suggesting that additional factors are required for the accumulation of these cells in cancer patients. Nevertheless, GM-CSF stands out as an important MDSC-polarizing cytokine produced by many cancer cells.57,70,71
The addition of recombinant GM-CSF and IL-4 to culture media conditioned by various cancer cell lines (including EL4 lymphoma, LLC lung carcinoma, B16-F10 melanoma and C3 cervical carcinoma cells) reportedly induces the differentiation of MDSCs, correlating with the immunosuppressive effects exerted by tumor cells in vivo. However, also in this case the differentiation and proliferation of MDSCs ex vivo were rather inefficient. Indeed, relative amounts of MDSCs higher than 25–30% were hardly reached after 5 d of culture.9
A recent study has identified GM-CSF, granulocyte colony-stimulating factor (G-CSF) and IL-6 as factors that drive the differentiation of MDSCs from the bone marrow. In this context, the co-administration of GM-CSF and IL-6 combination generated MDSCs exerting robust immunosuppressive functions. Nonetheless, also the authors of this study pointed out that the recovery rate of MDSCs was comparable with the number of bone marrow cells initially plated.72 Thus, in this setting myeloid cell precursors had lost their proliferative capacity, differentiated and acquired potent immunosuppressive functions. Along similar lines, the differentiation of human MDSCs has been achieved upon 1 wk of incubation of CD33+ mononuclear cells with GM-CSF and IL-6.73 Other cytokines found within neoplastic lesions and in the supernatants of cultured cancer cells have been shown to contribute to MDSC differentiation, although to a minor extent.73 The authors of this study did not comment on the efficiency of differentiation, but pointed out that their cytokine cocktails also promoted the expansion of other cell lineages. Moreover, the expression of the transcription factor CCAAT/enhancer binding protein β (C/EBPβ) was shown not to correlate with the immunosuppressive activity of MDSCs, in sheer contrast with previously published results.72
The administration of recombinant GM-CSF coupled to macrophage colony-stimulating factor (M-CSF) drives the differentiation of murine MDSCs from the bone marrow in the absence of cancer cell-conditioned culture medium.74 Interestingly, the addition of IL-13 appears to increase the immunosuppressive activity of these cells, which have been shown to efficiently inhibit manifestations of the graft-vs-host disease in an arginase 1-dependent fashion. Nonetheless, also in this case the proportion of MDSCs obtained from bone marrow cell cultures did not exceed 40–50%.74 Such a yield approaches those that would be required for clinical applications.
Another recent study developed a method to obtain large amounts of MDSCs from murine embryonic stem cells (ESCs) for therapeutic purposes.75 Such ESC-derived MDSCs successfully inhibited graft-vs-host disease and were more immunosuppressive than tumor-derived MDSCs. However, the authors of this study used a mouse ESC line that overexpressed homeobox B4 (HoxB4), resulting in a net enhancement of myeloid differentiation. By this means, highly immunosuppressive MDSCs were obtained, including a Ly6Cneg subset that had never been described before in vivo.75 This method to drive the differentiation of MDSCs is not straightforward and depends on a 3-stage process that involves complex cytokine cocktails. Still, this work demonstrated for the first time modified ESCs might constitute a source for high numbers of MDSCs.
Other protocols for the expansion of MDSCs ex vivo do not rely exclusively on recombinant GM-CSF, but also involve prostaglandin E2 (PGE2) and TGFβ, both of which are abundant in tumor-derived exosomes.76,77 Indeed, antibody blocking either of these 2 molecules has been shown to abrogate the capacity of tumor-derived exosomes to drive the differentiation of MDSCs in vivo. Still, the production of MDSCs ex vivo by means of tumor-derived exosomes fail to yield murine MDSCs in percentages higher than 29% of cultured bone marrow cells.76 PGE2 in combination with GM-CSF, IL-4 and LPS has also been shown to promote the differentiation of human MDSCs ex vivo, presumably via a feedforward amplification loop resulting in the robust activation of cyclooxygenase 2 (COX2).78
Putative Advantages of Using Large-Scale Ex Vivo MDSC-T Cell Assays
Many research groups, including us, routinely use bone marrow-derived or monocyte-derived DCs to assess the immunostimulatory potential of a range of interventions. To this aim, DCs are mainly employed in ex vivo presentation assays involving antigen-specific T cells (Fig. 3). These assays heavily rely on the presentation of model antigens such as OVA, as both CD4+ and CD8+ T-cell epitopes of OVA are well characterized. Other antigens including hemagglutinin and the nucleoprotein from influenza virus, as well as human molecules such as melan-A (MLANA, also known as MART1), are sometimes used in these assays.30,79 Frequently, we choose to engineer bone marrow-derived DCs with lentiviral vectors that drive the co-expression of OVA and DC maturation stimuli, allowing for the use OVA-specific transgenic CD4+ and CD8+ T cells in presentation assays.29,30,35 We have used this system to evaluate immunostimulatory as well as immunosuppressive treatments.4 Although the results of ex vivo antigen presentation assays correlate with therapeutic activity in some tumor models,29 this does not always hold true. These inconsistencies are probably due to the strong immunosuppressive microenvironment generated by some neoplasms, which completely inactivate tumor-specific T cells. This might also explain discrepancies between the results of ex vivo assays and in vivo therapeutic profile of DC-based vaccines, as discussed above.
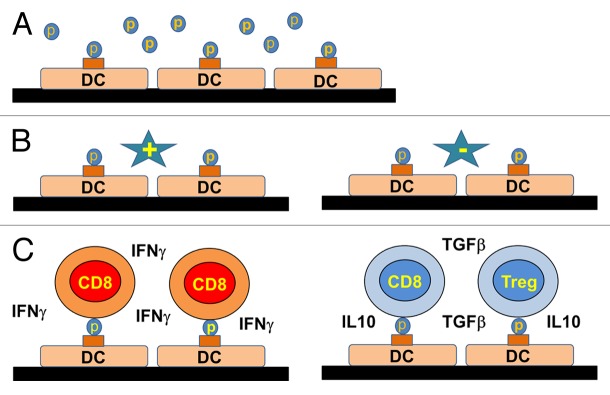
Figure 3. Dendritic cell-T cell antigen presentation assays for the preclinical evaluation of immunotherapeutic regimens. (A) Myeloid dendritic cells (DCs) differentiated ex vivo are loaded with the antigen of interest by overnight incubation. (B) Antigen-loaded DCs are then treated with a potential immunostimulatory (+) or immunosuppressive (−) agent. (C) Antigen-loaded DCs exposed to the agent of interest are incubated with transgenic antigen-specific CD8+ T cells. The elicitation of antigen-specific T-cell responses is then monitored by quantifying the production of interferon γ (IFNγ) by T cells and/or their proliferation. If necessary, the conversion of antigen-specific CD4+ T cells into regulatory T cells (Tregs) can also be monitored.
Based on these premises, we propose that the use MDSCs in antigen presentation assays would make up a more faithful model to assess the efficacy of immunotherapeutic anticancer regimens than that of DCs. Successful immunotherapeutic and chemotherapeutic approaches are known to inhibit the expansion and immunosuppressive activity of MDSCs.12,59,80 A range of treatments could therefore be systematically assessed in MDSC-T cell antigen presentation assays to evaluate their ability to overcome MDSC-dependent immunosuppression. An experimental setup comparable to that generally employed for DC-T cell presentation assays (involving similar model antigens) could be applied to MDSC-T cell tests. However, as discussed above, the generation of cancer-specific MDSCs ex vivo is by far more cumbersome than that of DCs. In addition, the MDSCs obtained in this manner might lose their proliferative potential and/or plasticity, rendering their preservation in culture for the entire duration of the assay a challenge.
At least in part, these problems could be overcome with immortalized MDSC lines, as recently described by Apolloni and collagues.81 One could argue that the genetic alterations required to immortalize these cells could significantly modify their phenotypic and functional profile. In particular, these authors employed v-myc and v-raf, which are indeed expected to affect several signal transduction cascades involved in proliferation and survival. As a matter of fact, immortalized MDSCs exhibited a modified phenotype, expressing increased levels of MHC class II molecules and no GR1.
Thus, what should we be looking for? Basically, a system that allows for the large-scale production of cancer-specific MDSCs ex vivo, similar to the protocols that have already been established for DCs. These MDSCs, which ideally should conserve their proliferative potential and differentiation plasticity, would constitute the basis for the development of an ex vivo test system that would faithfully resemble the tumor microenvironment. Moreover, a large-scale MDSC production system would allow for the high-throughput assessment of the effects of chemotherapeutic agents on MDSCs we well as for their proteomic/genomic profiling. All these strategies would accelerate the preclinical development of antineoplastic drugs, by favoring the early identification of agents that specifically target cancer-derived MDSCs.
In conclusion, the development of a protocol for the differentiation of DCs ex vivo has revolutionized immunotherapy, setting up the basis for their application to neoplastic, infectious, and autoimmune disorders. We propose that a similar production system for MDSCs would be invaluable for the preclinical assessment of novel immunotherapeutic and chemotherapeutic agents. Such a system could be complementary to DC-T cell antigen presentation assays, in particular by mimicking robust immunosuppressive conditions. A significant effort is currently being devoted to the development of protocols for the expansion of cancer-specific MDSCs. Nonetheless, the methods published so far are quite heterogeneous, complicated, and generally characterized by low MDSC yields. To overcome this problem, some research groups have used modified ESCs that are particularly prone to myeloid proliferation or immortalized MDSC lines. However, the cells obtained with these systems may not resemble closely enough those that accumulate in the course of tumor progression and infiltrate neoplastic lesions in vivo. On a positive note, the area of tumor immunology specifically dealing with MDSCs is now speeding up and new isolation/production techniques may lead to the development of routinely applicable high-throughput MDSC-based T-cell assays.
Author Disclosure
The authors declare no conflicts of interests.
Acknowledgments
TL is funded by a University College London Overseas PhD Scholarship. NPJ is funded from an APPICS scholarship from the Health Department and European Social Fund (PO Navarra 2007–2013) and the Basque Country Foundation for Health Innovation and Research (Bioef). AL is funded by a University College London Bench-to-Bedside PhD Scholarship. Karine Breckpot is funded by the Fund for Scientific Research- Flandes. DE has been funded by an Arthritis Research UK Career Development Fellowship (18433) and currently by a Miguel Servet Fellowship from the Instituto de Salud Carlos III, Spain.
Glossary
Abbreviations:
- APC
antigen presenting cell
- CTL
cytotoxic T lymphocyte
- CTLA4
cytotoxic T lymphocyte-associated protein 4
- DAMP
damage-associated molecular pattern
- DC
dendritic cell
- ESC
embryonic stem cell
- G-CSF
granulocyte colony-stimulating factor
- GM-CSF
granulocyte macrophage colony-stimulating factor
- IDO
indoleamine 2,3 deoxygenase
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- Ly6C1
lymphocyte antigen 6 complex, locus C1
- Ly6G
lymphocyte antigen 6 complex, locus G
- MAMP
microbe-associated molecular pattern
- M-CSF
macrophage colony-stimulating factor
- MDSC
myeloid-derived suppressor cell
- OVA
ovalbumin
- PBMC
peripheral blood mononuclear cell
- PD-1
programmed cell death 1
- PD-L1
PD-1 ligand 1
- PGE2
prostaglandin E2
- PRR
pattern recognition receptor
- TAA
tumor-associated antigen
- TCR
T-cell receptor
- TGFβ
transforming growth factor β
- Treg
regulatory T cell
Citation: Liechtenstein T, Perez-Janices N, Breckpot K, Dufait I, Breckpot K, Lanna A, Arce F, Blanco-Luquin I, Kochan G, Guerrero-Setas D, et al. Assessing T cell responses for cancer immunotherapy: dendritic cells or myeloid-derived suppressor cells?. OncoImmunology 2013; 2:e26148; 10.4161/onci.26148
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/26148
References
- 1.Liechtenstein T, Dufait I, Lanna A, Breckpot K, Escors D. Modulating Co-Stimulation During Antigen Presentation to Enhance Cancer Immunotherapy. Immunol Endocr Metab Agents Med Chem. 2012;12:224–35. doi: 10.2174/187152212802001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nurieva R, Thomas S, Nguyen T, Martin-Orozco N, Wang Y, Kaja MK, Yu XZ, Dong C. T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J. 2006;25:2623–33. doi: 10.1038/sj.emboj.7601146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liechtenstein T, Dufait I, Bricogne C, Lanna A, Pen J, Breckpot K, Escors D. PD-L1/PD-1 co-stimulation, a brake for T cell activation and a T cell differentiation signal. J Clin Cell Immunol. 2012;S12(S12:006) doi: 10.4172/2155-9899.S12-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liechtenstein T, Perez-Janices N, Bricogne C, Lanna A, Dufait I, Goyvaerts C, Laranga R, Padella A, Arce F, Baratchian M, et al. Immune modulation by genetic modification of dendritic cells with lentiviral vectors. Virus Res. 2013;176:1–15. doi: 10.1016/j.virusres.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 5.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Tran T, Burt D, Eapen L, Keller OR. Spontaneous regression of metastatic melanoma after inoculation with tetanus-diphtheria-pertussis vaccine. Curr Oncol. 2013;20:e270–3. doi: 10.3747/co.20.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–9. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solito S, Bronte V, Mandruzzato S. Antigen specificity of immune suppression by myeloid-derived suppressor cells. J Leukoc Biol. 2011;90:31–6. doi: 10.1189/jlb.0111021. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother. 2013;62:909–18. doi: 10.1007/s00262-013-1396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–62. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinman RM, Witmer MD. Lymphoid dendritic cells are potent stimulators of the primary mixed leukocyte reaction in mice. Proc Natl Acad Sci U S A. 1978;75:5132–6. doi: 10.1073/pnas.75.10.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–8. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–79. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breckpot K, Escors D. Dendritic cells for active anti-cancer immunotherapy: targeting activation pathways through genetic modification. Endocr Metab Immune Disord Drug Targets. 2009;9:328–43. doi: 10.2174/187153009789839156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou LJ, Tedder TF. CD14+ blood monocytes can differentiate into functionally mature CD83+ dendritic cells. Proc Natl Acad Sci U S A. 1996;93:2588–92. doi: 10.1073/pnas.93.6.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janeway CA, Jr., Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–85. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 21.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 22.Arce F, Kochan G, Breckpot K, Stephenson H, Escors D. Selective activation of intracellular signalling pathways in dendritic cells for cancer immunotherapy. Anticancer Agents Med Chem. 2012;12:29–39. doi: 10.2174/187152012798764679. [DOI] [PubMed] [Google Scholar]
- 23.Dullaers M, Breckpot K, Van Meirvenne S, Bonehill A, Tuyaerts S, Michiels A, Straetman L, Heirman C, De Greef C, Van Der Bruggen P, et al. Side-by-side comparison of lentivirally transduced and mRNA-electroporated dendritic cells: implications for cancer immunotherapy protocols. Mol Ther. 2004;10:768–79. doi: 10.1016/j.ymthe.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 24.Gruber A, Kan-Mitchell J, Kuhen KL, Mukai T, Wong-Staal F. Dendritic cells transduced by multiply deleted HIV-1 vectors exhibit normal phenotypes and functions and elicit an HIV-specific cytotoxic T-lymphocyte response in vitro. Blood. 2000;96:1327–33. [PubMed] [Google Scholar]
- 25.Dyall J, Latouche JB, Schnell S, Sadelain M. Lentivirus-transduced human monocyte-derived dendritic cells efficiently stimulate antigen-specific cytotoxic T lymphocytes. Blood. 2001;97:114–21. doi: 10.1182/blood.V97.1.114. [DOI] [PubMed] [Google Scholar]
- 26.Arrighi JF, Pion M, Wiznerowicz M, Geijtenbeek TB, Garcia E, Abraham S, Leuba F, Dutoit V, Ducrey-Rundquist O, van Kooyk Y, et al. Lentivirus-mediated RNA interference of DC-SIGN expression inhibits human immunodeficiency virus transmission from dendritic cells to T cells. J Virol. 2004;78:10848–55. doi: 10.1128/JVI.78.20.10848-10855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu Q, Kovacs C, Yue FY, Ostrowski MA. The role of the p38 mitogen-activated protein kinase, extracellular signal-regulated kinase, and phosphoinositide-3-OH kinase signal transduction pathways in CD40 ligand-induced dendritic cell activation and expansion of virus-specific CD8+ T cell memory responses. J Immunol. 2004;172:6047–56. doi: 10.4049/jimmunol.172.10.6047. [DOI] [PubMed] [Google Scholar]
- 28.Klein C, Bueler H, Mulligan RC. Comparative analysis of genetically modified dendritic cells and tumor cells as therapeutic cancer vaccines. J Exp Med. 2000;191:1699–708. doi: 10.1084/jem.191.10.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, Escors D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. 2011;3:581–92. doi: 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Escors D, Lopes L, Lin R, Hiscott J, Akira S, Davis RJ, Collins MK. Targeting dendritic cell signaling to regulate the response to immunization. Blood. 2008;111:3050–61. doi: 10.1182/blood-2007-11-122408. [DOI] [PubMed] [Google Scholar]
- 31.Hu B, Dai B, Wang P. Vaccines delivered by integration-deficient lentiviral vectors targeting dendritic cells induces strong antigen-specific immunity. Vaccine. 2010;28:6675–83. doi: 10.1016/j.vaccine.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akazawa T, Shingai M, Sasai M, Ebihara T, Inoue N, Matsumoto M, Seya T. Tumor immunotherapy using bone marrow-derived dendritic cells overexpressing Toll-like receptor adaptors. FEBS Lett. 2007;581:3334–40. doi: 10.1016/j.febslet.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 33.Breckpot K, Aerts-Toegaert C, Heirman C, Peeters U, Beyaert R, Aerts JL, Thielemans K. Attenuated expression of A20 markedly increases the efficacy of double-stranded RNA-activated dendritic cells as an anti-cancer vaccine. J Immunol. 2009;182:860–70. doi: 10.4049/jimmunol.182.2.860. [DOI] [PubMed] [Google Scholar]
- 34.Chiang CL, Hagemann AR, Leskowitz R, Mick R, Garrabrant T, Czerniecki BJ, Kandalaft LE, Powell DJ, Jr., Coukos G. Day-4 myeloid dendritic cells pulsed with whole tumor lysate are highly immunogenic and elicit potent anti-tumor responses. PLoS One. 2011;6:e28732. doi: 10.1371/journal.pone.0028732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arce F, Breckpot K, Stephenson H, Karwacz K, Ehrenstein MR, Collins M, Escors D. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. 2011;63:84–95. doi: 10.1002/art.30099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006;108:1435–40. doi: 10.1182/blood-2006-03-006403. [DOI] [PubMed] [Google Scholar]
- 37.Kabelitz D, Wesch D, Oberg HH. Regulation of regulatory T cells: role of dendritic cells and toll-like receptors. Crit Rev Immunol. 2006;26:291–306. doi: 10.1615/CritRevImmunol.v26.i4.10. [DOI] [PubMed] [Google Scholar]
- 38.Toscano MG, Delgado M, Kong W, Martin F, Skarica M, Ganea D. Dendritic cells transduced with lentiviral vectors expressing VIP differentiate into VIP-secreting tolerogenic-like DCs. Mol Ther. 2010;18:1035–45. doi: 10.1038/mt.2009.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ilarregui JM, Croci DO, Bianco GA, Toscano MA, Salatino M, Vermeulen ME, Geffner JR, Rabinovich GA. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat Immunol. 2009;10:981–91. doi: 10.1038/ni.1772. [DOI] [PubMed] [Google Scholar]
- 40.Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, Yang H, Suthanthiran M, Mojsov S, Steinman RM. Dendritic cell-expanded, islet-specific CD4+ CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med. 2007;204:191–201. doi: 10.1084/jem.20061631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 42.Liu LN, Shivakumar R, Allen C, Fratantoni JC. Delivery of whole tumor lysate into dendritic cells for cancer vaccination. Methods Mol Biol. 2008;423:139–53. doi: 10.1007/978-1-59745-194-9_9. [DOI] [PubMed] [Google Scholar]
- 43.Schnurr M, Galambos P, Scholz C, Then F, Dauer M, Endres S, Eigler A. Tumor cell lysate-pulsed human dendritic cells induce a T-cell response against pancreatic carcinoma cells: an in vitro model for the assessment of tumor vaccines. Cancer Res. 2001;61:6445–50. [PubMed] [Google Scholar]
- 44.Hegmans JP, Hemmes A, Aerts JG, Hoogsteden HC, Lambrecht BN. Immunotherapy of murine malignant mesothelioma using tumor lysate-pulsed dendritic cells. Am J Respir Crit Care Med. 2005;171:1168–77. doi: 10.1164/rccm.200501-057OC. [DOI] [PubMed] [Google Scholar]
- 45.Herbert N, Haferkamp A, Schmitz-Winnenthal HF, Zöller M. Concomitant tumor and autoantigen vaccination supports renal cell carcinoma rejection. J Immunol. 2010;185:902–16. doi: 10.4049/jimmunol.0902683. [DOI] [PubMed] [Google Scholar]
- 46.Gitlitz BJ, Belldegrun AS, Zisman A, Chao DH, Pantuck AJ, Hinkel A, Mulders P, Moldawer N, Tso CL, Figlin RA. A pilot trial of tumor lysate-loaded dendritic cells for the treatment of metastatic renal cell carcinoma. J Immunother. 2003;26:412–9. doi: 10.1097/00002371-200309000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Zarour HM, Kirkwood JM. Melanoma vaccines: early progress and future promises. Semin Cutan Med Surg. 2003;22:68–75. doi: 10.1053/sder.2003.50006. [DOI] [PubMed] [Google Scholar]
- 48.Abdel-Wahab Z, Cisco R, Dannull J, Ueno T, Abdel-Wahab O, Kalady MF, Onaitis MW, Tyler DS, Pruitt SK. Cotransfection of DC with TLR4 and MART-1 RNA induces MART-1-specific responses. J Surg Res. 2005;124:264–73. doi: 10.1016/j.jss.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 49.Xu Y, Darcy PK, Kershaw MH. Tumor-specific dendritic cells generated by genetic redirection of Toll-like receptor signaling against the tumor-associated antigen, erbB2. Cancer Gene Ther. 2007;14:773–80. doi: 10.1038/sj.cgt.7701073. [DOI] [PubMed] [Google Scholar]
- 50.Win SJ, McMillan DG, Errington-Mais F, Ward VK, Young SL, Baird MA, Melcher AA. Enhancing the immunogenicity of tumour lysate-loaded dendritic cell vaccines by conjugation to virus-like particles. Br J Cancer. 2012;106:92–8. doi: 10.1038/bjc.2011.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamanaka R, Honma J, Tsuchiya N, Yajima N, Kobayashi T, Tanaka R. Tumor lysate and IL-18 loaded dendritic cells elicits Th1 response, tumor-specific CD8+ cytotoxic T cells in patients with malignant glioma. J Neurooncol. 2005;72:107–13. doi: 10.1007/s11060-004-3550-9. [DOI] [PubMed] [Google Scholar]
- 52.Bonehill A, Tuyaerts S, Van Nuffel AM, Heirman C, Bos TJ, Fostier K, Neyns B, Thielemans K. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol Ther. 2008;16:1170–80. doi: 10.1038/mt.2008.77. [DOI] [PubMed] [Google Scholar]
- 53.Jackson AM, Mulcahy LA, Zhu XW, O’Donnell D, Patel PM. Tumour-mediated disruption of dendritic cell function: inhibiting the MEK1/2-p44/42 axis restores IL-12 production and Th1-generation. Int J Cancer. 2008;123:623–32. doi: 10.1002/ijc.23530. [DOI] [PubMed] [Google Scholar]
- 54.Van Lint S, Goyvaerts C, Maenhout S, Goethals L, Disy A, Benteyn D, Pen J, Bonehill A, Heirman C, Breckpot K, et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res. 2012;72:1661–71. doi: 10.1158/0008-5472.CAN-11-2957. [DOI] [PubMed] [Google Scholar]
- 55.Hardwick N, Ledermann JA, Aitkens E, Chain B. Pre-clinical assessment of autologous DC-based therapy in ovarian cancer patients with progressive disease. Cancer Immunol Immunother. 2012;61:1929–39. doi: 10.1007/s00262-012-1252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prokopowicz ZM, Arce F, Biedroń R, Chiang CL, Ciszek M, Katz DR, Nowakowska M, Zapotoczny S, Marcinkiewicz J, Chain BM. Hypochlorous acid: a natural adjuvant that facilitates antigen processing, cross-priming, and the induction of adaptive immunity. J Immunol. 2010;184:824–35. doi: 10.4049/jimmunol.0902606. [DOI] [PubMed] [Google Scholar]
- 57.Lutz MB, Kukutsch NA, Menges M, Rössner S, Schuler G. Culture of bone marrow cells in GM-CSF plus high doses of lipopolysaccharide generates exclusively immature dendritic cells which induce alloantigen-specific CD4 T cell anergy in vitro. Eur J Immunol. 2000;30:1048–52. doi: 10.1002/(SICI)1521-4141(200004)30:4<1048::AID-IMMU1048>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 58.Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96:3838–46. [PMC free article] [PubMed] [Google Scholar]
- 59.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25. doi: 10.1016/j.it.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. 2013;14:211–20. doi: 10.1038/ni.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Small EJ, Reese DM, Um B, Whisenant S, Dixon SC, Figg WD. Therapy of advanced prostate cancer with granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 1999;5:1738–44. [PubMed] [Google Scholar]
- 64.Mastrangelo MJ, Maguire HC, Jr., Eisenlohr LC, Laughlin CE, Monken CE, McCue PA, Kovatich AJ, Lattime EC. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6:409–22. doi: 10.1038/sj.cgt.7700066. [DOI] [PubMed] [Google Scholar]
- 65.Dranoff G. GM-CSF-secreting melanoma vaccines. Oncogene. 2003;22:3188–92. doi: 10.1038/sj.onc.1206459. [DOI] [PubMed] [Google Scholar]
- 66.Kushner BH, Cheung NK. GM-CSF enhances 3F8 monoclonal antibody-dependent cellular cytotoxicity against human melanoma and neuroblastoma. Blood. 1989;73:1936–41. [PubMed] [Google Scholar]
- 67.Rössner P, Bubeník J, Sobota V, Indrová M, Hájková R, Mendoza L, Jandlová T, Símová J. Granulocyte-macrophage colony-stimulating factor-producing tumour vaccines. Folia Biol (Praha) 1999;45:173–7. [PubMed] [Google Scholar]
- 68.Gutschalk CM, Yanamandra AK, Linde N, Meides A, Depner S, Mueller MM. GM-CSF enhances tumor invasion by elevated MMP-2, -9, and -26 expression. Cancer Med. 2013;2:117–29. doi: 10.1002/cam4.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez M, Ono N, Planutiene M, Planutis K, Nelson EL, Holcombe RF. Granulocyte-macrophage stimulating factor (GM-CSF) increases circulating dendritic cells but does not abrogate suppression of adaptive cellular immunity in patients with metastatic colorectal cancer receiving chemotherapy. Cancer Cell Int. 2012;12:2. doi: 10.1186/1475-2867-12-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat. 2010;123:39–49. doi: 10.1007/s10549-009-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40:22–35. doi: 10.1002/eji.200939903. [DOI] [PubMed] [Google Scholar]
- 72.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 73.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010;185:2273–84. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116:5738–47. doi: 10.1182/blood-2010-06-287839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou Z, French DL, Ma G, Eisenstein S, Chen Y, Divino CM, Keller G, Chen SH, Pan PY. Development and function of myeloid-derived suppressor cells generated from mouse embryonic and hematopoietic stem cells. Stem Cells. 2010;28:620–32. doi: 10.1002/stem.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, Cheng Z, Shah SV, Wang GJ, Zhang L, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. 2009;124:2621–33. doi: 10.1002/ijc.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, Corbelli A, Fais S, Parmiani G, Rivoltini L. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res. 2006;66:9290–8. doi: 10.1158/0008-5472.CAN-06-1819. [DOI] [PubMed] [Google Scholar]
- 78.Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011;118:5498–505. doi: 10.1182/blood-2011-07-365825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Macdonald DC, Singh H, Whelan MA, Escors D, Arce F, Bottoms SE, Barclay WS, Maini M, Collins MK, Rosenberg WC. Harnessing alveolar macrophages for sustained mucosal T-cell recall confers long-term protection to mice against lethal influenza challenge without clinical disease. Mucosal Immunol. 2013 doi: 10.1038/mi.2013.27. In press; PMID:23715172. [DOI] [PubMed] [Google Scholar]
- 80.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506–13. doi: 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Apolloni E, Bronte V, Mazzoni A, Serafini P, Cabrelle A, Segal DM, Young HA, Zanovello P. Immortalized myeloid suppressor cells trigger apoptosis in antigen-activated T lymphocytes. J Immunol. 2000;165:6723–30. doi: 10.4049/jimmunol.165.12.6723. [DOI] [PubMed] [Google Scholar]