

Abstract
The oxysterols cholestan-3β, 5α, 6β-triol (Triol) and 3-keto-cholest-4-ene (3K4) are increased in Opisthorchis viverrini-associated hamster cholangiocarcinoma and induce DNA damage and apoptosis via a mitochondria-dependent mechanism in MMNK-1 human cholangiocytes. Based on these observations, we hypothesized that chronic exposure of cholangiocytes to these pathogenic oxysterols may allow a growth advantage to a subset of these cells through selection for resistance to apoptosis, thereby contributing to cholangiocarcinogenesis. To test this hypothesis, we cultured MMNK-1 cells long-term in the presence of Triol. Alteration in survival and apoptotic factors of Triol-exposed cells were examined. Cells cultured long-term in the presence of Triol were resistant to H2O2-induced apoptosis, and demonstrated an increase in the phosphorylation of p38-α, CREB, ERK1/2 and c-Jun. Elevations in the ratio of Bcl-2/Bax and in the protein levels of anti-apoptotic factors including cIAP2, clusterin, and survivin were detected. These results show that long-term exposure of MNNK-1 cells to low doses of Triol selects for kinase-signaling molecules with regulate resistance to apoptosis and thereby enhance cell survival. Clonal expansion of such apoptosis-resistant cells may contribute to the genesis of cholangiocarcinoma.
Keywords: Oxysterols, Cholestan-3β, 5α, 6β-triol, cholangiocarcinoma, apoptosis, MMNK-1 cells
1. Introduction
Oxysterols are oxygenated derivatives of cholesterol derived from enzymatic pathways mediated by CYP450 enzymes or from non-enzymatic mechanisms via reactive oxygen and/or nitrogen species (ROS/RNS) [1, 2]. Numerous studies have demonstrated pathological effects of oxysterols such as 7-ketocholesterol, 24-hydroxycholesterol and Triol in a variety of diseases including cardiovascular diseases, neurological diseases and cancer [1–3]. Oxysterols also appear to play critical roles in multiple stages of carcinogenesis [3], such as in tumor initiation via enhancement of oxidative stress. Oxysterols increase intracellular ROS levels by inducing NADPH oxidase [4, 5]. Persistent production of pro-oxidative molecules including ROS/RNS induced by oxysterols may perturb cellular stress responses [6, 7] that can impair the DNA repair system and inhibit apoptosis [8, 9]. Apoptosis induction by oxysterols may select a subset of cells that are resistant to apoptosis and thereby enhance carcinogenesis [10, 11]. Infection with the liver fluke Opisthorchis viverrini (Ov) induces the generation of ROS/RNS in bile duct epithelia [12, 13]. We have previously reported a possible role for oxysterols in the genesis of Ov-associated cholangiocarcinoma (CCA), a malignant tumor of bile duct epithelia [14]. Two oxysterols, cholestan-3β, 5α, 6β-triol (Triol) and 3-keto-cholest-4-ene (3K4), were increased in Ov-induced hamster CCA. Triol and 3K4 induced DNA damage and apoptosis in the human cholangiocyte MMNK-1 cell line [14]. Moreover, the expression of cytosolic oxysterol-binding proteins was increased in Ov-induced hamster CCA [15].
Mechanisms that explain the role of oxysterols in the initiation and progression of CCA remain undefined. Certain oxysterols can stabilize cyclooxygenase-2 (COX-2) mRNA via a p38 MAPK-dependent mechanism resulting in COX-2 protein accumulation in a CCA cell line [16]. Previous experiments, however, have examined the effects of high doses of oxysterols over short time courses, a situation that is diametrically opposed to that encountered in clinical Ov-induced CCA development where long term exposure to the liver fluke is present, and low concentrations of oxysterols are present in the biliary tract [14, 17]. Indeed, culturing cholangiocytes with high concentration of oxysterols is expected to enhance their cytotoxicity. Interestingly, Gregorio-King and colleagues demonstrated that long-term culture of hematopoietic HL60 cells in the presence of low doses of the oxysterol 25-hydroxycholesterol (25-OHC) led to increased survival but it did not show resistance to apoptosis [18]. On the other hand, repeated exposure to oxysterols induced resistance to apoptosis in rat colon crypt cells and in a colon cell line [19, 20]. Such resistance, was stable after at least 4 weeks growth in the absence of the oxysterol [20], and thus must have been due to somatically inherited changes in the cells, such as epigenetic or mutational alterations. Based on these observations, we hypothesized that chronic exposure of cholangiocytes to low doses of oxysterols may allow a growth advantage to a subset of these cells through selection for resistance to apoptosis, thereby contributing to cholangiocarcinogenesis. To test this hypothesis, we cultured MMNK-1 cells long-term in the presence of low doses of Triol, and investigated the molecular signaling pathways that have been associated with apoptosis resistance.
2. Materials and Methods
2.1. Cell culture
The immortalized human cholangiocyte MMNK-1 cell line, transduced with SV40T and hTERT, was generated and supplied by Professor Naoya Kobayashi (Okayama University, Japan). Cells were cultured in Hams F12 (Invitrogen, CA, USA) supplemented with 10% fetal calf serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C and 5% CO2.
2.2. Establishment of a cholangiocyte cell line chronically exposed to low doses of Triol
MMNK-1 cells were cultured by step-wise exposure to cholestan-3β, 5α, 6β-triol (Triol) (Steraloids Inc., USA). In brief, MMNK-1 cells were cultured repeatedly in the presence of 15 μM Triol. When the viability of Triol-treated cells was similar to that observed in untreated control cells, the concentration of Triol in the culture media was increased to 18 μM and 20 μM consecutively. Cell viability was determined using sulforhodamine B as described below.
2.3. Cell viability assay
MMNK-1 cells (2×103/100 μl) were seeded into 96-well plates and incubated overnight at 37°C and 5% CO2. Cells were treated with Triol at designated concentrations for 48 h. Triol was dissolved in 100% ethanol and added to culture media at different concentrations; the final concentration of ethanol was 0.5%. The number of viable cells was determined using sulforhodamine B (SRB, Sigma-Aldrich, MO, USA) as described previously [21]. Briefly, cells were fixed with 10% cold trichloroacetic acid for 1 h at 4°C and stained with 0.4% SRB in 1% acetic acid for 30 min. Excess dye was washed with 1% acetic acid and stained cells were solubilized with 200 μl of 10 mM unbuffered Tris-base. The absorbance was measured with a microplate reader (Sunrise, TECAN Trading, Switzerland) at 540 nm. The results were expressed as a percentage of cell viability relative to control (cells treated with 0.5% ethanol). The concentration that inhibited cell growth to 50% of control (IC50) was calculated by plotting the percentage of cell growth inhibition against oxysterol concentration.
2.4. Determination of apoptosis resistance
To determine resistance to apoptosis in MMNK-1 cells cultured in the presence of Triol, cells were treated with 300–1000 μM of hydrogen peroxide (H2O2), an apoptosis inducer [22], for 24 h. The number of viable cells was determined using sulforhodamine B as described above. Parental MMNK-1 cells were used as controls. DNA ladder formation was analyzed in Triol-exposed cells compared to MMNK-1 control cells. Cells were exposed to 10–30 μM Triol for 48 h, and then adherent cells were trypsinized and cell pellets were collected. Briefly, 106 cells were lysed twice with 1 ml lysis buffer (1% (w/v) Triton X-100, 0.32 M saccharose, 5 mM MgCl2, 10 mM Tris-HCl) and centrifuged for 20 sec at 10,000 x g. Cell pellets were collected and treated with 200 μl of enzyme reaction solution (1% (w/v) SDS, 5 mM EDTA-Na2, 10 mM Tris-HCl) and 8 μl of 1 mg/ml of RNaseA for 20 min at 37°C. Subsequently, 10 μl of 10 mg/ml Proteinase K was added to the sample and incubated for 1 h at 37°C. Then, 300 μl of 7.6 M NaI solution (7.6 M NaI, 20 mM EDTA-Na2, 40 mM Tris-HCl) and 500 μl of isopropanol were added to the sample. After centrifugation, the pellet was mixed with 40% isopropanol, washed with 70% ethanol and centrifuged. The pellet was dried and resuspended with 100 μl of TE buffer (10 mM Tris–HCl and 1 mM EDTA) and loaded onto 1% agarose gel in 1 x TBE buffer (89 mM Tris Base, 89 mM boric acid, and 2 mM EDTA). Electrophoresis was done at 50V for 1 h and the gel then stained with ethidium bromide for 15 min. Visualization was by ImageQuant 400 (GE Healthcare Bio-Science, UK).
2.5. Proteomic profiles of phospho-kinase and apoptosis proteins
Analysis of phosphorylation profiles of kinase proteins and expression profiles of apoptosis-related proteins were performed using Proteome Profiler Human Phospho-Kinase and Apoptosis Array Kits (R&D System, USA) according the manufacturer’s instructions using protein extracts from MMNK-1 cells cultured in the presence of Triol in comparison to MNNK-1 cells cultured in the absence of Triol. Briefly, Triol-exposed MMNK-1 and control MMNK-1 cells were cultured in serum free media without oxysterol for 24 h. Cell lysates (800 μg per membrane) were incubated overnight with nitrocellulose membrane containing antibodies against 46 kinase phosphorylation sites and 35 apoptotic proteins printed in duplication. Then membranes were incubated with Detection Antibody Cocktail and subsequently with streptavidin-horseradish peroxidase. Signals were detected with ECL Plus Western blotting Detection Reagents (GE Healthcare UK Ltd., UK). The array images were analyzed and quantified using ImageQuant™ Imager (GE Healthcare UK Ltd., UK).
2.6. Western blot analysis
Cell lysate (30 μg) of Triol-exposed MMNK-1 and unexposed control cells were separated by 10% SDS-polyacrylamide electrophoresis and blotted to PVDF membrane (GE Healthcare Bio-Science, UK). Blots were probed with 1:100 of goat anti-Bax antibody (BD Biosciences, USA), 1:100 of rabbit anti-Bcl-2 (C21) antibody (Santa Cruz, Biochemical, USA), 1:1000 of rabbit anti-phospho-p44/42 MAPK (ERK1/2) antibody (Cell Signaling, USA), 1:1000 of rabbit anti-p44/42 MAPK (ERK1/2) antibody (Cell Signaling, USA), 1:1000 of rabbit anti-phospho-p38 MAPK (Thr180/Tyr182) antibody (Cell Signaling, USA), 1:1000 of rabbit anti-p38 MAPK (Thr180/Tyr182) antibody (Cell Signaling, USA) or 1:10,000 of the mouse anti-β-actin antibody (Sigma Aldrich, USA). Then, membranes were incubated in horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biochemical, Santa Cruz, CA) at room temperature for 1 h. After that, detection was performed using the chemiluminescence for western blotting system (ECL, GE Healthcare Bio-Science, UK). Signals were analyzed by using Imagequant 400 and quantified by using ImageQuant™ Imager (GE Healthcare UK Ltd., UK).
2.7. Statistics
The results are presented as a mean ± SD for at least two individual experiments. Data were compared using Student’s t-test. A P-value less than 0.05 was considered statistically significant. All analyses were performed using IBM SPSS 19 (Chicago, IL, USA) software.
3. Results
3.1. Triol-exposed MMNK-1 cells are resistant to H2O2-induced apoptosis
Triol-tolerant cholangiocytes were established by repeated exposure of MMNK-1 cells to Triol. Typically, only 20–30 % of MMNK-1 cells survived within 5 days after exposure to 15 μM Triol. The surviving cells were cultured and repeatedly exposed to Triol until 80–90 % viable cells were attained. The dose of Triol was then increased consecutively to 18 and 20 μM. The total time required to establish a Triol tolerance in MMNK-1 cell lines was approximately 12 months. After the 12-month exposure, Triol-exposed MMNK-1 cells showed a significant increase in IC50 (22.7±1.1) when compared to the parental cells (16.7±0.5 μM) as shown in Fig. 1A. To investigate whether Triol-exposed cells were resistant to apoptosis, cells were treated with 300–1000 μM H2O2 for 24 h. Triol-exposed MMNK-1 cells showed significantly higher cell viability than did the parental cells that were not exposed to Triol (Fig. 1B). Moreover, a marked decrease DNA laddering was found in Triol-exposed cells upon treatment with 30 μM of Triol (Fig. 2)
Fig 1.

Triol-exposed MMNK-1 cells are resistant to Triol and H2O2 treatment.(A) Both control and Triol-exposed cells were treated with Triol at designated concentrations for 48 h. After repeated exposure to Triol, cells were more resistant to Triol than control. (B) Control and Triol-exposed cells were treated with H2O2 at designated concentrations for 24 h. Increased numbers of viable cells were observed in Triol-exposed cells compared to control cells. Results are expressed as percent of control. Two individual experiments were performed. * indicates P< 0.05
Fig. 2.
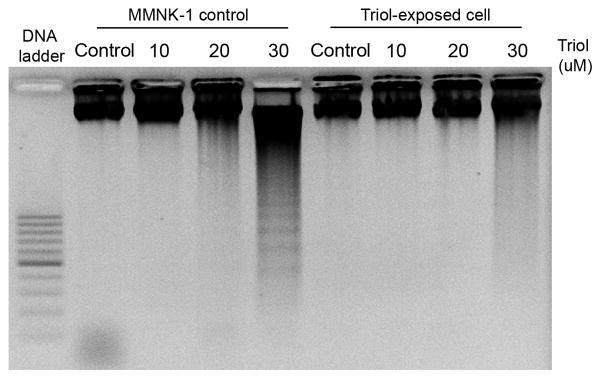
Triol-exposed cells are resistant to apoptosis induction by Triol. Cells were treated with 10–30 μM Triol for 48 h. First lanes were 100 bp DNA ladder. 0.5% ethanol-treated cells were used as control.
3.2. Profiling of kinase phosphorylation and apoptosis proteins in Triol-exposed cells
Patterns of activated kinases and apoptotic-associated proteins were investigated in Triol-exposed cells in comparison to the parental cells not exposed to Triol. Triol-exposed cells demonstrated increased phosphorylation of several kinases, including p38 mitogen-activated protein kinase α (p38-α), cAMP response element-binding (CREB), extracellular signal-regulated kinase (ERK1/2), mitogen- and stress-activated protein kinases 1 and 2 (MSK1/2) and c-Jun (S63) when compared to the parental cells not exposed to Triol (Fig. 3A and Fig. 3B).
Fig. 3.
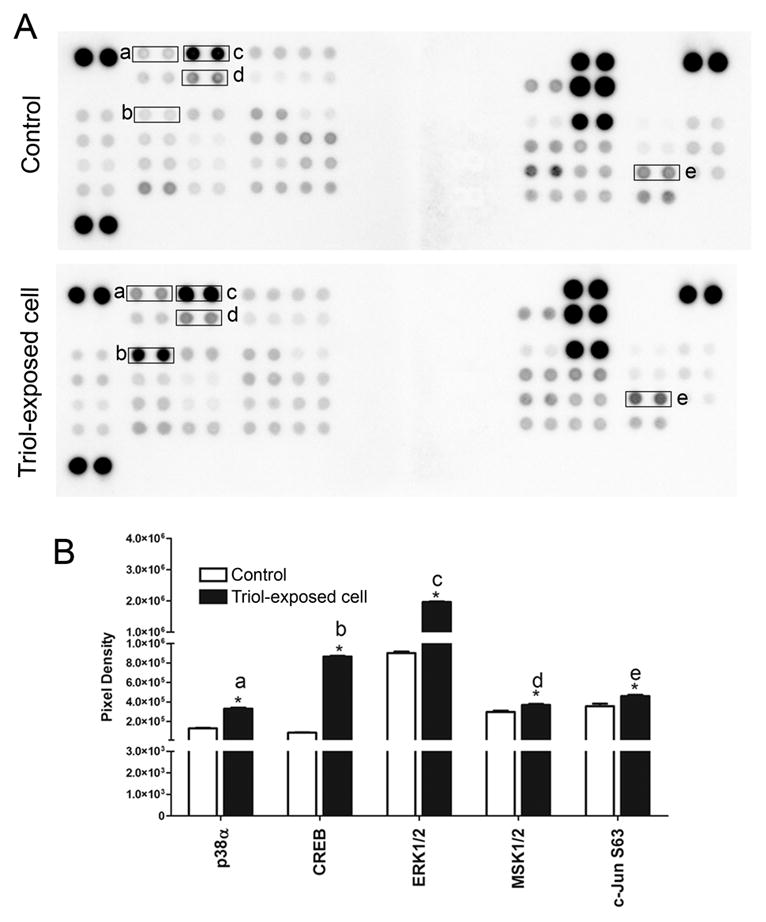
Activation of kinase signaling cascade in Triol-exposed cells compares to MMNK-1 control cells.(A) Protein kinase phosphorylation in control and Triol-exposed cells were performed by using the phospho-kinase array. (B) Bar graph comparison of phosphorylated kinase protein expression in MMNK-1 control and Triol-exposed cells. Spot pixel densities were analyzed using ImageQuant™ and expressed as mean ± SD from two individual experiments. Results indicated the activation of kinase signaling cascade in Triol-exposed cells. * indicates P< 0.05.
The results also revealed a significant increase in anti-apoptotic proteins including Bad, cIAP-2, Claspin and Survivin in Triol-exposed cells compared to the parental cells (Fig. 4A and Fig. 4B). A marked reduction in pro-apoptotic proteins including Fas-associated protein with death domain (FADD) and Fas receptor (Fas/TNFRSF6) was detected in Triol-exposed cells when compared to the parental cells (Fig. 4A and Fig. 4B).
Fig. 4.

Activation of apoptosis-related proteins in Triol-exposed cells compares to MMNK-1 control cells. (A) Profiling of apoptosis-related proteins expression in control and Triol-exposed cells was done by using the apoptosis array. (B)The quantification of mean spot pixel densities were analyzed using ImageQuant™ from two individual experiments. * indicates P< 0.05.
3.3. Western blot analysis confirmed the phosphorylation status of kinase proteins and expression of apoptosis-related proteins
To confirm the phosphorylation status of kinase proteins and expression of apoptosis-related proteins in Triol-exposed cells, we examined the protein levels of key-targeted proteins that included total-ERK1/2, phospho-ERK1/2, total-p38MAPK, phospho-p38MAPK, Bcl-2 and Bax. A significant induction in the ratio of phosphorylated to total forms of ERK1/2 and p38MAPK and Bcl-2 to Bax was observed for Triol-exposed cells when compared to the parental cells that were not exposed to Triol (Fig. 5A and Fig. 5B).
Fig. 5.

Activation of survival signaling cascade in Triol-exposed cells. (A) Western blot analysis confirmed the protein expression status of kinase and apoptosis-related proteins in MMNK-1 control and Triol-exposed cells. (B) Bar graphs show relative densitometric values of western blot of Bcl-2, the ratio of Bcl-2/Bax, the ratio of phosphorylated ERK/total ERK and the ratio of phosphorylated p38α/total p38α, respectively. * indicates P< 0.05.
4. Discussion
Oxysterols have been proposed to contribute to multiple stages of carcinogenesis [3]. We reported increased levels of the pathogenic oxysterols Triol and 3K4 in Ov-induced hamster CCA tumor tissue. In addition, the cytotoxic effects of Triol and 3K4 were studied in human MMNK-1 cholangiocytes. Both Triol and 3K4 induced DNA damage and apoptosis via a mitochondria-dependent mechanism [14]. Studies have described the mechanisms of oxysterol-induced cytotoxicity in different cell systems [4, 23]. Interestingly, those experiments were performed using single and/or high doses of oxysterols. High concentrations of oxysterols are cytotoxic to many cell types. Moreover, our previous study showed that low concentrations of oxysterols induce DNA damage without necessarily inducing cell death [14]. As previously suggested in bile-acid-induced gastrointestinal cancer [20], cells with an apoptosis-resistant phenotype are selected after long-term repeated exposure to pulses of bile acids, resulting in the survival of cells with unrepaired DNA damage, and a consequent increase in genomic instability leading to cancer progression. Long-term exposure to bile acids allowed cells to become resistant to apoptosis in vitro and in vivo [19, 20]. Our study demonstrates that chronic exposure of cholangiocytes to low doses of the pathogenic oxysterol Triol leads cell to survival and resistance to apoptosis. Interestingly, we could not maintain 3K4-tolerant cells in a similar experiment. This may be due to greater cytotoxicity of Triol than 3K4 caused by higher polarity [14]. Another possible explanation is that a more extended treatment period may be required for establishing 3K4-tolerant cells. We proved that Triol-exposed cells were more resistant to Triol and H2O2 than the parental cells by using SRB assay. Moreover, a decrease in DNA ladder formation after treatment with Triol confirms that Triol-exposed cells are more resistant to apoptosis. This suggests that Triol-induced cytotoxicity may operate via similar mechanisms with apoptosis induction-related oxidative stress [24, 25]. Nevertheless, cell death of 70% in Triol-exposed cells after treatment with 30 μM Triol using SRB with undetectable DNA laddering could happen due to a discrepancy between the two methods. Apoptotic and necrotic cells were excluded by collection of only adherent cells for assays.
Previous studies reported that oxysterols exert their survival effect through the activation of signal transduction pathways, especially kinase-dependent pathways [16]. An oxysterol-activated MAPK cascade exists in various cell types including in a human cholangiocarcinoma (KMBC) cell line [16, 26]. We found that the Triol-exposed cholangiocytes exhibited pro-survival and anti-apoptotic properties in association with activation of signaling molecules in the MAPK pathway. The increase in activation of extracellular signal activated kinases including p38α MAPK and ERK1/2 were found in Triol-exposed cells. Activation of the MAPK cascade results in stimulation of a wide range of cellular responses including metabolism, survival, apoptosis and differentiation [27]. Moreover, activation of the MAPK cascade has profound effects on the regulation of the cell cycle and apoptosis [28]. An increase in phosphorylation of the transcription factors CREB and c-Jun, which are downstream molecules in the MAPK pathway, was also found in Triol-exposed cells. The activation of these downstream transcription factors may regulate the expression of survival-related proteins [28]. Induction of CREB stimulates a survival pathway in alveolar epithelial cells [29]. MAP kinase that activates c-Jun may underlie the common stimulation of this transcription factor by growth factors and oncogenes [30].
Oxysterols can induce apoptosis through both death receptor-dependent and mitochondrial pathways [14, 31]. We found the alteration of both pathways in Triol-exposed cells. We demonstrated that cholangiocytes exposed to Triol exert anti-apoptotic properties by increasing the ratio of Bcl-2/Bax, enhanced expression of cIAP-2 and survivin. This finding contrasts to two other reports [18, 32]. These contrasting findings may be due to the different types of oxysterols used and varied mechanisms of oxysterol-induced cytotoxicity in different cell types. Moreover, decreased expression of the death receptor Fas was found in our model. Even though previous reports have implicated Fas receptor mediated apoptosis by certain oxysterols, the mechanisms involved remain obscure [31, 33].Hence, the suppression of Fas ligand induced by Triol treatment should be investigated further. Notably, the activation of pro-apoptotic related proteins, Bad and Claspin, was observed in our study but did not trigger apoptosis.
In conclusion, we established a human cholangiocyte MMNK-1 cell line that was selected for resistance to cytotoxicity by Triol through long term, low dose exposure. The clonally selected cells expressed activated tyrosine kinases and their effector proteins allowed these cells to evade apoptosis. These findings support a model in which exposure of cholangiocytes to pathogenic oxysterols at a low dose and for a long duration (as occurs with Ov infection in humans) contributes to cancer development. Reduced DNA repair and ongoing exposure to DNA damaging agents cause an accumulation of DNA damage. Higher level of DNA damage leads to either increased mutation due to error-prone translational processes [34] or increased epimutations due to epigenetic alterations at the sites of DNA damage when repair is error-prone [35–37]. Such mutations and/or epimutations enable the selection of resistant cells. These resistant cells constitutively activate down-stream effectors of receptor tyrosine kinase signaling that may result in regulation of gene expression of apoptosis-related proteins, which are involved in the stimulation of cell survival and evasion of apoptosis. Clonal expansion of apoptosis-resistant cells may enhance the malignant transformation of biliary cells leading to the genesis of CCA (as depicted in Fig. 6). Pathogenic oxysterols found in the biliary tract, such as Triol, could play a key role in driving this process in CCA. Long term chronic inflammation due to Ov infection would allow the generation of reactive oxygen and nitrogen species, DNA damage, selection for apoptosis resistance, and increased mutation leading to carcinogenesis.
Fig. 6.
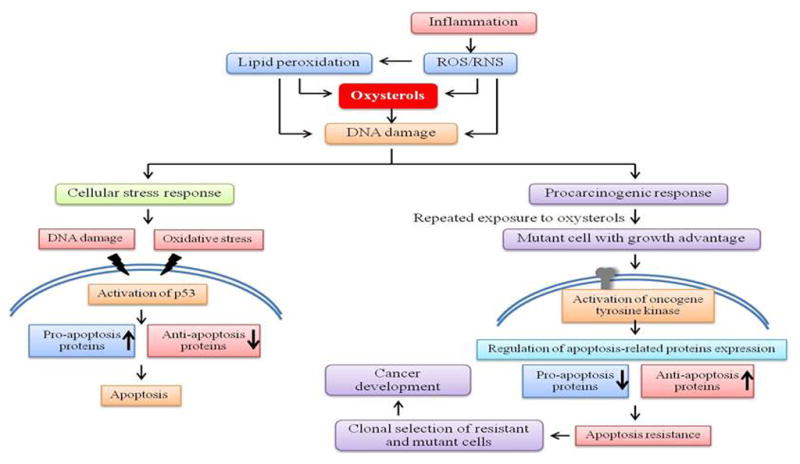
Proposed scheme showing the mechanisms of action of oxysterols in cholangiocarcinogenesis.
Highlights.
We examine the mechanisms of oxysterol-induced apoptosis resistance.
Long-term exposure to Triol causes apoptosis resistant cells.
Kinase signaling cascade was activated in Triol-exposed cells.
Increased expression of anti-apoptosis proteins was found in Triol-exposed cells.
Acknowledgments
This study was supported by the Thailand Research Fund through the Royal Golden Jubilee Ph.D. Program (Grant No. PHD/0177/2549) to AJ and PY, Thailand Research Fund through Window II (BRG 5280020); the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, through the Health Cluster (SHeP-GMS) Khon Kaen University to PY; and NIH grant CA114403 to RK. We thank Dr. Naoya Kobayashi (Department of Surgery, Okayama University Graduate School of Medicine and Dentistry, Okayama, Japan) for kindly providing the MMNK-1 cell line.
Abbreviations
- CCA
Cholangiocarcinoma
- Triol
cholestan-3β, 5α, 6β-triol
- 3K4
3-keto-cholest-4-ene
- p38-α
p38 mitogen-activated protein kinase α
- CREB
cAMP response element-binding
- ERK 1/2
extracellular signal-regulated kinase 1/2
- cIAP2
cellular inhibitor of apoptosis 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brown AJ, Jessup W. Oxysterols and atherosclerosis. Atherosclerosis. 1999;142:1–28. doi: 10.1016/s0021-9150(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 2.Bjorkhem I, Cedazo-Minguez A, Leoni V, Meaney S. Oxysterols and neurodegenerative diseases. Mol Aspects Med. 2009;30:171–9. doi: 10.1016/j.mam.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Jusakul A, Yongvanit P, Loilome W, Namwat N, Kuver R. Mechanisms of oxysterol-induced carcinogenesis. Lipids Health Dis. 2011;10:44. doi: 10.1186/1476-511X-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biasi F, Mascia C, Astegiano M, Chiarpotto E, Nano M, Vizio B, Leonarduzzi G, Poli G. Pro-oxidant and proapoptotic effects of cholesterol oxidation products on human colonic epithelial cells: a potential mechanism of inflammatory bowel disease progression. Free Radic Biol Med. 2009;47:1731–41. doi: 10.1016/j.freeradbiomed.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 5.Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, Marie JC, Pouzet C, Samadi M, Elbim C, et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol. 2004;24:10703–17. doi: 10.1128/MCB.24.24.10703-10717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vejux A, Malvitte L, Lizard G. Side effects of oxysterols: cytotoxicity, oxidation, inflammation, and phospholipidosis. Braz J Med Biol Res. 2008;41:545–56. doi: 10.1590/s0100-879x2008000700001. [DOI] [PubMed] [Google Scholar]
- 7.Monier S, Samadi M, Prunet C, Denance M, Laubriet A, Athias A, Berthier A, Steinmetz E, Jurgens G, Negre-Salvayre A, et al. Impairment of the cytotoxic and oxidative activities of 7 beta-hydroxycholesterol and 7-ketocholesterol by esterification with oleate. Biochem Biophys Res Commun. 2003;303:814–24. doi: 10.1016/s0006-291x(03)00412-1. [DOI] [PubMed] [Google Scholar]
- 8.Jaiswal M, LaRusso NF, Shapiro RA, Billiar TR, Gores GJ. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology. 2001;120:190–9. doi: 10.1053/gast.2001.20875. [DOI] [PubMed] [Google Scholar]
- 9.Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184–90. [PubMed] [Google Scholar]
- 10.Powell AA, Akare S, Qi W, Herzer P, Jean-Louis S, Feldman RA, Martinez JD. Resistance to ursodeoxycholic acid-induced growth arrest can also result in resistance to deoxycholic acid-induced apoptosis and increased tumorgenicity. BMC Cancer. 2006;6:219. doi: 10.1186/1471-2407-6-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. 2005;589:47–65. doi: 10.1016/j.mrrev.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Pinlaor S, Hiraku Y, Ma N, Yongvanit P, Semba R, Oikawa S, Murata M, Sripa B, Sithithaworn P, Kawanishi S. Mechanism of NO-mediated oxidative and nitrative DNA damage in hamsters infected with Opisthorchis viverrini: a model of inflammation-mediated carcinogenesis. Nitric Oxide. 2004;11:175–83. doi: 10.1016/j.niox.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Ohshima H, Bandaletova TY, Brouet I, Bartsch H, Kirby G, Ogunbiyi F, Vatanasapt V, Pipitgool V. Increased nitrosamine and nitrate biosynthesis mediated by nitric oxide synthase induced in hamsters infected with liver fluke (Opisthorchis viverrini) Carcinogenesis. 1994;15:271–5. doi: 10.1093/carcin/15.2.271. [DOI] [PubMed] [Google Scholar]
- 14.Jusakul A, Loilome W, Namwat N, Haigh WG, Kuver R, Dechakhamphu S, Sukontawarin P, Pinlaor S, Lee SP, Yongvanit P. Liver fluke-induced hepatic oxysterols stimulate DNA damage and apoptosis in cultured human cholangiocytes. Mutat Res. 2012;731:48–57. doi: 10.1016/j.mrfmmm.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Loilome W, Wechagama P, Namwat N, Jusakul A, Sripa B, Miwa M, Kuver R, Yongvanit P. Expression of oxysterol binding protein isoforms in opisthorchiasis-associated cholangiocarcinoma: a potential molecular marker for tumor metastasis. Parasitol Int. 2012;61:136–9. doi: 10.1016/j.parint.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Yoon JH, Canbay AE, Werneburg NW, Lee SP, Gores GJ. Oxysterols induce cyclooxygenase-2 expression in cholangiocytes: implications for biliary tract carcinogenesis. Hepatology. 2004;39:732–8. doi: 10.1002/hep.20125. [DOI] [PubMed] [Google Scholar]
- 17.O’Callaghan YC, Woods JA, O’Brien NM. Comparative study of the cytotoxicity and apoptosis-inducing potential of commonly occurring oxysterols. Cell biology and toxicology. 2001;17:127–37. doi: 10.1023/a:1010914306375. [DOI] [PubMed] [Google Scholar]
- 18.Gregorio-King CC, Gough T, Van Der Meer GJ, Hosking JB, Waugh CM, McLeod JL, Collier FM, Kirkland MA. Mechanisms of resistance to the cytotoxic effects of oxysterols in human leukemic cells. J Steroid Biochem Mol Biol. 2004;88:311–20. doi: 10.1016/j.jsbmb.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 19.Magnuson BA, Shirtliff N, Bird RP. Resistance of aberrant crypt foci to apoptosis induced by azoxymethane in rats chronically fed cholic acid. Carcinogenesis. 1994;15:1459–62. doi: 10.1093/carcin/15.7.1459. [DOI] [PubMed] [Google Scholar]
- 20.Crowley-Weber CL, Payne CM, Gleason-Guzman M, Watts GS, Futscher B, Waltmire CN, Crowley C, Dvorakova K, Bernstein C, Craven M, et al. Development and molecular characterization of HCT-116 cell lines resistant to the tumor promoter and multiple stress-inducer, deoxycholate. Carcinogenesis. 2002;23:2063–80. doi: 10.1093/carcin/23.12.2063. [DOI] [PubMed] [Google Scholar]
- 21.Namwat N, Amimanan P, Loilome W, Jearanaikoon P, Sripa B, Bhudhisawasdi V, Tassaneeyakul W. Characterization of 5-fluorouracil-resistant cholangiocarcinoma cell lines. Chemotherapy. 2008;54:343–51. doi: 10.1159/000151541. [DOI] [PubMed] [Google Scholar]
- 22.Cerella C, Coppola S, Maresca V, De Nicola M, Radogna F, Ghibelli L. Multiple mechanisms for hydrogen peroxide-induced apoptosis. Ann N Y Acad Sci. 2009;1171:559–63. doi: 10.1111/j.1749-6632.2009.04901.x. [DOI] [PubMed] [Google Scholar]
- 23.Seo DW, Choi HS, Lee SP, Kuver R. Oxysterols from human bile induce apoptosis of canine gallbladder epithelial cells in monolayer culture. American journal of physiology. 2004;287:G1247–56. doi: 10.1152/ajpgi.00013.2004. [DOI] [PubMed] [Google Scholar]
- 24.Ogawa Y, Takahashi T, Kobayashi T, Kariya S, Nishioka A, Mizobuchi H, Noguchi M, Hamasato S, Tani T, Seguchi H, et al. Mechanism of hydrogen peroxide-induced apoptosis of the human osteosarcoma cell line HS-Os-1. Int J Mol Med. 2003;12:459–63. [PubMed] [Google Scholar]
- 25.Vejux A, Lizard G. Cytotoxic effects of oxysterols associated with human diseases: Induction of cell death (apoptosis and/or oncosis), oxidative and inflammatory activities, and phospholipidosis. Molecular aspects of medicine. 2009;30:153–70. doi: 10.1016/j.mam.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Werneburg NW, Yoon JH, Higuchi H, Gores GJ. Bile acids activate EGF receptor via a TGF-alpha-dependent mechanism in human cholangiocyte cell lines. American journal of physiology. 2003;285:G31–6. doi: 10.1152/ajpgi.00536.2002. [DOI] [PubMed] [Google Scholar]
- 27.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barlow CA, Kitiphongspattana K, Siddiqui N, Roe MW, Mossman BT, Lounsbury KM. Protein kinase A-mediated CREB phosphorylation is an oxidant-induced survival pathway in alveolar type II cells. Apoptosis. 2008;13:681–92. doi: 10.1007/s10495-008-0203-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353:670–4. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 31.Lordan S, Mackrill JJ, O’Brien NM. Involvement of Fas signalling in 7beta-hydroxycholesterol-and cholesterol-5beta,6beta-epoxide-induced apoptosis. Int J Toxicol. 2008;27:279–85. doi: 10.1080/10915810802208616. [DOI] [PubMed] [Google Scholar]
- 32.Nishio E, Watanabe Y. Oxysterols induced apoptosis in cultured smooth muscle cells through CPP32 protease activation and bcl-2 protein downregulation. Biochem Biophys Res Commun. 1996;226:928–34. doi: 10.1006/bbrc.1996.1452. [DOI] [PubMed] [Google Scholar]
- 33.Lordan S, Mackrill JJ, O’Brien NM. Oxysterols and mechanisms of apoptotic signaling: implications in the pathology of degenerative diseases. J Nutr Biochem. 2009;20:321–36. doi: 10.1016/j.jnutbio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 34.Makridakis NM, Reichardt JK. Translesion DNA polymerases and cancer. Front Genet. 2012;3:174. doi: 10.3389/fgene.2012.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR, et al. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007;3:e110. doi: 10.1371/journal.pgen.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, Washburn MP, Florens L, Ladurner AG, Conaway JW, et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc Natl Acad Sci U S A. 2009;106:13770–4. doi: 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]