

Abstract
Aging and the presence of cerebrovascular disease are associated with increased incidence of Alzheimer’s disease (AD). A common feature of aging and cerebrovascular disease is decreased endothelial nitric oxide (NO). We studied the effect of a loss of endothelium derived NO on amyloid precursor protein (APP) related phenotype in late middle aged (LMA) (14–15 month) endothelial nitric oxide synthase deficient (eNOS−/−) mice. APP, β-site APP cleaving enzyme (BACE) 1, and amyloid beta (Aβ) levels were significantly higher in the brains of LMA eNOS−/− mice as compared to LMA wild type controls. APP and Aβ1-40 were increased in hippocampal tissue of eNOS−/− mice as compared to wild type mice. LMA eNOS−/− mice displayed an increased inflammatory phenotype as compared to LMA wild type mice. Importantly, LMA eNOS−/− mice performed worse in a radial arm maze test of spatial learning and memory as compared to LMA wild type mice. These data suggest that chronic loss of endothelial NO may be an important contributor to both Aβ related pathology and cognitive decline.
Keywords: Amyloid precursor protein, endothelium, nitric oxide, Alzheimer’s disease, memory, endothelial nitric oxide synthase
Introduction
Deposition of extracellular amyloid plaques and intracellular neurofibrillary tangles in the central nervous system are hallmarks of Alzheimer’s disease (AD). Other components of the disease are inflammation, neuronal loss, and progressive cognitive decline. Furthermore, vascular abnormalities and changes in blood flow are also a feature of AD (de la Torre 1997, Faraci 2011). Indeed, it has been demonstrated that endothelial dysfunction is an early event in AD transgenic mouse models and this precedes overt plaque development (Iadecola et al. 1999, Niwa et al. 2002). The cause of sporadic AD, which accounts for approximately 95% of AD cases, is unknown. Several risk factors have been identified. Of these risk factors, aging is the single greatest risk factor for the development of AD (Kukull et al. 2002). Cardiovascular risk factors are also associated with an increased incidence of AD. Importantly, one commonality between aging and cardiovascular risk factors is the decreased bioavailability of endothelial nitric oxide (NO) (Dudzinski et al. 2006).
Amyloid beta (Aβ) is the primary component of the extracellular plaques. Aβ is generated by the sequential cleavages of amyloid precursor protein (APP) by β-site APP cleaving enzyme (BACE) 1 and γ-secretase. We recently demonstrated that loss of endothelial NO led to increased expression of APP and BACE1 protein and Aβ levels in brain tissue suggesting that endothelial NO may play a role in the modulation of neuronal APP expression and amyloidogenic processing (Austin et al. 2012, Austin et al. 2010). We also demonstrated that supplementation of NO in endothelial nitric oxide synthase deficient (eNOS−/−) mice via nitroglycerin treatment was able to attenuate the upregulation of APP and BACE1 protein levels in the cerebral microvasculature (Austin et al. 2012). Furthermore, Pak and colleagues reported that NO downregulated BACE1 in human neuroblastoma cells (Pak et al. 2005). Lastly, Kwak et al demonstrated that treatment of cultured neurons with low concentrations of NO suppressed BACE1 levels (Kwak et al. 2011). These data support the hypothesis that endothelial NO modulates Aβ levels in the brain.
In this study, we sought to determine the effect of chronic loss of endothelial NO on several AD related pathologies using late middle aged (LMA) (14–15 month old) eNOS−/− mice. Our studies provide evidence that chronic loss of endothelial NO in LMA animals leads to increased APP expression and amyloidogenic processing, increased microglial activation, and impaired performance in a radial arm maze spatial memory test.
Materials and Methods
Animals
Male wild type (C57BL6) and eNOS−/− (Nos3tm1Unc/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME). All mice used in these experiments were on the C57BL/6J background. Mice had free access to food and water. Mice were sacrificed by lethal dose of pentobarbital at 4 months or 14–15 months of age. It has been published that age-dependent cardiac abnormalities, including: perturbations in left ventricular function, decreased ejection fraction, and septum thickening, occur in 18 month and older eNOS−/− mice (Li et al. 2004). To avoid this complication, we performed our studies on 14–15 months old animals. All animal care and use were approved by Mayo institutional Animal Care and Use Committee.
Tissue collection
Brains were carefully removed and immediately placed in ice cold modified Krebs-Ringer bicarbonate solution plus protease inhibitors. Large arteries, including the basilar and cerebral arteries, were removed. When noted, the hippocampus was removed from the brain to analyze hippocampal tissue alone.
Microvessel isolation
Cerebral microvessels were isolated from brain tissue as previously described (Austin et al. 2010). Briefly, brain tissue, devoid of large vessels, was homogenized in ice cold PBS with Dounce homogenizer. Microvessels were isolated by layering over 15% Dextran/PBS solution and filtering using a 40 μm filter. In the case of hippocampal microvessels, the hippocampus was dissected from the brain prior to isolation as described.
Confocal Microscopy
Mice were perfused with 4% paraformaldehyde. Brains were fixed in a 4% paraformaldehyde solution and frozen in OCT under isopentane on dry ice and stored at −80°C. Histological examinations were performed on sagittal sections (5 μm) mounted onto glass slides. Tissue was permeabilized using 0.1% Triton X-100 in 10% normal goat serum. Sections were incubated with primary antibodies and then incubated with Cy5-conjugated secondary antibody (Jackson Immuno Research). 4′,6′-diamiddino-2-phenylindole dilactate (DAPI) was used to visualize nuclei. Sections were visualized using a Zeiss LSM 510 laser scanning confocal microscope.
Western blot
Brain, hippocampal or microvessel tissue homogenates were lysed in ice cold Triton lysis buffer. Equal protein amounts were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Blots were probed with primary antibodies.
ELISA
SDS-soluble Aβ1-40 and Aβ1-42, granulocyte macrophage-colony stimulating factor (GM-CSF), interleukin (IL) 1α, and macrophage inflammatory protein (MIP) 1β were measured using a commercially available colorimetric ELISA kit per manufacturer’s instructions (Aβ1-40 and Aβ1-42: Invitrogen, Camarillo, CA; GM-CSF: Biosensis via Novus Biologicals, Littleton, CO; IL-1α and MIP-1β: R&D Systems, Minneapolis, MN)
Cytokine array
A commercially available mouse cytokine array (Mouse Cytokine Array Panel A) was used per manufacturer’s instructions (R&D Systems, Minneapolis, MN).
8-arm radial arm maze behavioral testing
Behavioral testing was modeled after Dubreuil et al (Dubreuil et al. 2003). Mice were progressively food-deprived until their weight reached 85% of their “ad libitum weight”. Mice were allowed to familiarize to the maze for 3 days. The maze was fully baited which required the mice to learn to visit each arm only once and retrieve a chocolate flavored food reinforcement. Daily sessions were performed over 7 days. Mouse was placed in a central area with all arms blocked. All arms were opened simultaneously, once a mouse selected a given arm, the other 7 arms closed. Upon entering the central area again all arm doors were closed and the mouse was confined for 5 seconds. Following the confinement, all doors reopened allowing the mouse to again select an arm. This procedure was repeated until all 8 food reinforcements were correctly collected or the time allotted to complete the task (10 minutes) was achieved. Data was automatically collected via the Med-PC IV program.
Open-field locomotor activity test
Mice were allowed to acclimate to the testing room for one hour prior to being placed in the open field chambers. Mice were placed in an open field box and locomotor activity was recorded automatically. Mouse activity was observed and total distance traveled was recorded during one hour sessions, daily, for 5 days.
Statistical analysis
Data are represented as mean ± SD. Statistical analysis was performed using the unpaired Student’s t-test or two-way ANOVA followed by Tukey-Kramer post hoc test for individual comparison.
Results
Metabolic phenotype in late middle aged wild type and eNOS−/− mice
LMA eNOS−/− mice displayed a phenotype consistent with metabolic syndrome (Supplemental Table 1). Systolic blood pressure, total cholesterol, HDL, triglycerides and glucose were all significantly elevated in LMA eNOS−/− mice as compared to LMA wild type mice (Supplemental Table 1). Body weight of LMA eNOS−/− mice was significantly lower than aged wild type mice.
Increased APP expression and processing in the brains of late middle aged eNOS−/− mice
APP and BACE1 protein levels were elevated in whole brain tissue of LMA eNOS−/− animals as compared to LMA wild type mice (Figure 1A–B, P<0.05). Consistent with increased BACE1 and thus amyloidogenic processing of APP, levels of Aβ1-40 and Aβ1-42 were significantly increased in whole brain tissue of LMA eNOS−/− mice as compared to LMA wild type mice (Figure 1A, 1C–D, P<0.05).
Figure 1.

Levels of APP, BACE1 and Aβ were increased in the brains of LMA eNOS−/− mice. (A) Fixed tissue sections from the brains of wild type and eNOS−/− animals were immunolabeled with anti-APP, anti-BACE1 and anti-Aβ. Representative images of the cortex are shown. Magnification 40x; bar is representative of 20 μm. (B) Brain tissue from eNOS−/− and wild type animals was analyzed via Western blotting using anti-APP, anti-BACE1, and anti-Actin (loading control) antibodies. Representative image and densitometric analysis is shown (n=12 animals). (C) Aβ1-40 and (D) Aβ1-42 levels from 8 individual brain lysates (250 μg total protein/sample) from LMA eNOS−/− and LMA wild type control were analyzed via commercially available ELISA kits. Data are represented as mean ± SD (*P<0.05 compared to wild type control mice).
To determine if loss of endothelial NO led to any compensatory expression of other NOS isoforms we examined protein levels of inducible NOS (iNOS) and neuronal NOS (nNOS) in whole brain tissue from LMA wild type and eNOS−/− animals. No compensatory increases were observed (Supplemental Figure 1A).
Next, we looked for other AD related changes that might be present in our model of chronic loss of endothelial-derived NO. We examined protein levels and phosphorylation of Tau. No changes were observed between LMA eNOS−/− animals and LMA wild type animals (Supplemental Figure 1B). Whole brain tissue derived from eNOS−/− mice demonstrated significantly higher levels of the microglial marker, cluster of differentiation (CD) 68, while markers for astrocytes (GFAP) and neurons (neuronal nuclei [NeuN]) were unaltered (Figure 2, P<0.05). To expand on this observation, we examined expression of several other microglial markers: CD68, ionized calcium-binding adaptor molecule (Iba) 1, and MHC II, in 4 month and 15 month old eNOS−/− mice. There was a significant increase based on genotype of all three microglial markers (Figure 3, eNOS−/− as compared to wild type control mice; P<0.05).
Figure 2.

Levels of the microglial marker, CD68, are increased in the brains of LMA eNOS−/− mice. (A) Fixed tissue sections from the brains of LMA eNOS−/− and LMA wild type mice were immunolabeled with anti-CD68. Representative images of the cortex are shown. Magnification 40x; bar is representative of 20μm. (B) Brain tissue from LMA eNOS−/− and LMA wild type animals was Western blotted using anti-CD68, anti-GFAP, anti-NeuN, and anti-Actin (loading control) antibodies. Representative image is shown. Densitometric analysis was performed for (C) CD68, (D) GFAP, and (E) NeuN. Data are presented as mean ±SD (*P<0.05 compared to wild type control mice, n=10–12 animals).
Figure 3.

Levels of CD68, Iba-1, and MHC II were increased in the brains of eNOS−/− mice. (A) Brain tissue from 4 month or 15 month old eNOS−/− and wild type mice was Western blotted using anti-CD68, anti-Iba-1, anti-MHC II, or anti-Actin (loading control) antibodies. Representative image is shown. Densitometric analysis was performed for (B) CD68, (C) Iba-1, and (D) MHC II. Data are presented as mean ± SD (n=8 animals, *P<0.05 compared to age-matched wild type control; #P<0.05 based on genotype: 2-way ANOVA, followed by Tukey-Kramer post hoc tests for individual comparisons).
Elevated APP expression and processing in the hippocampus of late middle aged eNOS−/− mice
We next examined the hippocampus, a brain region dramatically affected during AD, of LMA eNOS−/− animals. APP protein levels were significantly higher in the hippocampus of LMA eNOS−/− mice as compared to LMA wild type mice (Figure4A–B, P<0.05). While BACE1 levels were unchanged (Figure 4A–B), there were significantly higher levels of Aβ1-40 in hippocampal tissue from LMA eNOS−/− animals as compared to wild type mice (Figure 4A, 4C, P<0.01). No changes were observed in Aβ1-42 levels (Figure 4D).
Figure 4.

APP and Aβ1-40 levels were increased in the hippocampus of LMA eNOS−/− mice. (A) Fixed tissue sections from the brains of wild type and eNOS−/− animals were immunolabeled with anti-APP, anti-BACE1 and anti-Aβ. Representative images of the hippocampus are shown. Magnification 40x; bar is representative of 20 μm. (B) Hippocampal tissue from LMA eNOS−/− and LMA wild type animals was Western blotted using anti-APP, anti-BACE1, and anti-Actin (loading control) antibodies. Representative image and densitometric analysis is shown. (C) Aβ1-40 and (D) Aβ1-42 levels from hippocampal lysates (200 μg total protein/sample) from LMA eNOS−/− and wild type control mice were analyzed via commercially available ELISA kits. Data are represented as mean ± SD (n=6 animals, *P<0.05, **P<0.01 compared to wild type control mice).
No alterations in iNOS or nNOS were observed in the hippocampus of LMA eNOS−/− as compared to LMA wild type mice (Supplemental Figure 2A). Furthermore, no changes in Tau protein expression or phosphorylation were observed between hippocampal tissue from LMA eNOS−/− and wild type mice (Supplemental Figure 2B). Lastly, no changes were observed in cell specific markers for astrocytes, microglia or neurons in the hippocampal region of LMA eNOS−/− as compared to the LMA wild type mice (Supplemental Figure 3).
Cerebral microvessels from late middle aged eNOS−/− and wild type mice
We next examined microvessels isolated from the whole brain or hippocampal region of LMA eNOS−/− mice. No changes in APP or BACE1 expression were seen in either the brain microvessels or hippocampal microvessels of LMA eNOS−/− mice as compared to wild type mice (Supplemental Figure 4A, Supplemental Figure 5A, respectively). Importantly, no compensatory upregulation of iNOS protein expression occurred in LMA eNOS−/− brain or hippocampal microvessels as compared to LMA wild type microvessels (Supplemental Figure 4B, Supplemental Figure 5B, respectively). nNOS was not detected in microvascular tissue (Supplemental Figures 4B and 5B).
Inflammatory phenotype of late middle aged eNOS−/− mice
To follow up on our observation of statistically higher expression of several microglial markers, we next performed a cytokine array which detected 40 cytokines. Brain tissue from LMA eNOS−/− mice displayed higher levels of GM-CSF, IL-1α, and MIP-1β as compared to LMA wild type brain tissue (Figure 5A). No other obvious differences were observed in the array measured cytokines between LMA eNOS−/− and wild type brain tissue. To confirm, we next performed individual ELISA assays for GM-CSF, IL-1α, and MIP-1β. Levels of GM-CSF, IL-1α, and MIP-1β were significantly higher in brain tissue from LMA eNOS−/− mice as compared to LMA wild type mice (Figure 5B–5D).
Figure 5.
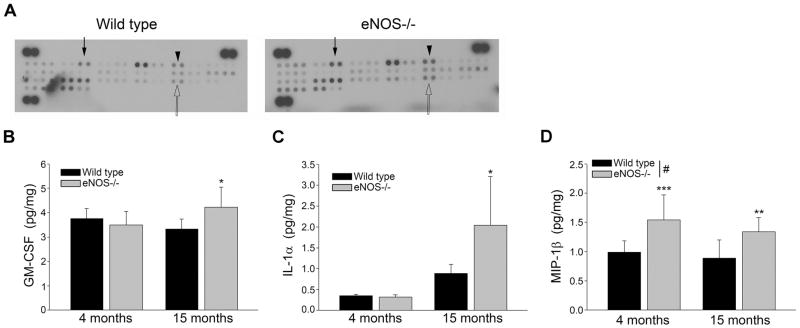
Levels of GM-CSF, IL-1α, and MIP-1β are increased in the brains of LMA eNOS−/− mice. (A) Brain lysates from LMA eNOS−/− and age-matched wild type control mice were analyzed via a commercially available cytokine array (300μg total protein loaded for each sample). A representative image is shown. Closed arrow depicts GM-CSF, arrowhead depicts IL-1α, and open arrow depicts MIP-1β. (B) GM-CSF (C) IL-1α, and (D) MIP-1β levels from 8 individual brain lysates (500 μg) from 4 month and 15 month old eNOS−/− and wild type control mice were analyzed via commercially available ELISA kits. Data are represented as mean ± SD (n=7–8 animals, *P<0.05, **P<0.01, and ***P<0.001 compared to age-matched wild type control mice; #P<0.05 based on genotype: 2-way ANOVA, followed by Tukey-Kramer post hoc tests for individual comparisons).
eNOS−/− mice performance in an 8-arm radial arm maze
Finally, we observed LMA eNOS−/− and wild type mice in an 8-arm radial arm maze to assess performance in a spatial learning and memory task. LMA eNOS−/− completed significantly more incorrect runs than did the LMA wild type mice (Figure 6A). When examining the number of revisiting errors on day 1, wild type mice had significantly more revisiting errors than did eNOS−/− mice (Figure 6B); however, by day 5 LMA wild type mice had a significantly lower number of revisiting errors as compared to the LMA eNOS−/− mice (Figure 6B). There was no difference in the time it took LMA eNOS−/− mice and wild type mice to complete the maze (Figure 6C). We also performed an open-field test to determine if the differences seen in the radial arm maze were simply due to a difference in locomotor activity. No difference in the distance traveled was detected between genotypes (Supplemental Figure 6).
Figure 6.
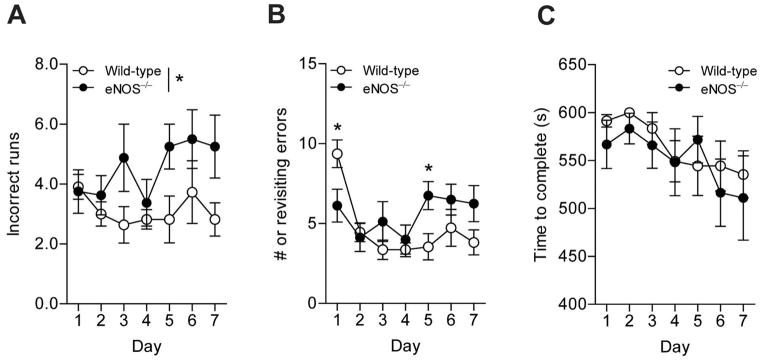
LMA eNOS−/− animals committed more errors than LMA wild type mice in an 8-arm radial arm maze. The number of (A) incorrect runs, (B) revisiting errors, and (C) time to complete were observed over the course of 7 days of testing. Data are presented as ±SD (*P<0.05, n=8–11 animals).
Discussion
Our results support the hypothesis that endothelial dysfunction, and specifically loss of endothelial NO, may be an important contributor to the pathogenesis of sporadic AD. We previously demonstrated increased amyloidogenic processing of APP in young eNOS−/− mice as compared to age matched wild type mice (Austin et al. 2012, Austin et al. 2010). Here, we confirmed that APP and BACE1 expression and production of Aβ were significantly higher in LMA eNOS−/− brains as compared to LMA wild type animals. However, we also report several important and novel observations in LMA eNOS−/− mice. First, we believe this is the first report of inflammatory changes found in the brains of LMA eNOS−/− animals under basal conditions. Several microglial markers (CD68, Iba–1, and MHC II) were significantly higher in the brains of LMA eNOS−/− mice and this was accompanied by higher levels of the cytokines: GM-CSF, IL-1α, and MIP-1β, as compared to LMA wild type mice. Second, we report significantly increased APP and Aβ1-40 levels in the hippocampus, a region critically involved in the pathogenesis of AD. Third, we report behavioral alterations in LMA eNOS−/− mice as compared to LMA wild type mice.
Several studies have suggested an association between NO and the development of AD; however, the literature is controversial. Kummer et al demonstrated that treating APP/PS1 mice, a transgenic model of AD, with an iNOS inhibitor decreased Aβ deposition and cognitive dysfunction by limiting tyrosine nitrosylation of Aβ (Kummer et al. 2011). Furthermore, several groups examined the role of iNOS in the pathogenesis of AD by utilizing AD mouse models which also lacked iNOS (Colton et al. 2008, Kummer et al. 2011, Nathan et al. 2005). Nathan et al and Kummer et al reported that genetic inactivation of iNOS protected AD mice from plaque formation, Aβ accumulation and microgliosis (Nathan et al. 2005) as well as memory deficits (Kummer et al. 2011). In contrast, Colton and colleagues reported that loss of iNOS activity worsened all symptoms in their AD mouse model (Colton et al. 2008). Similarly, several studies also report on nNOS and AD with conflicting findings (Lahiri et al. 2003, Martin et al. 2006, Simic et al. 2000). Simic et al demonstrated increased nNOS in the hippocampus and entorhinal cortex in areas around plaques and overt cell loss in AD patients as compared to age-matched controls (Simic et al. 2000). In addition, Martin et al reported increased nNOS activity in Tg2576 mice, a murine model of AD (Martin et al. 2006) conversely, Lahiri et al reported no statistical difference in nNOS activity in APP/PS1 transgenic mice as compared to control mice (Lahiri et al. 2003). While numerous studies have examined the role of iNOS and nNOS in relation to AD a systematic examination of the role of eNOS in the pathogenesis of AD is lacking. In the present study the rationale for focusing on eNOS was based on the established differences among the three isoforms of NOS in terms of biochemical and functional properties.
In our studies, we have utilized a murine model deficient in eNOS to help determine the effects of a chronic loss of endothelial NO in AD-related pathologies, including: APP expression and processing, Aβ generation, inflammation, and cognitive function. Notably, this is not a traditional mouse model for Alzheimer’s disease, in that eNOS−/− mice express unaltered wild type murine APP and secretase enzymes; however, it allows for the measurements of physiological alterations of APP and Aβ in response to a loss of endothelial NO. It is also important to note that, murine Aβ behaves much differently than human Aβ. Most importantly, murine Aβ does not form fibrils and therefore an elevated concentration of murine Aβ does not result in the formation of plaques (Dyrks et al. 1993). Future studies will include examination of AD mouse models that lack eNOS to determine the role of endothelial NO in the progression of AD.
Due to their close proximity, endothelial NO appears to be an important signaling molecule, between the vascular wall and neurons (Hopper & Garthwaite 2006, Garthwaite et al. 2006). Consistent with our previous findings in young eNOS−/− mice, we demonstrated that loss of endothelial NO led to increased expression of APP and BACE1 protein in brain tissue of LMA eNOS−/− mice (Austin et al. 2012, Austin et al. 2010). In line with our observations, Gutsaeva and colleagues detected changes in the expression of mitochondrial proteins within neurons in eNOS−/− mice demonstrating an important role of endothelial NO in control of protein expression in neuronal tissue (Gutsaeva et al. 2008). However, we have yet to determine the mechanism by which NO is able to suppress APP and BACE1 expression. Of note, the APP promoter contains a stimulating protein (Sp)1 site and the BACE1 promoter and 5′ untranslated region contains binding sites for cAMP response element binding protein (CREB), nuclear factor κB (NFκB), Sp1, and yin yang (YY)1 (Hoffman & Chernak 1995, Sambamurti et al. 2004) which can be modified by nitric oxide (Berendji-Grun et al. 2001, Hongo et al. 2005, Lu et al. 1999, Peng et al. 1995).
As previously reported, we detected significant changes in blood pressure, lipid profile, and glucose levels in LMA eNOS−/− mice as compared to LMA wild type controls (Huang 2009). These metabolic changes could contribute to the phenotype of the LMA eNOS−/− mice we report here; however, it is important to note that we previously reported that inhibition or loss of eNOS derived NO led to increased APP, BACE1, and Aβ levels in vitro in the absence of hemodynamic forces or alterations in metabolic factors (Austin et al. 2010). Furthermore, we reported increased brain APP, BACE1, and Aβ in young eNOS−/− which did not display significant differences in their metabolic profile as compared to young wild type mice (Austin et al. 2012, Austin et al. 2010).
To the best of our knowledge, this is the first report of an altered inflammatory response, specifically an increase in the microglial markers CD68, Iba-1, and MHCII in the brains of LMA eNOS−/− mice. As the immunocompetent cell of the central nervous system, microglia are constantly contacting neurons, other glia and the cells of the vasculature to survey and sample the environment (Nimmerjahn et al. 2005). Thus, it appears that microglia could detect alterations in the production of endothelial NO although it is unclear whether the increased microglial activation reported here is due to loss of NO per se. We observed increased GM-CSF, IL-1α, and MIP-1β, in the brains of LMA eNOS−/− mice as compared to wild type mice. As of yet, we do not know the source or the consequence of the increased GM-CSF, IL-1α, and MIP-1β we observed in the brains of eNOS−/− mice and this will require further investigation.
In the hippocampus, we observed an increase in Aβ1-40 levels while Aβ1-42 levels were unchanged. The mechanism or consequence of this preferential generation of Aβ1-40 remains to be determined. Numerous studies have demonstrated that administration of either Aβ1-40 or Aβ1-42 in rodents leads to a number of deleterious effects, such as: LTP dysfunction, neuronal loss, cellular stress, inflammation, and cognitive deficits [reviewed in (Chambon et al. 2011)]. Furthermore, it has been demonstrated that in vitro treatment of hippocampal neuronal cultures with Aβ1-40 and Aβ1-42 is highly neurotoxic; however, Aβ1-42 treatment exhibited significantly higher levels of toxicity (Dore et al. 1997). Other studies suggest that Aβ1-40 may be neuroprotective or at least require seeding of Aβ1-42 to produce toxicity (Abramowski et al. 2012, Vasilevko et al. 2010, Viet & Li 2012, Zou et al. 2002, Zou et al. 2003). Lastly, numerous studies suggest that both the concentration and conformation of Aβ peptides determines the neurotrophic or neurotoxic effects of the Aβ peptide (Busciglio et al. 1992, Hoshi et al. 2003, Yankner et al. 1990).
The hippocampal region is one of the earliest and most greatly altered regions of the brain affected by AD (Hyman et al. 1984). The hippocampus in humans plays a prominent role in the formation of new memories or newly learned information and thus early symptoms of AD include difficulty remembering recent events or newly acquired information (Hyman et al. 1984). In rodents, the hippocampal region is responsible for spatial learning and memory (O’Keefe & Dostrovsky 1971). Therefore, we used the 8-arm radial arm maze to assess the spatial memory and learning abilities of LMA eNOS−/− and wild type mice (Dubreuil et al. 2003). We report a spatial memory task deficit in LMA eNOS−/− animals as these mice made more incorrect runs as compared to LMA wild type mice in the radial arm maze. Differences were also observed in the number of revisiting errors. At first, LMA eNOS−/− mice appeared to perform better than the LMA wild type mice as eNOS−/− mice made less revisiting errors on the first day of the test. However, by day 5, LMA eNOS−/− mice were actually committing more revisiting errors than their wild type counterparts. These data illustrate that the LMA eNOS−/− mice have a deficit in spatial learning. It is unclear exactly why LMA eNOS−/− mice initially made less revisiting errors than wild type mice in the radial arm maze experiment; however, previous behavioral studies on young wild type and eNOS−/− mice have reported conflicting findings. Dere et al reported no change in performance of young eNOS−/− and wild type in the radial arm maze while Frisch and colleagues reported superior performance by eNOS−/− mice in a water maze as compared to wild type (Dere et al. 2001, Frisch et al. 2000). One possible explanation is that while both the water maze and the radial arm maze are testing spatial memory, the water maze is a negative reinforcement test while the radial arm maze is a positive reinforcement test.
We did not observe any differences in locomotor activity between LMA eNOS−/− and LMA wild type control mice; however, some groups have reported decreased activity in the open field test by eNOS−/− mice as compared to wild type control animals at both a young and old age (Dere et al. 2002, Frisch et al. 2000). We only examined LMA eNOS−/− and LMA wild type mice in this study. While we do not have an exact explanation for the difference there are several considerations. Dere et al examined locomotor activity of eNOS−/− and wild type mice that were 18–22 months old whereas our studies were performed on mice 14–15 months old (Dere et al. 2002). Importantly, it has been published that age-dependent cardiac abnormalities, including: perturbations in left ventricular function, decreased ejection fraction, and septum thickening, occur in 18 month and older eNOS−/− mice (Li et al. 2004). These changes in cardiac phenotype could lead to altered locomotor activity. We also wish to point out that the eNOS−/− mice used in the above mentioned studies were generated by disrupting the NADPH binding site (exons 24 and 25) whereas the eNOS−/− mice we used were generated by disrupting the calmodulin binding domain (exon 12) (Dere et al. 2002, Dere et al. 2001, Frisch et al. 2000, Godecke et al. 1998).
The exact cause of the spatial memory deficit observed in our study remains to be elucidated; however, several reports demonstrate the importance of the NO pathway in the formation of LTP and hippocampal-derived cognitive function (Arancio et al. 1996, Lu et al. 1999). Indeed, several groups have suggested modulation of the NO/cGMP to be used as a therapeutic target for the treatment of cognitive dysfunction in AD (Domek-Lopacinska & Strosznajder 2010, Puzzo et al. 2009, Reneerkens et al. 2009, Thatcher et al. 2004). Further studies will be needed to determine the exact changes in the brain causing the decreased performance by the LMA eNOS−/− mice in this particular maze.
In summary, our studies demonstrate that NO produced by the vasculature modulates APP expression and processing in brain tissue of LMA mice. The present studies also report several novel observations regarding the chronic loss of endothelial derived NO and microglia and cognitive performance. Taken together, our studies suggest that preservation of endothelial NO production and biological signaling are important potential therapeutic targets in the treatment of MCI and AD.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants HL-111062 and HL-91867, the Mayo Alzheimer’s Disease Research Center (Z.S.K.), AHA scientist development grant (0835436N, A.V.S.), AHA postdoctoral fellowship 12POST8550003 (S.A.A.), Clinical Pharmacology Training Grant (T32 GM08685) (Trainee S.A.A.), the Samuel Johnson Foundation for Genomics of Addiction Program at Mayo Clinic (D.-S.C.) and the Mayo Foundation.
Glossary
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- Aβ
amyloid beta
- BACE1
β-site APP cleaving enzyme 1
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- nNOS
nitric oxide synthase
- NO
nitric oxide
- MIP1-β
macrophage inflammatory protein 1 beta
- GM-CSF
granulocyte macrophage-colony stimulating factor
- IL-1α
interleukin 1 alpha
- CD68
cluster of differentiation 68
- Iba-1
ionized calcium-binding adaptor molecule 1
- MHC II
major histocompatibility complex II
- HDL
high density lipoprotein
- GFAP
glial fibrillary acidic protein
- NeuN
neuronal nuclei
- LTP
long term potentiation
- LMA
late middle aged
- Sp1
stimulating protein 1
- CREB
cAMP response element binding protein
- NFκB
nuclear factor κB
- YY1
yin yang 1
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to disclose.
Supplemental information is available at the Journal of Neurochemistry website at http://onlinelibrary.wiley.com.
References
- Abramowski D, Rabe S, Upadhaya AR, et al. Transgenic expression of intraneuronal Abeta42 but not Abeta40 leads to cellular Abeta lesions, degeneration, and functional impairment without typical Alzheimer’s disease pathology. J Neurosci. 2012;32:1273–1283. doi: 10.1523/JNEUROSCI.4586-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio O, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. Nitric oxide acts as a retrograde messenger during long-term potentiation in cultured hippocampal neurons. J Physiol Paris. 1996;90:321–322. doi: 10.1016/s0928-4257(97)87907-7. [DOI] [PubMed] [Google Scholar]
- Austin SA, d’Uscio LV, Katusic ZS. Supplementation of Nitric Oxide Attenuates AbetaPP and BACE1 Protein in Cerebral Microcirculation of eNOS-Deficient Mice. J Alzheimers Dis. 2012 doi: 10.3233/JAD-2012-121351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin SA, Santhanam AV, Katusic ZS. Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ Res. 2010;107:1498–1502. doi: 10.1161/CIRCRESAHA.110.233080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendji-Grun D, Kolb-Bachofen V, Kroncke KD. Nitric oxide inhibits endothelial IL-1[beta]-induced ICAM-1 gene expression at the transcriptional level decreasing Sp1 and AP-1 activity. Mol Med. 2001;7:748–754. [PMC free article] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yankner BA. Methodological variables in the assessment of beta amyloid neurotoxicity. Neurobiol Aging. 1992;13:609–612. doi: 10.1016/0197-4580(92)90065-6. [DOI] [PubMed] [Google Scholar]
- Chambon C, Wegener N, Gravius A, Danysz W. Behavioural and cellular effects of exogenous amyloid-beta peptides in rodents. Behav Brain Res. 2011;225:623–641. doi: 10.1016/j.bbr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Colton CA, Wilcock DM, Wink DA, Davis J, Van Nostrand WE, Vitek MP. The effects of NOS2 gene deletion on mice expressing mutated human AbetaPP. J Alzheimers Dis. 2008;15:571–587. doi: 10.3233/jad-2008-15405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC. Cerebromicrovascular pathology in Alzheimer’s disease compared to normal aging. Gerontology. 1997;43:26–43. doi: 10.1159/000213834. [DOI] [PubMed] [Google Scholar]
- Dere E, De Souza Silva MA, Topic B, Fiorillo C, Li JS, Sadile AG, Frisch C, Huston JP. Aged endothelial nitric oxide synthase knockout mice exhibit higher mortality concomitant with impaired open-field habituation and alterations in forebrain neurotransmitter levels. Genes Brain Behav. 2002;1:204–213. doi: 10.1034/j.1601-183x.2002.10402.x. [DOI] [PubMed] [Google Scholar]
- Dere E, Frisch C, De Souza Silva MA, Godecke A, Schrader J, Huston JP. Unaltered radial maze performance and brain acetylcholine of the endothelial nitric oxide synthase knockout mouse. Neuroscience. 2001;107:561–570. doi: 10.1016/s0306-4522(01)00382-7. [DOI] [PubMed] [Google Scholar]
- Domek-Lopacinska KU, Strosznajder JB. Cyclic GMP and nitric oxide synthase in aging and Alzheimer’s disease. Mol Neurobiol. 2010;41:129–137. doi: 10.1007/s12035-010-8104-x. [DOI] [PubMed] [Google Scholar]
- Dore S, Kar S, Quirion R. Insulin-like growth factor I protects and rescues hippocampal neurons against beta-amyloid- and human amylin-induced toxicity. Proc Natl Acad Sci U S A. 1997;94:4772–4777. doi: 10.1073/pnas.94.9.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil D, Tixier C, Dutrieux G, Edeline JM. Does the radial arm maze necessarily test spatial memory? Neurobiol Learn Mem. 2003;79:109–117. doi: 10.1016/s1074-7427(02)00023-0. [DOI] [PubMed] [Google Scholar]
- Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- Dyrks T, Dyrks E, Masters CL, Beyreuther K. Amyloidogenicity of rodent and human beta A4 sequences. FEBS Lett. 1993;324:231–236. doi: 10.1016/0014-5793(93)81399-k. [DOI] [PubMed] [Google Scholar]
- Faraci FM. Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol. 2011;300:H1566–1582. doi: 10.1152/ajpheart.01310.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch C, Dere E, Silva MA, Godecke A, Schrader J, Huston JP. Superior water maze performance and increase in fear-related behavior in the endothelial nitric oxide synthase-deficient mouse together with monoamine changes in cerebellum and ventral striatum. J Neurosci. 2000;20:6694–6700. doi: 10.1523/JNEUROSCI.20-17-06694.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite G, Bartus K, Malcolm D, Goodwin D, Kollb-Sielecka M, Dooldeniya C, Garthwaite J. Signaling from blood vessels to CNS axons through nitric oxide. J Neurosci. 2006;26:7730–7740. doi: 10.1523/JNEUROSCI.1528-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Godecke S, Schrader J. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H, Piantadosi CA. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci. 2008;28:2015–2024. doi: 10.1523/JNEUROSCI.5654-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PW, Chernak JM. DNA binding and regulatory effects of transcription factors SP1 and USF at the rat amyloid precursor protein gene promoter. Nucleic Acids Res. 1995;23:2229–2235. doi: 10.1093/nar/23.12.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongo F, Garban H, Huerta-Yepez S, Vega M, Jazirehi AR, Mizutani Y, Miki T, Bonavida B. Inhibition of the transcription factor Yin Yang 1 activity by S-nitrosation. Biochem Biophys Res Commun. 2005;336:692–701. doi: 10.1016/j.bbrc.2005.08.150. [DOI] [PubMed] [Google Scholar]
- Hopper RA, Garthwaite J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J Neurosci. 2006;26:11513–11521. doi: 10.1523/JNEUROSCI.2259-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, Sato K. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci U S A. 2003;100:6370–6375. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab. 2009;20:295–302. doi: 10.1016/j.tem.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Niwa K, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, van Belle G, Jolley L, Larson EB. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, Delekarte A, et al. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron. 2011;71:833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Kwak YD, Wang R, Li JJ, Zhang YW, Xu H, Liao FF. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener. 2011;6:17. doi: 10.1186/1750-1326-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri DK, Chen D, Ge YW, Farlow M, Kotwal G, Kanthasamy A, Ingram DK, Greig NH. Does nitric oxide synthase contribute to the pathogenesis of Alzheimer’s disease?: effects of beta-amyloid deposition on NOS in transgenic mouse brain with AD pathology. Ann N Y Acad Sci. 2003;1010:639–642. doi: 10.1196/annals.1299.117. [DOI] [PubMed] [Google Scholar]
- Li W, Mital S, Ojaimi C, Csiszar A, Kaley G, Hintze TH. Premature death and age-related cardiac dysfunction in male eNOS-knockout mice. J Mol Cell Cardiol. 2004;37:671–680. doi: 10.1016/j.yjmcc.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BL, Tokheim AM, McCarthy PT, Doms BS, Davis AA, Armitage IM. Metallothionein-3 and neuronal nitric oxide synthase levels in brains from the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Biochem. 2006;283:129–137. doi: 10.1007/s11010-006-2390-7. [DOI] [PubMed] [Google Scholar]
- Nathan C, Calingasan N, Nezezon J, et al. Protection from Alzheimer’s-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005;202:1163–1169. doi: 10.1084/jem.20051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol. 2002;283:H315–323. doi: 10.1152/ajpheart.00022.2002. [DOI] [PubMed] [Google Scholar]
- O’Keefe J, Dostrovsky J. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res. 1971;34:171–175. doi: 10.1016/0006-8993(71)90358-1. [DOI] [PubMed] [Google Scholar]
- Pak T, Cadet P, Mantione KJ, Stefano GB. Morphine via nitric oxide modulates beta-amyloid metabolism: a novel protective mechanism for Alzheimer’s disease. Med Sci Monit. 2005;11:BR357–366. [PubMed] [Google Scholar]
- Peng HB, Libby P, Liao JK. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J Biol Chem. 1995;270:14214–14219. doi: 10.1074/jbc.270.23.14214. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Staniszewski A, Deng SX, et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. J Neurosci. 2009;29:8075–8086. doi: 10.1523/JNEUROSCI.0864-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reneerkens OA, Rutten K, Steinbusch HW, Blokland A, Prickaerts J. Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology (Berl) 2009;202:419–443. doi: 10.1007/s00213-008-1273-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambamurti K, Kinsey R, Maloney B, Ge YW, Lahiri DK. Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. 2004;18:1034–1036. doi: 10.1096/fj.03-1378fje. [DOI] [PubMed] [Google Scholar]
- Simic G, Lucassen PJ, Krsnik Z, Kruslin B, Kostovic I, Winblad B, Bogdanovi nNOS expression in reactive astrocytes correlates with increased cell death related DNA damage in the hippocampus and entorhinal cortex in Alzheimer’s disease. Exp Neurol. 2000;165:12–26. doi: 10.1006/exnr.2000.7448. [DOI] [PubMed] [Google Scholar]
- Thatcher GR, Bennett BM, Dringenberg HC, Reynolds JN. Novel nitrates as NO mimetics directed at Alzheimer’s disease. J Alzheimers Dis. 2004;6:S75–84. doi: 10.3233/jad-2004-6s614. [DOI] [PubMed] [Google Scholar]
- Vasilevko V, Passos GF, Quiring D, Head E, Kim RC, Fisher M, Cribbs DH. Aging and cerebrovascular dysfunction: contribution of hypertension, cerebral amyloid angiopathy, and immunotherapy. Ann N Y Acad Sci. 2010;1207:58–70. doi: 10.1111/j.1749-6632.2010.05786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viet MH, Li MS. Amyloid peptide Abeta40 inhibits aggregation of Abeta42: evidence from molecular dynamics simulations. J Chem Phys. 2012;136:245105. doi: 10.1063/1.4730410. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- Zou K, Gong JS, Yanagisawa K, Michikawa M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci. 2002;22:4833–4841. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou K, Kim D, Kakio A, et al. Amyloid beta-protein (Abeta)1-40 protects neurons from damage induced by Abeta1-42 in culture and in rat brain. J Neurochem. 2003;87:609–619. doi: 10.1046/j.1471-4159.2003.02018.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.