

Summary
AMP-Activated protein kinase (AMPK) monitors cellular energy, regulates genes involved in ATP synthesis and consumption, and is allosterically activated by nucleotides and synthetic ligands. Analysis of the intact enzyme by hydrogen/deuterium exchange mass spectrometry reveals conformational perturbations of AMPK in response to binding of nucleotides, cyclodextrin and a synthetic small molecule activator, A769662. Results from this analysis clearly show that binding of AMP leads to conformational changes primarily in the γ subunit of AMPK and subtle changes in the α and β subunits. In contrast, A769662 causes profound conformational changes in the glycogen binding module of the β subunit and in the kinase domain of the α subunit suggesting that the molecular binding site of latter resides between the α and β subunits. The distinct short and long-range perturbations induced upon binding of AMP and A769662 suggest fundamentally different molecular mechanisms for activation of AMPK by these two ligands.
Keywords: diabetes, anti-diabetic agent, allosteric modulation, AMPK activation, synthetic ligands, structure, HDX
Introduction
Mammals use energy derived from the oxidation of carbon present in food sources to generate ATP from ADP. However, the balance between energy-storing and energy-consuming processes in the body is constantly changing, and the ability to sense changes in energy stores is essential for survival and thus evolutionarily conserved (Kahn et al., 2005). Classic genetic experiments in lower eukaryotes demonstrate that the AMP-activated protein kinase (AMPK) is a critical sensor of energy stores (Celenza and Carlson, 1984; Momcilovic et al., 2006; Schimmack et al., 2006). At times of metabolic stress such as during exercise, hypoxia and cell proliferation, AMPK becomes activated in response to the increased AMP:ATP ratio resulting from higher ATP consumption (Carling et al., 2012; Hardie et al., 2011; Hardie et al., 2012; Johnson et al., 2010; Oakhill et al., 2012; Steinberg and Kemp, 2009). As a result, AMPK is widely appreciated as a critical metabolic regulator and hence is an attractive target for the treatment of metabolic diseases. In particular, AMPK has been shown to play a role in life-span extension in response to calorie restriction and mediates glucose flux in cells. AMPK partially mediates the metabolic action of the anti-diabetes drug metformin, as well as those of compounds with known anti-cancer properties such as resveratrol (Hwang et al., 2009; Vingtdeux et al., 2011). This latter finding has implications for cancer research, and recent studies show that AMPK is involved in basic cellular processes such as growth and proliferation, inflammation, autophagy, and maintenance of cell polarity (Chen et al., 2011; Marx et al., 2010; Mihaylova and Shaw, 2011; Shang and Wang, 2011).
In muscle, activation of AMPK stimulates fatty acid oxidation, mitochondrial biosynthesis, and glucose uptake while the precise role of AMPK activation in liver has been controversial. Activation of AMPK also results in inhibition of de novo synthesis of fatty acid in adipose tissue and liver. Physical exercise has also been shown to activate AMPK in heart (Cacicedo et al., 2011) and muscle (Itani et al., 2003). Given the increasingly global epidemic of obesity and diabetes, the biomedical community has been seeking novel pharmacological targets for therapeutic intervention.
Mammalian AMPK is a 134–150 kDa heterotrimeric complex composed of a catalytic α subunit that harbors a protein kinase module (two isoforms) and regulatory β (two isoforms) and γ (three isoforms) subunits, each of which is encoded by a separate gene. The β subunit is considered a scaffolding subunit and contains a glycogen binding module (GBD). The γ subunit contains four nucleotide-binding sites, across two Bateman domains (Zhu et al., 2011). AMPK is in part regulated by allosteric binding of small-molecule ligands which induce conformational changes and control the phosphorylation state of a threonine residue in the activation loop, an event which switches on AMPK’s enzymatic activity. The nucleotide binding sites can bind AMP, ADP or ATP and mediate effects on either allosteric activation or protection from dephosphorylation of its activation loop phospho-threonine. Although one of these sites (site 4) was previously believed to be a non-exchangeable AMP site, recent work has revealed that ATP can also bind with functional consequences (Chen et al., 2012). The structure of each of the subunits is evolutionarily conserved (Hardie, 2007).
Our knowledge of the regulation of AMPK activity has been greatly improved by the availability of yeast and mammalian AMPK crystal structures (Chen et al., 2012; Chen et al., 2013; Koay et al., 2010; Rudolph et al., 2010; Xiao et al., 2007; Xiao et al., 2011; Zhu et al., 2011). These structures have revealed that the C-terminal portion of the β subunit serves as a structural anchor which holds together the γ subunit and the C-terminal region of the α subunit as a rigid ‘core’ module (Xiao et al., 2007). The kinase domain of the α subunit adopts a typical protein kinase fold and is somewhat isolated from the remainder of the structure except for the close interactions formed by its activation loop (Xiao et al., 2011). The precise details of the conformation and the location of the intervening polypeptide segment between the kinase module and the C-terminal portion of the α subunit have recently been challenged (Chen et al., 2013). Also, the location of glycogen binding module of the β subunit in mammalian AMPK structures is still unknown. While models have been proposed for the molecular mechanism responsible for AMPK activation by AMP, the dynamics of communication among the subunits are largely unknown, as are the details of the conformational changes induced by adenine nucleotides.
AMPK has a selected pool of cellular targets in adipose tissue, liver, heart, muscle, kidney and brain (reviewed in (Hardie, 2007)). During times of low energy reserves, AMPK inhibits cell growth and the biosynthesis of proteins, lipids, and carbohydrates, while also stimulating catabolic processes that generate ATP. AMPK is activated by phosphorylation mediated by upstream kinases, LKB1(Hawley et al., 2003), and CaMKKβ (Hawley et al., 2005; Hurley et al., 2005). Although it has been reported that AMPK could be activated by TAK1, the physiological role of this is not well understood (Momcilovic et al., 2006). Once activated, AMPK mediates rapid cellular responses to alterations in systemic energy status by phosphorylating downstream transcription factors, enzymes, and coactivators. Given the role of AMPK in cellular metabolism, structural insights into the mechanisms of AMPK activation could inform the design of AMPK-specific drugs for the treatment of diseases such as diabetes and obesity. Several indirect AMPK activators have already been reported such as biguanide (metformin) and thiazolidinediones (rosiglitazone) which are used to treat type 2 diabetes (Fryer et al., 2002); natural products such as resveratrol, catechins, theaflavins, triterpinoids are also known to activate AMPK indirectly by elevation of AMP levels. In addition, Cool et al. have reported direct activation of AMPK by compounds belonging to the thienopyridone chemical scaffold exemplified by A769662 (Cool et al., 2006). Since then, studies from several laboratories have suggested that the mechanism of AMPK activation by A769662 is likely to be very different from that by AMP (Scott et al., 2008). Pang et al. have reported the discovery of another class of direct AMPK activators represented by PT1 which are believed to target the α subunit (Pang et al., 2008). A recent review article on the AMPK patent literature describes a number of other small molecule ligands that directly activate AMPK (Giordanetto and Karis, 2012).
Previously, we have applied hydrogen/deuterium exchange (HDX) coupled with mass spectrometry to classify selective estrogen receptor α modulators (SERMS) and correlate the HDX signatures of each ligand class with their pharmacological effects (Dai et al., 2008), and have used HDX to characterize the protein-ligand interaction of other nuclear hormone receptors such as PPARγ (Bruning et al., 2007; Choi et al., 2010; Choi et al., 2011) and VDR-RXR (Zhang et al., 2011). Previous solution X-ray scattering studies suggested conformational changes of AMPK heterotrimer induced by the binding of AMP (Riek et al., 2008). In addition, recent crystal structures of the AMPK core complex show that nucleotide binding can lead to changes in the conformation of the Bateman domains in the γ subunit (Chen et al., 2012; Xiao et al., 2007). This alteration of the structure likely involves positional changes in the α and β subunits outside the core complex, but this has not been confirmed. In this study, we employ HDX-MS technique to investigate the conformational mobility of the kinase and dynamics of subunit communication during the binding of small molecule ligands to AMPK. In addition, the impact of phosphorylation of the threonine residue in the activation loop on the conformational mobility of the kinase was also monitored. The results of these combined methods show that while binding of AMP causes primarily conformational changes in the γ subunit with subtle effects on the α and β subunits, significant stabilization of the GBD of β subunit and the kinase module of the α subunit occurs upon binding of A769662. These studies also suggest that the molecular binding site of A769662 is located between the GBD of the β subunit and the kinase module of the α subunit and reveal distinct patterns of long range conformational changes induced by binding of A769662 and AMP.
RESULTS
Sequence coverage of 134 kDa trimeric AMPK
A bottom-up approach to AMPK was first investigated by optimizing conditions for protein digestion. Supplementary Figure S1a shows the MS/MS sequence coverage obtained for each subunit of the heterotrimer. Highlighted in yellow are the areas included in the chimeric structure of AMPK (PDB ID: 2Y94) which represents the most complete AMPK structure known (Xiao et al., 2011). Also, highlighted in red are key sites of phosphorylation. As the 2Y94 structure lacks the glycogen-binding domain of the β subunit (GBD, amino acids 77–156), we constructed a chimeric atomic model of mammalian AMPK composed of the 2Y94 structure along with the GBD based on its known position in the yeast AMPK structure (Amodeo et al., 2007). We have mapped our HDX results onto this chimeric model throughout the paper. The 3-dimensional representations of these two structures are shown in Supplementary Figure S1b. The percent sequence coverage for each subunit under MS/MS, t0, and on-exchange experimental conditions, is given in the Supplementary Figure S1c. Under MS/MS conditions, the coverage for the α, β and γ subunits are 80%, 98%, and 95%; under t0 conditions, the coverage is 78%, 95%, and 91%, and under on-exchange conditions the coverage is 70%, 82%, and 81%, respectively. A decrease in sequence coverage is expected for on-exchange due to increased overlap of peaks caused by expanding isotopic envelopes with increasing amounts of deuterium; however, the sequence coverage is substantial for a 134 kDa protein.
Conformational dynamics of unliganded protein
We investigated the conformational dynamics that occur over time in the heterotrimer prior to the addition of any exogenous ligand (Fig. 1a). The α subunit has a highly dynamic helix (Fig. 1b) from residues 60–70 (KIRREIQNLKL) which corresponds to the Cα-helix of protein kinases. The C-helix is a well-known regulatory module in protein kinases that controls their activation states. We also observed substantial perturbation of the DFG motif (highly conserved region consisting of characteristic aspartic acid, phenylalanine and glycine residues immediately N terminal to the activation loop) involved in inhibitor and substrate binding. A peptide spanning the activation loop (170–189: LRTSCGSPNYAAPEVISGRL) which includes the threonine residue important for enzyme activation, T172, exhibits differences in dynamics based upon its phosphorylation. As seen in the build-up curves for each peptide (Fig. 1c), the extent of deuterium incorporation is consistently less in the phosphorylated peptide compared to that in the non-phosphorylated peptide, implying restricted dynamics of the former. The mass spectral population of the phosphorylated peptide is roughly 30 fold greater than the non-phosphorylated peptide consistent with the previous mass spec data suggesting that the vast majority of the protein is in the activated (phosphorylated) state (Supplementary Figure S2). Similar behavior is observed for a peptide in the β subunit (101–112: SKLPLTRSHNNF) that contains a key serine residue, S108. The phosphorylated peptide exhibits a lower degree of deuterium incorporation (Fig. 1c) and the mass spectral population was approximately 10 fold greater than the non-phosphorylated peptide (data not shown).
Figure 1.

Dynamics of apo AMPK. (a) The percentage of corrected deuterium uptake for all peptides of apo AMPK is mapped onto the PDB: 2Y94. A separate model of GBD is built based on yeast AMPK structure and shown alongside 2Y94. Three different time points (10, 300 and 3600 s) are shown here. The percentage of deuterium uptake values, an average of 6 time points (0 s to 3600 s), is depicted by color code explained at the bottom of the figure. Regions colored as white represent peptides that are not consistently resolved in the study. (b) Deuterium uptake curve of Cα helix from kinase domain. (c, d) differential deuterium uptake of phosphorylated and non-phosphorylated peptide spanning the activation loop of α subunit (d) and the Ser108 loop of the β subunit. (e) Peptides belonging to two different CBS motifs of γ subunit show different levels of deuterium incorporation.
In addition to the aforementioned effects on the α and β subunits, we also observed significant changes in the dynamics of two cystathionine beta-synthase (CBS) motifs in the γ subunit responsible for AMP binding. The antiparallel beta sheets spanning residues 228–243 are known to comprise an exchangeable AMP binding site (site 3 – the revised nomenclature from–(Kemp et al., 2007) to match the nucleotide binding sites 1–4 matches with the CBS motifs 1–4) and show a high rate of deuterium incorporation across all time points indicating very dynamic behavior (Figure 1e). In contrast, an adjacent set of antiparallel beta sheets spanning residues 298–315 (site 4) that is known to be a tight (weakly-exchangeable) nucleotide binding site show a much slower rate of deuterium incorporation consistent with this region being much less dynamic. Taken together, the observations regarding the dynamics of these two AMP-binding sites are consistent with their purported functions and with crystallographic and mutational studies (Chen et al., 2012; Oakhill et al., 2010; Xiao et al., 2011).
Allosteric effect of AMP/ZMP on AMPK 111 conformational dynamics
We next studied the allosteric effects of AMP/ZMP binding on AMPK conformational dynamics. In these studies, we observed that the binding of AMP to the heterotrimer induced protection from solvent exchange in the Cα-helix (60–70) and auto-inhibitory domain (288–298) of the α subunit compared to the unliganded enzyme (Fig. 2, Supplementary Table S2). Similarly, upon binding of AMP, a peptide from the C-terminal region of the β subunit (224–240) also exhibited increased protection from deuterium exchange. Moreover, significant protection from solvent exchange was also observed for the activation loop peptide that harbors the phosphorylated T172 (Supplementary Table S1). The largest change in deuterium protection pattern caused by AMP was observed in the γ subunit. As shown in Figure 2b, AMP binding causes significant protection of peptides from the CBS 3 and 4 motifs. Peptides around site 3 (228–243) show greater protection compared to peptides around the weakly-exchangeable site 4 (298–315). Also, binding of AMP leads to protection of peptides around the CBS 1 and 2 motifs.
Figure 2.

Effect of AMP and ZMP on AMPK 111. (a) Differential HDX data mapped onto the AMPK atomic structure (PDB accession code:2Y94) (b) Schematic representation of AMPK regions undergoing conformational changes upon AMP/ZMP binding. Percentage of deuterium differences are color coded according to the color key. Boxed area in the activation loop region represents the phosphorylated form as compared to the non-boxed area below that represents the non-phosphorylated form.
See also Table S1 and S2.
A similar protection pattern was observed in the auto-inhibitory domain of the α subunit upon binding of ZMP, an AMP analog and a cellular metabolite of AICAR. Binding of ZMP causes only very little perturbations in the β subunit, consistent with the observations with AMP. As expected, the largest effect of ZMP binding was observed in the γ subunit with greater protection seen in the exchangeable site 3 when compared to the weakly-exchangeable site 4. However, the degree of protection is less than that observed following binding of AMP (Fig. 2a, 2b, Supplementary Table S2). AMP and ZMP were added at 50 fold molar excess (500 μM) compared to 10 fold molar excess of other ligands as AMP affinities for the two readily exchangeable sites (sites 1 and 3) have been reported in the range of 2–80 μM (Xiao et al., 2011). However, it is clear from the HDX perturbation fingerprints of AMPK bound to either AMP or ZMP that both ligands display tight binding in the γ subunit that trigger allosteric communication across the other subunits.
Effect of A769662, beta-cyclodextrin and staurosporine binding to AMPK111; investigation of novel activator binding site
In an effort to understand the mechanism of action of the small molecule AMPK activator A769662 (supplementary Fig S3), we examined the dynamics of the enzyme in the presence and absence of this ligand. HDX studies revealed that the binding of A769662 resulted in significant perturbation in all three subunits of AMPK (Fig. 3a, 3b). A closer look at these results demonstrated a large degree of protection from deuterium exchange in the substrate binding cleft of the α subunit kinase module, particularly the Cα-helix. Moreover, slight but significant protection was also observed in the activation loop peptides containing the phosphorylated T172 while the equivalent non-phosphorylated peptide showed no degree of protection to exchange (Supplementary Table S1). Similar behavior was seen in the GBD of the β subunit (Fig. 3a, 3b). Here we observed nearly the entire region was protected from exchange when bound to A769662 including the region encompassing the phosphorylated S108 residue, which, similar to the T172 peptide in the α subunit, was not protected in the absence of phosphorylation (Supplementary Table S1). This is consistent with previous observations that mutation of S108 to alanine rendered AMPK virtually insensitive to activation by A769662 (Sanders et al., 2007). Finally, we observed an increase in exchange in site 3 of the γ subunit which consists of peptides spanning the antiparallel beta sheets of the AMP exchangeable site (228–243) (Fig. 3a, 3b and Supplementary Tables S1, S2). All together these data indicate that the binding of A769662 to AMPK leads to a stabilization of the protein indicated by reduced dynamics in both the activation loop and GBD domain within the α and β subunits and an increase in dynamics in site 3 of the γ subunit which contains an AMP exchangeable site. Most importantly, this HDX profile shows that interaction of A769662 with AMPK leads to allosteric changes in the enzyme that directly impact important sites known to be involved in enzyme activation.
Figure 3.

Conformational changes of AMPK 111 upon binding of A769662, beta-cyclodextrin and staurosporine. (a) Differential HDX data mapped onto the AMPK atomic structure (PDB:2Y94). (b) Schematic representation of AMPK regions undergoing conformational changes upon binding of A769662, b-cyclodextrin and staurosporine. Percentage of deuterium differences are color coded according to the color key. Boxed area in the activation loop region represents the phosphorylated form as compared to the non-boxed area below that represents the non-phosphorylated form.
We next examined the protein dynamics of AMPK with beta-cyclodextrin, a cyclic oligosaccharide known to bind the β subunit from x-ray crystallography studies (Polekhina et al., 2005). When bound to beta-cyclodextrin, the α and γ subunits of AMPK showed no significant perturbation compared to unliganded enzyme. As expected, we did observe protection from deuterium exchange in the β-subunit encompassing the majority of the GBD (Fig. 3a, 3b and Supplementary table S1, S2). However, no protection was seen for peptides containing S108 in either its phosphorylated or non-phosphorylated forms. Instead, the greatest protection is seen in peptides containing residues that are known to interact with glycogen (W100, K126, W133, L146, T148). Taken together, this profile suggests that beta-cyclodextrin is capable of binding the β subunit of AMPK but does not modulate the conformation of the other subunits that contain key residues involved in enzyme activation. This is consistent with previous observations that β-cyclodextrin binds to AMPK but does not augment or inhibit its catalytic activity (Bieri et al., 2012; Polekhina et al., 2003).
We next investigated the protein dynamics of AMPK in the presence of both A769662 and beta-cyclodextrin to determine whether these ligands compete for binding or occupy different binding sites on the enzyme. The protection profile observed in the α subunit of AMPK with simultaneous addition of A769662 and beta-cyclodextrin was very similar to that obtained with A769662 alone. Together, beta-cyclodextrin and A769662 protected the Cα-helix, DFG-motif, the activation and catalytic loops of the α subunit to a similar extent as observed with A769662 alone. In addition, the entire GBD of the β subunit exhibited protection from exchange and showed differential protection patterns around S108 depending on its precise phosphorylation state. Moreover, in presence of both β-cyclodextrin and A76962, we observed an additive effect for protection from solvent exchange in peptide segments that are known to be involved in interaction with glycogen. Taken together, the pattern of protection observed when both of these ligands were present was a combination of the patterns elicited by each ligand individually. The only increase in magnitude of protection occurred in regions of known glycogen interactions where both ligands individually showed protection compared to the unliganded enzyme. This suggests that A769662 and beta-cyclodextrin occupy different binding sites on the β subunit. As observed with beta-cyclodextrin alone, very little perturbation was observed in the γ subunit on combined addition of A769662 and beta-cyclodextrin. (Fig. 3a, 3b and Supplementary Tables S1, S2).
We also interrogated the differential HDX profile of AMPK bound to the known kinase inhibitor staurosporine. Protection from exchange was observed in the ATP binding site (94–101), catalytic loop (127–147) and the DFG motif (152–163) which corresponds to the observed binding site of staurosporine in the co-crystal structure (Xiao et al., 2011). No perturbation was seen in the Cα-helix or the activation loop and an increase in deuterium exchange was observed in a helix (288–298) in the auto-inhibitory region downstream of the catalytic module. Moreover, no significant perturbation was observed in either the β or γ subunits (Fig. 3a, 3b and Supplementary Table S1, S2). The deuterium exchange pattern of staurosporine shows that while this promiscuous kinase inhibitor bound to previously identified regions of the α subunit, there were no long-range effects observed either on the activation loop of the kinase or the β/γ subunits. Moreover, the differences in the HDX fingerprints between staurosporine and A769662 suggest that they share non-overlapping binding sites.
Probing binding mode; effect of AMP and A769662 on truncated AMPK
To further probe the binding sites of AMP and A769662, we employed a truncated AMPK reagent (AMPKt) similar to the trimeric complex used for the crystallographic study of the first mammalian AMPK structure (Xiao et al., 2007). While the entire γ1 subunit is present in the AMPKt construct, it contains only the C-terminal portions of the α1 (405–557) and β1 (185–270) subunits. Subunits were truncated to exclude the kinase module of the α subunit and the GBD of the β subunit to determine if these ligands could still interact with the enzyme that is devoid of these domains. The differential HDX profile of AMPKt obtained with AMP revealed significant protection around the AMP sites 3 and 4 on the γ subunit (Fig. 4a, 4b, Supplementary Table S2). In contrast, no perturbation was observed for any of the peptides from the α or β subunit upon binding of AMP. Moreover, comparable protection profiles were observed for the exchangeable (site 3) and weakly-exchangeable (site 4) AMP binding sites (Fig. 4a, 4b). The degree of protection of AMPKt by AMP was less than what was observed for the full length AMPK heterotrimer bound to AMP (Supplementary Table S2). This suggests that presence of other structural modules of the α and β subunits contributes to the overall allosteric conformational dynamics of full-length AMPK. Incubation of AMPKt with A769662 produced no significant HDX perturbation in any of the subunits suggesting that it no longer harbors a binding site for the ligand (Fig. 4a, 4b).
Figure 4.

Effect of AMP and A769662 binding on truncated AMPK. (a) Differential HDX data mapped onto the AMPK atomic structure (PDB accession code:2Y94). (b) Schematic representation of AMPK regions undergoing conformational changes upon AMP/ A769662 binding. Percentage of deuterium differences are color coded according to the color key. Boxed area in the activation loop region represents the phosphorylated form as compared to the non-boxed area below that represents the non-phosphorylated form.
Effect of AMP and A769662 on AMPK221; negative control for A769662 binding
To investigate the specificity of A769662 for β1-containing AMPK isoforms, as has been reported in the literature (Sanders et al., 2007; Scott et al., 2008), we studied the interaction of A769662 and AMP with the α2β2γ1 isoform. Several studies in the literature have reported that AMP is a more potent activator of α2β2γ1 compared to the α1β1γ1 isoform (Sanders et al., 2007). In contrast, A769662 is known to be a weak activator of β2-containing AMPK (Sanders et al., 2007; Scott et al., 2008). Binding of AMP resulted in protection of the Cα-helix (60–70), ATP site (94–101), catalytic loop (127–147) the DFG module (152–163) and an AID peptide (305–311) of the α subunit of α2β2γ1 isoform. Interestingly, the α2β2γ1 differential HDX profile with AMP is somewhat similar to what was observed on binding of A769662 to the α1β1γ1 isoform. Peptides in the activation loop containing the phosphorylated T172 exhibited protection as well. Slight protection was also seen in the C-terminal portion of the β subunit (215–243). In contrast, we observed significantly more protection across a longer segment of the γ subunit in α2β2γ1 when compared to the effect of AMP on the α1β1γ1 isoform with greater protection around the exchangeable AMP binding site (3) than the weakly-exchangeable site (Fig. 5a, 5b and Supplementary Tables S1 and S2). The increased stabilization of AMPK α2β2γ1 by AMP in the γ subunit where AMP binds as well as the important regions for activation in the α and β subunits are consistent with the observations that AMP is a more potent activator of α2β2γ1. Upon binding of A769662, we observed similar levels of protection induced in the kinase module of α2β2γ1 as was observed in the differential HDX experiments with α1β1γ1 isoform (Fig. 5a, 5b and Supplementary Tables S1, S2). However, slightly different peptides in the AID regions of the two isoforms were perturbed on binding of A769662. While the peptide 288–298 of the α1 subunit was stabilized by A769662, in the α2 subunit, a nearby peptide 305–311 showed a similar magnitude of stabilization. The significance of these subtle differences in the conformational dynamics of the two α subunits is unclear at present. Interestingly, significant protection was seen across most of the GBD of the β subunit, but no perturbation was observed for peptides containing S108 irrespective of the phosphorylation state. This indicated that while A769662 still bound to α2β2γ1, it did not have the same role in activation of the β2-containing AMPK isoform. It further suggests that for A769662 to activate AMPK, it binds to the β subunit in such a way as to directly stabilize the S108 phosphorylation site (on the β subunit) and also allosterically stabilize the activation loop and T172 site within the α subunit.
Figure 5.

Effect of AMP and A769662 binding on AMPK 221 isoform (a) Differential HDX data mapped onto the AMPK atomic structure (PDB:2Y94). (b) Schematic representation of AMPK regions undergoing conformational changes upon AMP/ A769662 binding. Percentage of deuterium differences are color coded according to the color key. Boxed area in the activation loop region represents the phosphorylated form as compared to the non-boxed area below that represents the non-phosphorylated form.
Discussion
In this study we have applied hydrogen/deuterium exchange (HDX) coupled with mass spectrometry to probe the protein dynamics of AMPK in the absence and presence of activators and inhibitors to better understand the allosteric conformational changes involved in activation of this master metabolic regulator. These HDX studies reveal allosteric communication across subunits of the heterotrimeric kinase trigged by the binding of small molecule ligands and point to the molecular binding site of A769662 on AMPK. These data also strongly suggest two fundamentally different modes of AMPK activation by the nucleotides and A769662 (summarized in Fig. 6). These data could provide a potential framework for the design and discovery of isoform-specific AMPK activators for the treatment of metabolic diseases.
Figure 6.
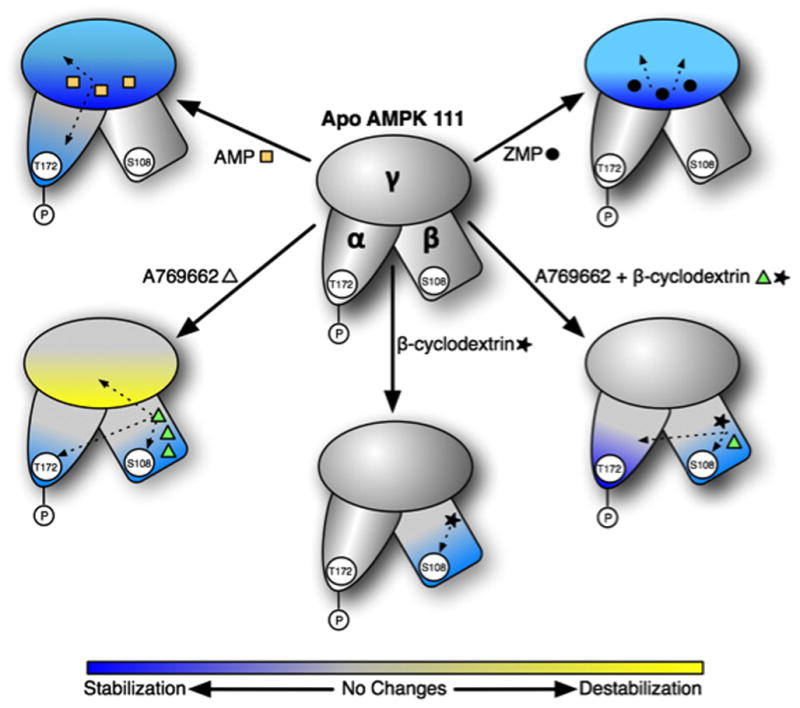
Schematic of AMPK conformational dynamics induced by different activators. Protein stabilization caused by ligand binding (as measured by the differential HDX profile) is shown in blue while destabilization is shown in yellow. Changes in stabilization are a result of altered conformation of the protein. Known or putative ligand binding sites on AMPK subunits are shown by orange squares (AMP), dark circle (ZMP), green triangle (A769662) and solid star (beta-cyclodextrin). These are just schematic representations of the general observations from the HDX experiments with different ligands but not an absolute mapping of the extent of deuterium exchange. AMP binding results in enhanced protection or stabilization of the γ subunit and the activation loop in the kinase module of the α subunit. Binding of ZMP, an AMP analogue and a cellular metabolite of AICAR, primarily alters the HDX profile in the γ subunit and to a smaller extent, the α subunit. The synthetic activator A769662 displays strong stabilization of the β subunit and portions of the α subunit involved in substrate binding and catalysis. Also, A769662 causes destabilization of the γ subunit the reasons for which are not completely understood. The effect of glycogen mimetic, beta-cyclodextrin, is mostly restricted to the β subunit. Combination of A769662 and beta-cyclodextrin elicits a profile that is a combination of the individual profiles obtained by each of the ligands. These results also suggest that A769662 and beta-cyclodextrin bind at non-overlapping sites on the glycogen binding module of the β subunit. The HDX data strongly suggest two fundamentally different modes of AMPK activation by A769662 and the nucleotides.
To perform these studies using our automated HDX-MS platform, we followed over 500 peptides of heterotrimeric AMPK (134kDa). First, we evaluated the differences in protein dynamics of apo AMPK (unliganded or free enzyme), specifically to examine the effect of phosphorylation on two important sites crucial for the activation of the enzyme, T172 (α subunit) and S108 (β subunit). Phosphopeptides around these two sites displayed attenuated dynamics compared to their unphosphorylated counterparts. This is consistent with the notion that phosphorylation of the activation loop of the α subunit and the S108 loop of the β subunit promotes a more stable conformation. In addition, we observed that the dynamics between the exchangeable and non-exchangeable AMP sites within the CBS domains of the γ subunit were significantly different in the unliganded enzyme. The exchangeable AMP site (site 3) clearly exhibited enhanced dynamics while the non-exchangeable AMP site (site 4) was much more stable. This suggests that the AMPK site 3 could have a more functional role in the activation mechanism of AMPK compared to site 4.
We then evaluated a number of known AMPK activators and inhibitors to see whether HDX profile could give new insights into its activation mechanism. Strong protection from deuterium exchange in the CBS domains of the γ subunit was elicited by AMP and its analog, ZMP (AICAR metabolite). As the γ subunit harbors the known AMP binding sites (exchangeable and non-exchangeable), it is not surprising that binding of these ligands to AMPK would lead to a stabilization of the dynamics of this region. We observed only limited perturbations on the α or β subunits on binding of AMP and ZMP with slightly enhanced effect observed for AMP. This is most likely a reflection of the enhanced binding affinity of AMP for AMPK compared to ZMP and not suggestive of any differences in their activation mechanisms.
We next investigated the HDX profile of A769662, the well-studied thienopyridone synthetic activator (Supplementary Figure S3). (Cool et al., 2006). Upon binding of A769662, there were decreases in dynamics of the activation loop and pT172 of the α subunit as well as most of the GBD module in the β subunit, including pS108. Moreover, there was increased dynamics observed in the γ subunit around the exchangeable AMP binding site (site 3). Thus, binding of A769662 induces a conformation that stabilizes regions containing both phosphorylation sites known to be important for activation and clearly displays allosteric effects across all three subunits. Some of the changes observed in the deuterium exchange profile such as those occurring in the activation loop of the α subunit can be attributed to conformational changes caused by the binding of the ligand. In contrast, the decreased dynamics observed in the GBD of the β subunit most likely reflects additive effects of conformational changes and those caused by direct binding of A769662 to the GBD module. However, it is unclear how binding of A769662 near the GBD module leads to perturbations in the γ subunit.
There are conflicting reports in the literature on how glycogen modulates the activity of AMPK. Earlier studies showed essentially no effect on AMPK activity upon binding of glycogen (Polekhina et al., 2003). Subsequent studies with small, branched oligosaccharides showed modest inhibition of AMPK activity (McBride et al., 2009). In our HDX-MS studies, addition of beta-cyclodextrin, a cyclized form of maltoheptaose, resulted in significant stabilization of the GBD module of AMPK β subunit. These results are consistent with previous reports that beta-cyclodextrin binds to the GBD module of AMPK (Polekhina et al., 2005). However, we observed no allosteric effects of beta-cyclodextrin on any of the important regions required for activation in either the α or γ subunit. This suggested that AMPK harbors a binding site for beta-cyclodextrin but is neither activated nor inhibited by it. Staurosporine, which is a well-known kinase inhibitor, displayed changes in dynamics of the α subunit around the ATP binding site nearly exclusively with very little allosteric effects seen in the β or γ subunits. This is consistent with previous crystallographic observations of the binding of staurosporine at the ATP site of AMPK (Xiao et al., 2011).
Ever since the discovery of A769662 as a direct AMPK activator, there has been considerable interest in deciphering its exact binding site on AMPK. The differential HDX experiments with A769662 described above demonstrated increased stabilization of the α and β subunits caused by ligand binding. In order to more precisely map the binding site of A769662, we analyzed the HDX profile of AMPK bound to both A769662 and beta-cyclodextrin. Combined addition of these two ligands elicited an HDX-MS profile that is a combination of the two individual profiles. The only region where we observed an increase in protein dynamic stability in the presence of both ligands was the GBD of the β subunit indicating that A769662 bound within the β subunit but in a different location than that of beta-cyclodextrin. This conclusion was further strengthened when we observed that a truncation construct of AMPK, which lacks the GBD and a large portion of the α subunit, was completely unable to interact with A769662. Furthermore, α2β2γ1 AMPK isoform, which is not known to be activated by A769662, displayed weaker stabilization of the β subunit compared to the α1β1γ1 isoform and specifically did not show any stabilization of the S108 phosphorylation site within the GBD of the β subunit. Moreover, all allosteric communication with the γ subunit appeared to be lost when A769662 bound to this β2-containing AMPK isoform. These results suggest that A769662 functions as an activator by binding directly to the GBD domain in the β subunit, specifically stabilizing the S108 phosphorylation site which then leads to allosteric changes to the α and γ subunits that activate the enzyme fully. These results are also consistent with previous observations that either mutation of Ser108 of the β subunit to alanine or truncation of the GBD module renders AMPK completely insensitive to activation by A769662 or salicylate (Hawley et al., 2012; Sanders et al., 2007; Scott et al., 2008).
The downstream biology elicited following activation of AMPK can differ from one tissue to another depending on the AMPK isoforms that are expressed and their protein substrates. Moreover, the precise conformational changes induced on AMPK following stimulation by endogenous ligands or pharmacological agents can alter the response further. Comparison of the HDX-MS profiles of α1β1γ1 versus α2β2γ1 show that AMP has a more profound effect on the dynamics of the γ subunit in the latter (Supplementary Table S2). Although both isoforms contain the same γ subunit where AMP binds, the increased protection observed in the γ subunit of α2β2γ1 isoform suggests a possible role for α and β subunits. This could also reflect the increased binding affinity of AMP for α2β2γ1compared to α1β1γ1. In addition, AMP has a larger effect in protection (from deuterium exchange) of some peptides from the α and β subunits of α2β2γ1 compared to α1β1γ1, consistent with its enhanced potency in biochemical activation of the former. Taken together, these results support observations that nucleotide binding has effects that extend beyond the γ subunit.
Although the synthetic activator A769662 is known to be a β1-specific activator, its differential HDX profile with α2β2γ1 shows clear evidence for binding and conformational changes. It induces similar conformational changes on the α subunits of α1β1γ1and α2β2γ1but a drastically different profile on the β subunits (Supplementary Table S2). The HDX profiles suggest that A769662 binds near the GBD modules of both α1β1γ1and α2β2γ1 isoforms, consistent with previous biochemical reports (Hawley et al., 2012; Sanders et al., 2007). This is further supported by the absence of any significant conformational changes upon addition of A769662 to a truncated α1β1γ1construct which lacks the GBD and kinase modules (Supplementary Table S2). Conformations of different regions of the GBD modules of α1β1γ1and α2β2γ1 are affected by A769662 which suggest that it binds at two distinct sites on the two β subunits. While binding of A7669662 leads to conformational changes around the pSer108 site in the β1 subunit, it is the beta-cyclodextrin binding site that is perturbed in the β2 subunit.
As a master regulator of metabolism, AMPK needs to quickly respond to changes in the cellular energy status. This requires long range communication between catalytic and regulatory domains. Through the application of HDX-MS technology, we have probed the intrinsic flexibility of different segments of AMPK and the changes induced by ligand binding. These HDX studies measure allosteric communication across subunits of the heterotrimeric kinase trigged by the binding of small molecule ligands and provide additional insight into the molecular binding site of A769662 on AMPK. These data also strongly suggest two fundamentally different modes of AMPK activation by the nucleotides and A769662 (summarized in Fig. 6).
Experimental procedure
Cloning of full-length human AMPK complex for bacterial expression
A tricistronic construct that included open reading frames encoding the full-length α, β and γ subunits of human AMPK was designed with a ribosome-binding site (RBS) ahead of each coding region (Neumann et al., 2003; Rajamohan et al., 2010). The construct was custom-synthesized with codons optimized for bacterial expression (GENEART Inc., Burlingame, CA 94010). The 22-residue N-terminal tag of the α subunit included six histidine residues and a cleavage site for thrombin (MGSSHHHHHHSSGLVPRGSMGT). The tricistronic construct was subcloned into the NcoI and XhoI sites of pET-14b expression vector (Novagen, Madison, Wisconsin) using standard molecular biology techniques (T. Maniatis, 1982). Similar protocols were used for the expression, purification and characterization of α1β1γ1, α2β2γ1 and α1β1γ1t AMPK reagents.
Expression and purification of recombinant AMPK complex from E. coli
AMPK tricistronic construct was transformed into E. coli BL21-CodonPlus™ (DE3)-RIPL strain (Stratagene) and transformants were selected on LB (Luria-Bertani) agar plates containing ampicillin (100 mg/ml). Single colonies were used to inoculate 10 ml of LB medium containing 100 mg/ml ampicillin. For large-scale expression and purification, an Erlenmeyer flask containing 1L of LB broth supplemented with 100 mg/ml ampicillin was inoculated with 25 ml of overnight culture and grown in a shaker incubator at 37 °C to an OD600 of 0.8. Protein expression was induced with 100 mM (final concentration) of isopropyl β-D-thiogalactopyranoside (IPTG), the temperature was reduced to 18 °C, and the cells were grown for an additional 18 h. Cells were harvested and resuspended in 50 ml lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol, 2 mM Tris-2-carboxyethyl phosphine (TCEP), 20 mM imidazole and 0.001% Triton X-100). The cell suspension was sonicated on ice with a Branson ultrasonic disintegrator (VWR Scientific Products, Chicago, IL) for 2–4 min at 50% duty cycle.
Insoluble material was removed by centrifugation at 15,000 rpm in a Sorvall® RC5 plus centrifuge for 30 min at 4 °C and the supernatant was loaded onto a 5 ml HisTrap™ HP column (GE Healthcare, Piscataway, NJ) and washed with five column volumes of lysis buffer. Bound proteins were eluted using a linear gradient (10 column volumes) with elution buffer (lysis buffer containing 300 mM imidazole). Fractions containing AMPK subunits were pooled based on SDS-10% PAGE analysis and dialyzed overnight in dialysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol, 2 mM TCEP, and 0.001% Triton X-100). The AMPK complex was further purified by gel filtration chromatography with a Superdex 200 HiLoad 16/60 column (GE Healthcare) in SEC buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol, 2 mM TCEP, and 0.001% Triton X-100).
Activation of AMPK heterotrimer
To determine the relevant amount of upstream kinase to be used in the phosphorylation reaction, the AMPK complex was incubated with increasing concentrations of upstream kinases and the concentration of upstream kinase which yielded the highest possible level of Thr172 (α2-subunit) phosphorylation was used in the subsequent reactions. To evaluate which upstream kinase was more effective at phosphorylating residue Thr172, 1.0 μM Ni2+-column purified AMPK complex was incubated in the presence or absence of 200 nM upstream AMPK Kinase (CaMKKβ or LKB; The University of Dundee, Scotland) in 100 μl of phosphorylation buffer (25 mM Tris, pH 7.5, 137 mM NaCl, 1 mM CaCl2, 5 mM MgCl2, 1 mM TCEP, 100 μM ATP and 100 nM calmodulin) for 30 min at 30 °C in a thermostated shaker. The reaction was stopped by freezing the samples at −80 °C. Activation Ni-purified recombinant AMPK heterotrimeric complexes was performed by incubating 1.0 μM AMPK complex in the presence of 200 nM CaMKKβ in phosphorylation buffer for 30 min at 30 °C in a thermostated shaker. The phosphorylated AMPK complex was re-purified on HisTrap™ HP column as before, dialyzed overnight in dialysis buffer. The phosphorylated AMPK complex was further purified by gel filtration chromatography with a Superdex 200 HiLoad 16/60 column (GE Healthcare) in SEC buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol, 2 mM TCEP, and 0.001% Triton X-100). The final samples were stored at −20 °C with 25% glycerol. Characterization by mass spectrometry showed that the recombinant enzyme is phosphorylated on Thr172 of the α subunit and Ser108 of the β subunit with no myristoylation on the β subunit.
Hydrogen/deuterium exchange mass spectrometry
Differential, solution phase HDX experiments were performed with a fully automated system using a LEAP Technologies Twin HTS PAL liquid handling robot interfaced to an Exactive Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany) (Chalmers et al., 2006). Each exchange reaction was initiated by incubating 4 μL of protein (with or without ligand) with 16 μL of D2O protein buffer for a predetermined time (10s, 30s, 60s, 300s, 900s and 3600s in a randomized order) at 4 °C. The exchange reaction was quenched by mixing the protein solution with 30 μL of 3 M Urea, 1% TFA at 1 °C. The mixture was passed across an in-house packed pepsin column (1 mm × 20 mm) at 50 ul/min and digested peptides were captured onto a 1 mm × 10 mm C8 trap column (Thermo Scientific) and desalted (total time for digestion and desalting was 2.5 min). Peptides were then separated across a 1 mm × 50 mm C18 column (5 μm Hypersil Gold, Thermo Scientific) with linear gradient of 5%–50% CH3CN, 0.3 % formic acid, over 5 min. Protein digestion and peptide separation were performed within a thermal chamber (Mécour) held at 15 °C and 1°C, respectively, to promote more efficient digestion and to reduce D/H back exchange. Electrospray ionization parameters were set as the following: sheath gas 30 au, auxiliary gas 15 au, spray voltage 3.7 V, capillary temperature 225 °C, capillary voltage 25 V, tube lens 95 V, skimmer 16 V. Mass spectrometric analyses were acquired with a measured resolving power of 100,000 at m/z 400. Three replicates were performed for each ion-exchange time point.
Peptide Identification and HDX data processing
MS/MS experiments were performed with a FinniganLTQ linear ion trap mass spectrometer (ThermoFinnigan, San Jose, CA). Product ion spectra were acquired in a data-dependent mode and the five most abundant ions were selected for the product ion analysis. The MS/MS *.raw data files were converted to *.mgf files and then submitted to Mascot (Matrix Science, London, UK) for peptide identification. Peptides included in the peptide set used for HDX had a MASCOT score of 20 or greater. The MS/MS MASCOT search was also performed against a decoy (reverse) sequence and ambiguous identifications were ruled out. The MS/MS spectra of all the peptide ions from the MASCOT search were further manually inspected and only those verifiable were used in the coverage. The intensity weighted average m/z value (centroid) of each peptide isotopic envelope was calculated with the latest version of our in-house developed software, MS Peptide Workbench.
Supplementary Material
Supplementary Figure S1. Sequence coverage of AMPK heterotrimer. (a) Sequence of AMPK 111 with blue horizontal bars beneath each sequence represents peptide fragments obtained during MSMS studies. The part of the sequence resolved in the crystal structure (PDB:2Y94) is colored yellow and the phosphorylation sites are marked red. (b) AMPK atomic structure (PDB:2Y94) along with the GBD domain generated based on yeast homologue. (c) Percentage of sequence coverage of different subunit of AMPK at different level of HDX experiment.
Supplementary Figure S2. Assessment of phosphorylation at activation loop residue Thr172 of the α-chain of AMPK α1β1γ1. Purified AMPK was precipitated with acetone, treated with dithiothreitol (reducing agent), S-alkylated with iodoacetamide and digested with endoproteinase Asp-N, and the resulting digest was analyzed by liquid chromatography-mass spectrometry (LC-MS). (A) LC-MS of activation loop peptide from unactivated AMPK, showing absence of the phosphorylated form. (NL, normalized level, indicates signal intensity in instrument units). (B) Activation loop peptide from AMPK activated by treatment with CamKKβ. The activation loop peptide is now recovered almost entirely in the phosphorylated form. (C) Activation loop peptide from activated AMPK treated with phosphatase 2α (PP2α). The activation loop peptide from this protein is recovered almost entirely in the unphosphorylated form. Comparison of peak areas indicated that ~97% of Thr172 was phosphorylated in activated AMPK.
Supplementary Figure S3. Chemical structure of A769662.
Supplementary table S1. Comparison of deuterium incorporation level in phosphorylated and non-phosphorylated peptides from α and β subunit of AMPK111 and AMPK221, upon binding of different ligands. Percentage of deuterium differences are color coded according to the color key.
Supplementary table S2. Deuterium incorporation level of important peptides of all AMPK constructs used in this study, upon binding of different ligands. Percentage of deuterium differences are color coded according to the color key.
Highlights.
Long range allosteric communication between AMPK subunits detected by HDX
Distinct mode of activation by nucleotides, carbohydrate and a synthetic activator
Comprehensive HDX analysis of a 130kDa hetrotrimeric kinase
Putative binding site of the synthetic activator, A769662, on AMPK is determined
Acknowledgments
We thank Kieran Geoghegan for critical reading of the manuscript and for his suggestions. We also thank Xiayang Qiu for his support and encouragement. R.K. and P.R.G. conceived of the project and designed the research; R.R.L., F.R., M.H., B.P., L.R.H., and R.M. conducted the research; R.L., B.P., M.C., R.K, D.G., M.J.C. and P.R.G. analyzed the data; and R.R.L., R.K., S.A.B. and P.R.G. wrote the paper, with contributions from all authors.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amodeo GA, Rudolph MJ, Tong L. Crystal structure of the heterotrimer core of Saccharomyces cerevisiae AMPK homologue SNF1. Nature. 2007;449:492–495. doi: 10.1038/nature06127. [DOI] [PubMed] [Google Scholar]
- Bieri M, Mobbs JI, Koay A, Louey G, Mok YF, Hatters DM, Park JT, Park KH, Neumann D, Stapleton D, et al. AMP-activated protein kinase beta-subunit requires internal motion for optimal carbohydrate binding. Biophys J. 2012;102:305–314. doi: 10.1016/j.bpj.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JB, Chalmers MJ, Prasad S, Busby SA, Kamenecka TM, He Y, Nettles KW, Griffin PR. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Cacicedo JM, Gauthier MS, Lebrasseur NK, Jasuja R, Ruderman NB, Ido Y. Acute Exercise Activates AMPK and eNOS in the Mouse Aorta. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.01279.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445:11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- Celenza JL, Carlson M. Cloning and genetic mapping of SNF1, a gene required for expression of glucose-repressible genes in Saccharomyces cerevisiae. Mol Cell Biol. 1984;4:49–53. doi: 10.1128/mcb.4.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, Griffin PR. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2006;78:1005–1014. doi: 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- Chen K, Xu X, Kobayashi S, Timm D, Jepperson T, Liang Q. Caloric Restriction Mimetic 2-Deoxyglucose Antagonizes Doxorubicin-induced Cardiomyocyte Death by Multiple Mechanisms. J Biol Chem. 2011;286:21993–22006. doi: 10.1074/jbc.M111.225805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wang J, Zhang YY, Yan SF, Neumann D, Schlattner U, Wang ZX, Wu JW. AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nat Struct Mol Biol. 2012;19:716–718. doi: 10.1038/nsmb.2319. [DOI] [PubMed] [Google Scholar]
- Chen L, Xin FJ, Wang J, Hu J, Zhang YY, Wan S, Cao LS, Lu C, Li P, Yan SF, et al. Conserved regulatory elements in AMPK. Nature. 2013;498:E8–10. doi: 10.1038/nature12189. [DOI] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Dai SY, Chalmers MJ, Bruning J, Bramlett KS, Osborne HE, Montrose-Rafizadeh C, Barr RJ, Wang Y, Wang M, Burris TP, et al. Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method. Proc Natl Acad Sci U S A. 2008;105:7171–7176. doi: 10.1073/pnas.0710802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- Giordanetto F, Karis D. Direct AMP-activated protein kinase activators: a review of evidence from the patent literature. Expert Opin Ther Pat. 2012;22:1467–1477. doi: 10.1517/13543776.2012.743994. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci. 2011;36:470–477. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- Hwang JT, Kwon DY, Yoon SH. AMP-activated protein kinase: a potential target for the diseases prevention by natural occurring polyphenols. N Biotechnol. 2009;26:17–22. doi: 10.1016/j.nbt.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Itani SI, Saha AK, Kurowski TG, Coffin HR, Tornheim K, Ruderman NB. Glucose autoregulates its uptake in skeletal muscle: involvement of AMP-activated protein kinase. Diabetes. 2003;52:1635–1640. doi: 10.2337/diabetes.52.7.1635. [DOI] [PubMed] [Google Scholar]
- Johnson EC, Kazgan N, Bretz CA, Forsberg LJ, Hector CE, Worthen RJ, Onyenwoke R, Brenman JE. Altered metabolism and persistent starvation behaviors caused by reduced AMPK function in Drosophila. PLoS One. 2010:5. doi: 10.1371/journal.pone.0012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kemp BE, Oakhill JS, Scott JW. AMPK structure and regulation from three angles. Structure. 2007;15:1161–1163. doi: 10.1016/j.str.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Koay A, Woodcroft B, Petrie EJ, Yue H, Emanuelle S, Bieri M, Bailey MF, Hargreaves M, Park JT, Park KH, et al. AMPK beta subunits display isoform specific affinities for carbohydrates. FEBS Lett. 2010;584:3499–3503. doi: 10.1016/j.febslet.2010.07.015. [DOI] [PubMed] [Google Scholar]
- Marx A, Nugoor C, Panneerselvam S, Mandelkow E. Structure and function of polarity-inducing kinase family MARK/Par-1 within the branch of AMPK/Snf1-related kinases. FASEB J. 2010;24:1637–1648. doi: 10.1096/fj.09-148064. [DOI] [PubMed] [Google Scholar]
- McBride A, Ghilagaber S, Nikolaev A, Hardie DG. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009;9:23–34. doi: 10.1016/j.cmet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- Neumann D, Woods A, Carling D, Wallimann T, Schlattner U. Mammalian AMP-activated protein kinase: functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expr Purif. 2003;30:230–237. doi: 10.1016/s1046-5928(03)00126-8. [DOI] [PubMed] [Google Scholar]
- Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK) Proc Natl Acad Sci U S A. 2010;107:19237–19241. doi: 10.1073/pnas.1009705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakhill JS, Scott JW, Kemp BE. AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol Metab. 2012;23:125–132. doi: 10.1016/j.tem.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Pang T, Zhang ZS, Gu M, Qiu BY, Yu LF, Cao PR, Shao W, Su MB, Li JY, Nan FJ, et al. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem. 2008;283:16051–16060. doi: 10.1074/jbc.M710114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polekhina G, Gupta A, Michell BJ, van Denderen B, Murthy S, Feil SC, Jennings IG, Campbell DJ, Witters LA, Parker MW, et al. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr Biol. 2003;13:867–871. doi: 10.1016/s0960-9822(03)00292-6. [DOI] [PubMed] [Google Scholar]
- Polekhina G, Gupta A, van Denderen BJ, Feil SC, Kemp BE, Stapleton D, Parker MW. Structural basis for glycogen recognition by AMP-activated protein kinase. Structure. 2005;13:1453–1462. doi: 10.1016/j.str.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Rajamohan F, Harris MS, Frisbie RK, Hoth LR, Geoghegan KF, Valentine JJ, Reyes AR, Landro JA, Qiu X, Kurumbail RG. Escherichia coli expression, purification and characterization of functional full-length recombinant alpha2beta2gamma3 heterotrimeric complex of human AMP-activated protein kinase. Protein Expr Purif. 2010;73:189–197. doi: 10.1016/j.pep.2010.04.022. [DOI] [PubMed] [Google Scholar]
- Riek U, Scholz R, Konarev P, Rufer A, Suter M, Nazabal A, Ringler P, Chami M, Muller SA, Neumann D, et al. Structural properties of AMP-activated protein kinase: dimerization, molecular shape, and changes upon ligand binding. J Biol Chem. 2008;283:18331–18343. doi: 10.1074/jbc.M708379200. [DOI] [PubMed] [Google Scholar]
- Rudolph MJ, Amodeo GA, Tong L. An inhibited conformation for the protein kinase domain of the Saccharomyces cerevisiae AMPK homolog Snf1. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:999–1002. doi: 10.1107/S1744309110028265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem. 2007;282:32539–32548. doi: 10.1074/jbc.M706543200. [DOI] [PubMed] [Google Scholar]
- Schimmack G, Defronzo RA, Musi N. AMP-activated protein kinase: Role in metabolism and therapeutic implications. Diabetes Obes Metab. 2006;8:591–602. doi: 10.1111/j.1463-1326.2005.00561.x. [DOI] [PubMed] [Google Scholar]
- Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, Iseli TJ, Koay A, Gooley PR, Stapleton D, et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008;15:1220–1230. doi: 10.1016/j.chembiol.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Shang L, Wang X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy. 2011;7:924–926. doi: 10.4161/auto.7.8.15860. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Maniatis T, EF, Sambrook J. Molecular Cloning: A Laboratory Manual. NY, Cold Spring Harbour Laboratory; Cold Spring Harbour, NY: 1982. [Google Scholar]
- Vingtdeux V, Chandakkar P, Zhao H, Davies P, Marambaud P. Small-molecule activators of AMP-activated protein kinase (AMPK), RSVA314 and RSVA405, inhibit adipogenesis. Mol Med. 2011;17:1022–1030. doi: 10.2119/molmed.2011.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Wang Y, Busby SA, Pascal BD, Garcia-Ordonez RD, Bruning JB, Istrate MA, et al. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat Struct Mol Biol. 2011;18:556–563. doi: 10.1038/nsmb.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Chen L, Zhou XM, Zhang YY, Zhang YJ, Zhao J, Ji SR, Wu JW, Wu Y. Structural insights into the architecture and allostery of full-length AMP-activated protein kinase. Structure. 2011;19:515–522. doi: 10.1016/j.str.2011.01.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Sequence coverage of AMPK heterotrimer. (a) Sequence of AMPK 111 with blue horizontal bars beneath each sequence represents peptide fragments obtained during MSMS studies. The part of the sequence resolved in the crystal structure (PDB:2Y94) is colored yellow and the phosphorylation sites are marked red. (b) AMPK atomic structure (PDB:2Y94) along with the GBD domain generated based on yeast homologue. (c) Percentage of sequence coverage of different subunit of AMPK at different level of HDX experiment.
Supplementary Figure S2. Assessment of phosphorylation at activation loop residue Thr172 of the α-chain of AMPK α1β1γ1. Purified AMPK was precipitated with acetone, treated with dithiothreitol (reducing agent), S-alkylated with iodoacetamide and digested with endoproteinase Asp-N, and the resulting digest was analyzed by liquid chromatography-mass spectrometry (LC-MS). (A) LC-MS of activation loop peptide from unactivated AMPK, showing absence of the phosphorylated form. (NL, normalized level, indicates signal intensity in instrument units). (B) Activation loop peptide from AMPK activated by treatment with CamKKβ. The activation loop peptide is now recovered almost entirely in the phosphorylated form. (C) Activation loop peptide from activated AMPK treated with phosphatase 2α (PP2α). The activation loop peptide from this protein is recovered almost entirely in the unphosphorylated form. Comparison of peak areas indicated that ~97% of Thr172 was phosphorylated in activated AMPK.
Supplementary Figure S3. Chemical structure of A769662.
Supplementary table S1. Comparison of deuterium incorporation level in phosphorylated and non-phosphorylated peptides from α and β subunit of AMPK111 and AMPK221, upon binding of different ligands. Percentage of deuterium differences are color coded according to the color key.
Supplementary table S2. Deuterium incorporation level of important peptides of all AMPK constructs used in this study, upon binding of different ligands. Percentage of deuterium differences are color coded according to the color key.