

SUMMARY
In cells, tRNAs are synthesized as precursor molecules bearing extra sequences at their 5′ and 3′ ends. Some tRNAs also contain introns which, in archaea and eukaryotes, are cleaved by an evolutionarily conserved endonuclease complex that generates fully functional mature tRNAs. In addition, tRNAs undergo numerous post-transcriptional nucleotide chemical modifications. In Trypanosoma brucei the single intron-containing tRNA (tRNATyrGUA) is responsible for decoding all tyrosine codons; therefore, intron removal is essential for viability. We show by molecular and biochemical approaches the presence of several non-canonical editing events, within the intron of pre-tRNATyrGUA, involving guanosine to adenosine transitions (G to A) and an adenosine to uridine transversion (A to U). The RNA editing described here is required for proper processing of the intron, establishing for the first time the functional significance of non-canonical editing with implications for tRNA processing in the deeply divergent kinetoplastid lineage and eukaryotes in general.
INTRODUCTION
Critical to the role of tRNAs in protein synthesis are a series of processing events that ensure their proper structure and function. In most eukarya, tRNAs are transcribed in the nucleus as precursor molecules that contain extra sequences: a 5′ leader, a 3′ trailer, and in fewer cases, introns. Removal of these extra sequences requires a number of enzymes, including RNase P for cleavage of the 5′ leader sequence, RNase Z and other endonucleases and exonucleases for 3′ end maturation (Mayer et al., 2000). Finally, a specialized tRNA-specific multi-protein splicing machinery removes the introns (Fan et al., 1998). A non-templated universally conserved CCA tail is also added at the 3′ end of the tRNA by a CCA nucleotidyl transferase. Following nuclear maturation, the tRNA is then exported into the cytoplasm where it can be used for aminoacylation and therefore translation (Phizicky, 2005; Rubio and Hopper, 2011; Wolin and Matera, 1999).
Although 5′ and 3′ end trimming is highly conserved, mechanistically, intron-removal varies. In bacteria, tRNA introns are autocatalytic and control their own removal (Reinhold-Hurek and Shub, 1992). In archaea and eukarya tRNA splicing is initiated by a protein endonuclease that recognizes and cleaves the intron, generating tRNA half-molecules that are then joined by a tRNA splicing ligase (Phizicky and Hopper, 2010). In eukarya, despite variations in the number of intron-containing tRNAs and their respective intron sizes, there exist two conserved features: 1) tRNATyr contains an intron in almost all sequenced eukaryotic genomes (Chan and Lowe, 2009); 2) most, if not all, introns interrupt the anticodon loop one nucleotide 3′ of the anticodon (Chan and Lowe, 2009). The former underscores an important, but not yet well-understood, aspect of tRNA intron maintenance and evolution; the latter implies that intron removal is essential for eukaryotic viability.
At various points during maturation, tRNAs also undergo numerous post-transcriptional chemical modifications placed on the sugar or at various positions of the base, producing a variety of nucleotides, each with slightly different chemical characteristics. To date, there are more than 100 different modified nucleotides found in tRNAs (Machnicka et al., 2013) and despite much progress on the role of some modifications in tRNA function, knowledge of the activity and mechanism of most modification enzymes in many organisms is far from complete.
In eukarya, a subset of post-transcriptional changes known as RNA editing may target non-coding RNAs and mRNAs. Editing alters genetic information at the RNA level beyond what can be found in the encoding genes and as such can increase genetic diversity. In tRNAs the most common editing mechanism involves base deamination; “programmed changes” of one canonical nucleotide for another that may impact both their overall structure and function. One type of deamination involves the conversion of adenosine (A) to inosine (I) and has been observed in archaea, bacteria and eukarya. Additionally, tRNAs may also undergo cytosine (C) to uridine (U) editing which has been described in archaea, marsupials, kinetoplastids, and plant organelles (Alfonzo et al., 1999; Fey et al., 2001; Janke and Paabo, 1993). The function of C to U editing of tRNAs varies depending on the position of the edited base in the tRNA. For example, C to U editing can fix stems or restore tertiary base pairing at positions where a nucleotide mismatch is genomically encoded, thus ensuring proper folding (Binder et al., 1994; Marechal-Drouard et al., 1993). In other instances, C to U editing (analogous to A to I editing) affects the anticodon, changing the decoding ability from one codon to another and effectively expanding the decoding properties of the edited tRNA (Alfonzo et al., 1999).
In total, the mechanisms that trim, splice, modify and edit tRNA molecules are required for the proper folding, recognition and function of mature tRNAs in translation. In the present study we are focusing on the identification of all the components of the T. brucei tRNA splicing endonuclease. Speaking to its degree of divergence, we could only identify a single putative subunit of the four highly conserved sub-units of other eukaryotes. Here, the role of this sub-unit in tRNA splicing is demonstrated by RNA interference. Surprisingly, analysis of the tRNA intron sequence led to the discovery of a number of non-canonical RNA editing events and these are shown to be important for tRNA splicing. Sequence comparisons presented here also show various degrees of representation of the edited introns in the genomes of trypanosomes, highlighting distinct evolutionary paths for the occurrence of such unusual non-canonical editing events.
RESULTS
The Putative T. brucei SEN34 is Essential for Splicing and Viability
Little is known about tRNA splicing in early divergent eukaryotes, such as kinetoplastids, which include Leishmania and Trypanosoma, responsible for devastating diseases worldwide. In T. brucei, only tRNATyr, a single-copy gene, contains an intron (Mottram et al., 1991; Schneider et al., 1993) (Figure 1A). Bioinformatic analysis of the kinetoplastid genomic databases (TriTrypDB), with various sub-units of the yeast and human tRNA splicing endonuclease (SEN2, SEN34, SEN54 and SEN15) (Paushkin et al., 2004; Trotta et al., 1997) revealed the presence of a potential SEN homolog in T. brucei (TbSEN) (Figure S1). To investigate the role of TbSEN in tRNA splicing in T. brucei, its protein-coding region was cloned into a T. brucei tetracycline-inducible RNAi vector (Wickstead et al., 2002). Following RNAi induction by tetracycline, we observed a growth phenotype and eventually cell death, thus TbSEN is essential for growth (Figure 1B). A substantial decrease of the putative TbSEN mRNA was also observed by RT-PCR analysis with TbSEN-specific oligonucleotide primers (Figure 1B, graph inset). These results are consistent with those reported in a high-throughput RNAi screen coupled to parallel sequencing that showed that down-regulation of the same gene led to a significant decrease in T. brucei fitness, although these authors did not assigned a function to the gene (Alsford et al., 2011).
Figure 1. The Putative SEN34 Homolog is Essential in T. brucei.
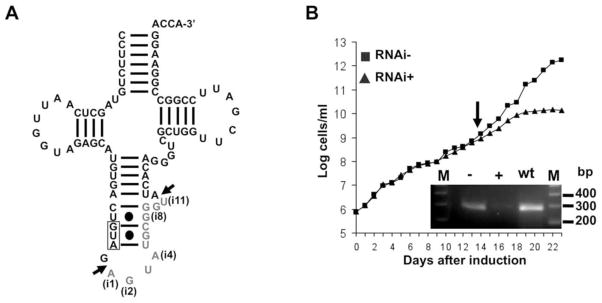
(A) The cloverleaf structure of the single copy intron-containing tRNA, tRNATyr, the only intron-containing tRNA in T. brucei. The 11-nucleotide intron is shown in gray letters. The anticodon sequence is boxed. Arrows denote the 5′ and 3′ cleavage sites. Nucleotide positions in the intron are denoted as “i” followed by a number, only the first (i1), last (i11) and the edited intron positions (i2, i4 and i8) are highlighted.
(B) A growth curve of tetracycline-induced RNA interference (RNAi+, black triangles) of TbSEN (endonuclease sub-unit) as compared to an uninduced control (RNAi−, black squares). The y-axis shows a log-scale of cumulative cell densities accounting for total dilutions and the X-axis shows the progression of the growth curve in days. The inset shows semi-quantitative RT-PCR with RNA isolated at the onset of the growth phenotype (day 14, arrow) from RNA+ (+), RNAi− (−) and wild type (wt) cells. M refers to a 100 base pair (bp) size marker (Invitrogen), the region between 200–400 bp is shown. See also Figure S1.
The Only tRNA Intron of T. brucei Undergoes Non-canonical Editing
To determine the level of intron-containing tRNATyr in these cells, Northern hybridization experiments were performed using an intron-specific probe and total RNA fractions from RNAi-induced and uninduced cells. Down-regulation of TbSEN should lead to accumulation of the intron-containing tRNA. Surprisingly, we were unable to detect significant hybridization signal using this probe in either RNA fraction (Figure 2A).
Figure 2. The Intron of tRNATyr Undergoes Non-canonical Editing.

(A) Control RNA transcripts and RNA extracted from the RNAi induced, uninduced and wild type cells (as above) analyzed by Northern hybridization with an intron-specific probe. The “control transcripts” (left panel) refer to in vitro-transcribed control tRNAs, that either contain (+) or lack (−) the intron, used as positive and negative controls for intron detection. RNA extracted from the RNAi induced, uninduced and wild type cells (as above) were transferred to a separate membrane and probe with the same probe (center panel). The same membrane was stripped and re-hybridized with a probe specific for tRNAGlu used as a positive control for hybridization (right panel).
(B) RT-PCR experiments with the same RNA samples as before but with oligonucleotide primers specific for the exons of tRNATyr, which do not discriminate between mature (mat) and intron-containing tRNA (pre), shown by arrows. No RT refers to a mock PCR reaction in the absence of reverse transcriptase used to control for DNA contamination in these samples.
(C) The “pre” band from the gel above was excise and the purified DNA cloned into plasmids and 25 independent clones analyzed by automated sequencing. Representative sequencing traces are shown; positions of non-canonical editing are marked by an asterisk and highlighted by gray boxes. The schematics on the right show the anticodon stem portion of tRNATyr with the intron in gray letters and the observed non-canonical edits, G to A (green letters) and A to U (in red). Percentages denote the number of clones of each type observed divided by the number of total cones sequenced (in parenthesis) times 100. The arrow marks the cleavage sites (as above). The data are representative of at least 4 independent experiments for each section. The edited nucleotide positions in the intron are labeled as above (i2, i4 and i8). See also Figure S2.
Possibly in uninduced cells, intron-containing tRNATyr is a short-lived intermediate that escapes detection by Northern analysis. Thus we took a more sensitive approach using reverse transcription polymerase chain reaction (RT-PCR) with the same RNA fractions and oligonucleotide primers specific for the exons of tRNATyr. Down regulation of TbSEN leads to the appearance of a slower migrating band on non-denaturing gels, consistent with accumulation of the intron-containing species (Figure 2B). This species also partially accumulated in the uninduced cells, likely due to the well-established leakiness of the T. brucei RNAi system (Kolev et al., 2011; Ullu et al., 2004). We cloned this fragment into a plasmid vector and sequenced plasmid DNA from 25 independent clones. None of the clones matched the genomic sequence; instead, we found two populations of intron sequences: one containing G to A and A to U alterations in the intron (16 out of 25 clones or 64%) and the other with an additional G to A change (9 out of 25 clones or 36%) (Figure 2C). Similar experiments with total DNA isolated from wild type cells showed that all clones (a total of 25 independent clones) contained sequences identical to those reported in the T. brucei genome database (TriTrypDB) (not shown). The data are consistent with the alteration of nucleotides in the sequence of the single tRNA intron occurring in at least two of the three sites with >96% efficiency (where 1/25 clones would represent a theoretical value for unedited sequences of 4%). The nature of the nucleotide changes is unusual and does not belong to any of the canonical RNA editing events described to date (deamination or nucleotide insertion/deletion).
To further substantiate tRNATyr intron editing, we probed the same Northern blot as before with intron-specific probes that include the editing changes. These could now detect the intron-containing tRNAs (Figure S2). This demonstrates that the observed nucleotide changes are not the result of either RT-PCR or sequencing artifacts. Interestingly, we observed higher levels with the probe specific for the tRNA edited at 2 positions (Figure S2, edited 2), compared to the other species (Figure S2, edited 3). These differences in the relative levels of the two edited species are consistent with our sequencing data.
In an attempt to further assess editing levels, RNA isolated from RNAi induced and uninduced cells were used in “poisoned” primer extension assays (Alfonzo et al., 1999), where editing would lead to incorporation of the chain terminator ddTTP (Figure 3A). Again with this approach, we could detect a “strong” stop at the position corresponding to the primer extended by two nucleotides, indicative of editing (Figure 3B, left panel). The only significant additional stop corresponded to a signal at primer plus 3 nucleotides, but this signal is due to mis-priming via the mature tRNA sequence (Figure 3B, right panel, mature control transcript). Mis-priming may occur due to the sequence similarity between the primer annealing sites for the precursor tRNA (intron-containing) and the mature sequence (Figure S3). In addition, several fainter bands were observed with nuclear and cytosolic RNA fractions from wild type cells (Figure 3B, right panel). These read-through products are visible because of incomplete termination with ddTTP, but their sizes corresponded to primer plus 6 nucleotides (i4 position of the intron) and primer +8 nucleotides the 5′-most editing site (i2 position of the intron), consistent with unedited and edit 3 transcripts respectively. The low-level signal with these additional bands is in agreement with the high-editing efficiency observed by RT-PCR/sequencing.
Figure 3. Efficient Non-canonical Editing is Localized in the Nucleus.

(A) Diagram describing the “poisoned” primer extension assay. A radioactive intron-specific primer that anneals two nucleotides 3′ of the editing site is shown (intron-specific primer). The reaction was performed in the presence of the chain terminator ddTTP (ddT), which if incorporated at the edited position yields a primer plus two-nucleotide product. A primer plus 6 nucleotides is indicative of lack of editing at the 3′-most editing site (G to A). Editing sites are indicated by arrows and the exonic sequences are boxed. The edited positions are shown in boldface letters. +8, +6 and +2 indicate the distance in nucleotides of each edited position from the last nucleotide of the primer.
(B) “Poisoned” primer extension reactions of total RNA isolated from the RNAi uninduced (−, lanes 1 and 2) and induced (+, lanes 3 and 4) cells, as before. Reactions were performed in the absence (−, lanes 1 and 3) or presence (+, lanes 2 and 4) of ddNTP (left panel). The right panel shows similar reactions as in the left but performed with sub-cellular RNA fractions: nuclear (nuc) and cytoplasmic (cyto), isolated from wild type cells. The arrows denoted the position expected for edited (primer+2 and primer+8) and unedited (primer+6) products in these reactions, as well as the primer. Control transcripts refer to RNA generated in vitro and used as markers for the different expected products (as indicated). Mature refers to an in vitro generated intronless transcript used as a control for mis-priming during the assay. See also Figure S3 and S4.
It is possible that the observed non-canonical changes are due to the general poor physiological state of the cells following RNAi of an essential gene. We also isolated nuclear RNA from wild-type cells to determine: 1) if editing occurs in wild-type cells and 2) where editing is localized within cells. In nuclear RNA fractions, we detected intron-containing tRNA edited to similar levels as the tRNA isolated from the RNAi strain (Figure 3B, right panel). These results support the view that the non-canonical editing described here occurs naturally in the nucleus of T. brucei. Edited tRNA was also detected in the cytoplasmic fractions, likely due to breakage of the nuclei during preparation, which leads to nuclear contamination of the cytoplasmic fractions as shown by significant hybridization of the cytoplasmic fractions with a probe specific for U6 snRNA (a nuclear marker) (Figure S4).
Because of the prevalence of post-transcriptional modifications in tRNAs, it is possible that the observed editing changes are due to some unusual modification which leads to misreading by the reverse transcriptase. To explore this possibility we purified native intron-containing and mature tRNATyr from the RNAi cell line (following induction by tetracycline). Total RNA from these cells was hybridized to a biotinylated oligonucleotide (as described previously) (Alfonzo et al., 1999). This oligonucleotide does not discriminate between the intron-containing and the mature tRNA. Following hybridization the tRNAs were gel-purified and subjected to post-labeling (Alfonzo et al., 1999). Although this approach does not reveal the specific location of modifications, it shows the total modification set for each species. We found that the intron-containing tRNA had negligible levels of modifications, while the mature was fully modified (Figure 4). Importantly, some of the modifications detected in the mature tRNA (for example acp3U and m7G) occur only once per tRNA molecule (Machnicka et al., 2013), arguing that the lack of modifications in the intron-containing tRNA is not due to the modifications existing at such low levels that they escape detection. This experiment suggests that a) in contrast to other systems, there are no intron-dependent modifications and b) no unusual modifications could account for the editing changes and these are likely due to replacement by canonical nucleotides.
Figure 4. Native pre-tRNATyr Has Negligible Modification Levels.
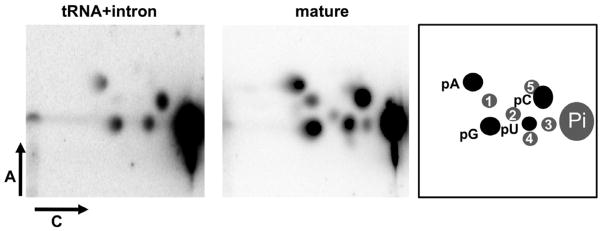
Two-dimentional thin-layer chromatography (2D-TLC) of native tRNATyr purified from the RNAi-induced cells. The purification procedure does not discriminate between mature and pre-tRNA (intron-containing). Post-labeling analysis of the two native species shows the presence of modified nucleotides only in the mature tRNA (center panel). These modifications are commonly found in tRNATyr from other eukaryotes and often found in most tRNAs. The left panel shows the lack of modified nucleotides in the intron-containing tRNA. The right panel shows a key with 5-monophosphorylated nucleotide assignments for the right and center panels, where: 1= pQ or pGm, queuosine or 2′-O-methylguanosine; 2= pΨm, 2′-O-methylpseudouridine; 3= acp3U, 3-amino-3-carboxypropyluridine; 4= pΨ, pseudouridine; 5= pm7G, 7-methylguanosine. pA, pC, pG and pU, refer to the unmodified 5′ ribonucleotides. Pi refers to inorganic phosphate released following incubation with P1 nuclease. Arrows labeled A and C represent the two dimensions used during Thin-layer Chromatography (TLC), as described in Experimental Procedures. All modification assignments are based on published maps (Grosjean et al., 2007; Grosjean et al., 2004). None of these modifications are known to alter base pairing during reverse transcription.
Non-canonical Editing is Important for tRNATyr Splicing
We next investigated the possibility that editing of the intron plays a role in tRNA splicing. We partly purified the endonucelase from T. brucei by following previously published detailed protocols for the enrichment of endonuclease activity from yeast (Peebles et al., 1983). We tested the efficiency of cleavage by this enriched T. brucei fraction on substrates site-specifically labeled at the first position of the intron (i1, the 5′-most nucleotide of the intron). Three different substrates were tested: either the two edited transcripts or an unedited version of the intron (representing the genomic sequence). The edited pre-tRNAs were efficiently cleaved as compared to the unedited (Figure 5A and 5B). Furthermore, we observed transfer of the label to the 3′-most nucleotide of the 5′ exon, allowing visualization of the free 5′ exon. This is consistent with the cleavage chemistry observed for other tRNA splicing endonucleases regardless of the organism (Abelson et al., 1998). This observation is also in line with previous work in T. brucei showing that in vitro, a single nucleotide mutation at the first position of the anticodon abrogates splicing (Schneider et al., 1993). Although this position is not precisely the 3′-most edited position described here, it would disrupt a canonical base pair between the anticodon and the intronic sequence. Taken together, these results suggest the importance of a specific structural requirement, formed by editing of nucleotides to create a fully base-paired intron stem in the T. brucei system.
Figure 5. The Edited Intron is the Prefered Substrate for In vitro Cleavage.

(A) Cleavage assays using an enriched endonuclease fraction from T. brucei and chemically synthesized intron-containing tRNATyr corresponding to either an intron with the unedited sequence (genomic) or edited at two (edited 2) and three positions (edited 3). These substrates were site-specifically labeled at the 5′-most nucleotide of the intron in the pre-tRNA and incubated with a constant and subsaturating concentration of enzyme fraction at increasing time. The size markers on the left (also shown pictorically on the right) correspond to mature (no intron), 5′ exon-intron intermediate and the mature 5′ exon.
(B) Progress curves of the cleavage reactions on (a) where the tRNA with the unedited intron is denoted by black squares while the tRNA substrates with the edited introns are shown by black triangles (edited 2) and black circles (edited 3). Each curve is the average of 3 independent assays and their R2 values are as indicated. Error bars represent the standard error from the mean.
We then constructed an intron-containing tRNA variant in which two base pairs of the anticodon stem were flipped (Figure 6A, schematic). This construct could allow similar RT-PCR analysis as before, while differentiating this variant from the endogenous wild-type tRNATyr. This construct was integrated in the genome where expression is still driven by the native RNA polymerase III. Total RNA from this strain was used in an RT reaction performed with an oligonucleotide primer that contains the 3′ tag and overlaps the intronic sequence as to bias the cDNA towards intron/“tag” containing tRNA (Figure 6B). The PCR reaction was then performed with the “tag”-specific primers, which yielded a product of a size consistent with that of the intron-containing tRNA. This product was analyzed by restriction digests diagnostic for either edited (cleaved by BfuAI) or unedited (cleaved by AciI, but not BfuAI) products. RT-PCR analysis showed that the mutations in this tRNA variant led to complete inhibition of editing (Figure 6B, left panel, compare AciI to BfuAI digests). We do not understand the reason for this, other than to suggest that those positions may be important for recognition by a factor important for editing.
Figure 6. Editing is Important for tRNA Splicing.

(A) Schematic diagram of the expected products for the intron-containing tRNA “tagged” variant generated by flipping the two base pairs of the anticodon stem of tRNATyr (as indicated) producing two complementary tags (Tag1 and 2). The sequence of the intron corresponding to the unedited tRNA contains an AciI restriction site, which upon editing becomes a BfuAI site. These restriction sites (boxed) were used as diagnostic for either product during RT-PCR reactions specific for the tagged constructs (below).
(B) RT-PCR reactions of only the “tagged” intron-containing tRNA variant (left panel). An oligonucleotide primer specific for the intron and that contains the Tag2 sequence was used to generate a cDNA specific for intron-containing tagged species. Two additional primers whose 3′ ends were anchored at the tags were then used for the PCR reaction. Reactions were performed in the presence (RT+) or absence (RT−) of reverse transcriptase, where the RT− reaction serves as a DNA contamination control. The products from the RT+ reaction were digested with either AciI, which only cleaves a DNA product corresponding to the unedited intron or BfuAI, which only cleaves the edited species but does not discriminate between the edited two and three species. A synthetic edited transcript was also used and the resulting RT-PCR product was digested with BfuAI as a positive control for digestion (BfuAI control) (left panel). The center panel shows an RT-PCR reaction with the tag-specific oligonucleotide primers, which do not discriminate between the mature and intron-containing tagged tRNAs. A full-length intron-containing product, shown by the black arrow (76 bp expected size), demonstrating the accumulation of the unspliced tRNA. The gray arrow denotes the expected position for the mature tagged tRNA product (65 bp expected size). The right panel shows a similar assay with a tagged construct containing a fully edited intron and showing that this variant is proficient in splicing. M, refers to a 10-bp size marker (Invitrogen) and “bp” indicates size in base pairs.
We also performed similar RT-PCR reactions but using only the primers specific for the exonic “tags”. In this variant, both editing and splicing were inhibited, suggesting a possible connection between the two events (Figure 6B, center panel). As a control, we generated a tRNA variant with the same sequence “tags” but in which a fully edited intron was provided (Figure 6B, right panel). A significant amount of tagged mature tRNA was observed with this variant, supporting the view that in vivo non-canonical editing is essential for splicing (Figure 6B, compare center and right panels). In this experiment, there is still a portion of the tRNA that remains unspliced, presumably due to the fact that the tag mutations also affect splicing. This last observation suggests that editing and splicing share common recognition motifs (Figure 6B, right panel). However, we cannot rule out the possibility that the accumulated unspliced “tagged” tRNA is simply due to the general over-expression of the tRNA when transcribed from a plasmid.
Evolutionary Conservation of Non-canonical Editing in Kinetoplastids
We also investigated the potential prevalence of intron editing in kinetoplastids in general. In all kinetoplastids, tRNATyr is the only intron-containing tRNA. We aligned the genomic intron sequences from various kinetoplastids (genus Crithidia, Leishmania, etc.), revealing that the edited nucleotides exist to various degrees at the DNA level in these genomes (Figure 7A). Most astonishing is that L. tarentolae encodes two copies of tRNATyr in the genome, precisely matching the sequence of each edited species found in T. brucei at the RNA level (Figure 7B).
Figure 7. Evolutionary conservation of the edited introns in kinetoplastids.

(A) Sequence alignment of the intron portion of tRNATyr from various kinetoplastids showing potential editing in closely related organisms.
(B) The closely related kinetoplastid Leishmania tarentolae encodes two copies of tRNATyr with intron sequences matching precisely those observed in T. brucei at the RNA level. The arrows mark the cleavage sites and the asterisks denote those positions found to undergo non-canonical editing in T. brucei. “cDNA” refers to the two edited sequences obtained from T. brucei. Genomic refers to sequences obtained from the genomic database (TriTryp). Tb, Trypanosoma brucei; Lm, Leishmania major; Cf, Crithidia fasciculata; Tcon, Trypanosoma congolense; Em, Endotrypanum monterogeii; Lp, Leishmania panamemsis; Lb, Leishmania braziliensis.
DISCUSSION
In all organisms, introns disrupt the sequence of tRNAs and their precise removal is essential to generate a mature molecule that can engage in protein synthesis. Archaea and eukarya share a common mechanism for tRNA splicing, which involves a protein endonuclease that cleaves the intron at the two spliced sites and initiates the splicing process (Calvin and Li, 2008; Hopper and Phizicky, 2003; Phizicky and Greer, 1993). All eukaryal tRNA endonucleases so far described, for example from plants, mammals and yeast, contain 4 distinct subunits (Calvin and Li, 2008). Two subunits contain the enzyme’s active site required for cleavage (SEN2 and SEN34) yet the other two sub-units (SEN15 and SEN54) are essential structural components. Our analysis of the kinetoplastids databases (which include T. brucei) revealed one bioinformatically recognizable sub-unit of the endonuclease, which despite divergence can still be assigned as a SEN34 homolog based on sequence comparative analysis (Figure S1). Additionally, the T. brucei genome encodes a single intron-containing tRNA. This intron is only 11 nucleotides long, which is close to the lower theoretical limit required for cleavage (Di Nicola Negri et al., 1997; Fabbri et al., 1998; Tocchini-Valentini et al., 1993).
Beyond splicing comes the question of the unusual editing observed in this intron. The RNA editing repertoire includes a number of disparate mechanisms, many of which serve to alter coding capacity. These include uridine insertions and deletions in trypanosomatid mitochondria, C insertions in Physarum, nucleotide deaminations (C to U and A to I) in various coding and non-coding RNAs (including tRNAs) (Aphasizhev and Aphasizheva, 2011; Blanc and Davidson, 2010; Chateigner-Boutin and Small, 2011; Gommans, 2012; Goringer et al., 2011; Jackman and Alfonzo, 2013; Jackman et al., 2012; Mallela and Nishikura, 2012; Paris et al., 2012; Sie and Kuchka, 2012; Smith et al., 2012). All of these reactions have been extensively studied and the various editing activities reconstituted in vitro. New sequencing technologies have now uncovered many new editing sites in a number of organisms including humans, the functions of which are not currently clear (Li et al., 2011; Rosenberg et al., 2011; Sakurai et al., 2010). In addition a number of editing events have been found essential for mRNA splicing (Castandet et al., 2010; Lamattina et al., 1989; Petschek et al., 1996; Rueter et al., 1999). Most recently, using deep sequencing approaches, potentially novel non-conical editing events were described as widespread in the human transcriptome (Li et al., 2011). However, it is still not clear to what extent these events exist. A number of these can be alternatively explained by known-difficulties and potential artifacts in interpreting the vast amounts of data derived from these approaches. In the present work for the first time, we demonstrate that the non-canonical editing described here is required for processing of a tRNA intron, which is in turn essential for viability. We have presented several lines of evidence using more classical sequencing and molecular approaches to substantiate our observations. Taken together, our data support the view that non-canonical editing occurs in at least one tRNA. Still outstanding is the nature of the editing enzyme. Currently no enzymatic activity has been described that could account for these non-canonical editing changes. Mechanistically, no deamination event could be at play and the activity likely involves either nucleotide or base replacement. Our attempts to recreate this editing activity in vitro, has yet to provide conclusive answers. Regardless, it is difficult to imagine that such an activity is only relegated to editing a single intron in kinetoplastids and likely has additional cellular targets.
An interesting question is why kinetoplastids still maintain an intron in a single tRNA. This fact points at some secondary function that has led to selection for, and maintenance of, the intron. Again, one reason may rest on previous work, where the presence of the intron is required for the modification of the anticodon (Grosjean et al., 1997; Johnson and Abelson, 1983; Szweykowska-Kulinska et al., 1994). This does not seem to be the case with T. brucei, given that no modifications were observed in the native intron-containing tRNA, with the exception of the edited positions shown in this work. It is also possible that the intron itself may play some chaperoning role in ensuring proper L-shape folding of the tRNA; an essential structure for splicing in eukaryotes (Calvin and Li, 2008). In this realm, intron editing may provide order to the folding process, while avoiding formation of non-functional undesirable alternative conformers.
The pivotal observation of both edited intron populations encoded in the genome of L. tarentolae is significant in at least two respects: 1) it indicates that this type of editing may occur in other kinetoplastids; 2) unlike T. brucei, intron editing may not be required in all kinetoplastids. However, it is apparent that the involvement of editing as an important splicing determinant is a naturally selected trait. As to what extent it appears in other organisms, only time will tell. Clearly, the possibility of altering RNAs to this degree by non-canonical editing opens a realm of possibilities and contributes greatly towards pushing the limits of the editing field and RNA biology in general.
EXPERIMENTAL PROCEDURES
RNAi Induction and Cell Cultivation
A 624-nucleotide long portion of the putative TbSEN34 gene (Tb927_11_v5) was PCR amplified from total genomic DNA of T. brucei strain 29–13. The amplicon was cloned into the p2T7-177 vector, which was linearized with Not I (for genome integration) and then used to transform procyclic T. brucei 29–13 cells and clonal lines were selected as described elsewhere (Wickstead et al., 2002). RNAi was triggered by the addition of 1 μg/ml of tetracycline to the growth medium (SDM-79). Cell density was measured every 24 h using the Beckman Z2 Coulter counter over a period of 22 days after the induction of double-stranded RNA synthesis.
Endonuclease Cleavage Assay
RNA halves were commercially synthesized, these corresponded to either the two edited pre-tRNAs (containing the intron) or a version corresponding to the genomic sequence (unedited). The RNA pieces were designed so that the 5′-most nucleotide of the 3′ fragment corresponded precisely to that of the first nucleotide of the intron. This fragment was radioactively end-labeled as previously described and joined to the 5′ fragment by splint ligation (Moore and Sharp, 1992). This generated the site-specifically labeled substrates used in the cleavage assays. These substrates were incubated, in yeast endonuclease cleavage buffer (Peebles et al., 1983), with a partially purified T. brucei endonuclease fraction generated by sequential column chromatography using the same protocol used to enrich the yeast enzyme (Peebles et al., 1983). The tRNA substrates were incubated, for various times under conditions where the substrate was saturating. Following incubation the resulting products were extracted with Tris-buffered phenol and precipitated by the addition of 0.3 M sodium acetate (pH 4.5) and 3 volumes of 100% ethanol followed by centrifugation at 13,000 RPM. The resulting pellet were resuspended in denaturing urea buffer and separated under denaturing conditions in a 10% acrylamide/7M urea gel. Following electrophoresis the gel was dried and the reaction products visualized and quantitated with a Storm PhosphoImager (Molecular Dynamics).
Northern Blot analysis and Primer Extension Assay
RNA was isolated using the guanidinium thiocyanate/phenol/chloroform extraction method (Chomczynski and Sacchi, 1987). 10 μg of total T. brucei RNA were separated on denaturating 8% polyacrylamide gel with 7 M urea and electroblotted to Zeta-probe (Bio-Rad) membranes, which were subsequently probed with [32P]-5′ end-labeled oligonucleotide specific for each RNA (5′-TGATACCCGCATACTCTAC-3′ for genomic; 5′-TGATACCTGCAAACTCTAC-3′ for edited 2 and 5′-TGATACCTGCAAATTCTAC-3′ for Edited 3). Hybridization procedures were carried out according to the manufacturer’s instructions (Bio-Rad). Images were taken with a Storm PhosphorImager (Molecular Dynamics).
Poisoned Primer extension analysis was carried out as described previously (Alfonzo et al., 1999). Briefly, a [32P]-5′ labeled oligonucleotide primer complementary to the pre-tRNA and anchored at the intron (5′-TCGAACCAGCGACCCTGTGATAC-3′) was used in reactions containing dATP, dGTP and dCTP and the chain terminator dideoxyTTP (ddTTP) in place of dTTP. Control reactions were also performed with all four standard nucleotides, these were used as controls to ensure that any “strong” stops seen in the “poisoned” primer extension reactions is not due to secondary structure of the tRNA. In vitro transcripts corresponding to the different edited and unedited species as well as mature tRNATyr were also used in control reactions and also served as size markers.
Reverse Transcription PCR and Sequencing
Total RNA from T. brucei (5 μg) was used for reverse transcription PCR reactions (RT-PCR) as described by the manufacturers (Invitrogen). Briefly, RNA samples were heated at 70 °C for 10 min in the presence of 2 pmoles of a reverse oligonucelotide primer (5′-AATCGAACCAGCGACCCTGTGA-3′) complementary to the 3′ exon sequence of wild type tRNA. This primer does not discriminate between mature and intron-containing tRNATyr. This was followed by addition of cDNA reverse transcription buffer and deoxynucleotides to a final concentration of 0.5 mM. The reaction was incubated at 42 °C for 2 minute at which point 200 units of Superscript II reverse transcriptase (Invitrogen) was added and then further incubated for 50 min at 42 °C. A similar reaction incubated in the absence of reverse transcriptase was used as a negative control (RT- control) and also as a control for DNA contamination of the RNA fraction. The resulting cDNA was PCR amplified as described previously (Alfonzo et al., 1999) using the same reverse oligonucleotide primer as above and a second primer (5′-CCTTCTGTAGCTCAATTGGTAG-3′) specific for the 5′ exon. The resulting PCR products were separated on a 10% non-denaturing polyacrylamide gel and the intron-containing band was excised and eluted. This product was then cloned into a pCRII-TOPO (Invitrogen) plasmid vector, transformed into E. coli and 25 independent clones sequenced by automated sequencing (ABI-Life Technologies). Reactions omitting the reverse transcription step were performed with total DNA isolated from T. brucei and the same PCR primers. Likewise 25 independent clones were sequenced and used as the genomic control.
Similar reactions with oligonucelotides specific for the tagged sequence were used to analyze both editing and splicing using the tagged tRNA variants described in Figure 6. For this reactions, the reverse transcription step was performed with an oligonucleotide anchored at the intron but that contains the Tag2 sequence (5′-TCGAACCAGCGACCCCATGATAC-3′, tag nucleotides are in boldface). The resulting cDNA was used in a PCR reaction with oligonucleotide primers that extend the length of the tRNA and which are anchored at the Tag1 and Tag2 sequences (Figure 6A).
Thin-layer Chromatography Analysis
Native and intron-containing tRNAs where purified from total RNA isolated from Tb-SEN RNAi-induced cells following 15 days of induction. The tRNAs were purified using an antisense biotinylated oligonucleotide (5′-CCTTCCGGCCGGAATCGAACCAGCGACCCTG-3′) and streptavidin beads (Sigma) using a procedure described previously (Alfonzo et al., 1999; Crain et al., 2002). This oligonucleotide does not discriminate between mature and pre-tRNA (intron-containing). The resulting products were isolated by purification on a 7 M urea–10% acrylamide gel. The gel-purified tRNAs were separately dephosphorylated with calf intestinal phosphatase (Invitrogen) and digested with RNase T2 for 5 h at 37°C in 10 mM ammonium acetate buffer (pH 4.5). The resulting 3′ NMPs were labeled with T4 polynucleotide kinase and [γ-32P]-ATP for 45 min at 37°C in the appropriate buffer (Invitrogen). To remove unincorporated radioactive ATP, the mixture was treated with 5 U of apyrase (Sigma), and unlabeled ATP was added to a final concentration of 1 mM. The mixture was incubated further at 37°C for 2 h. This treatment yields a mixture of 5′-labeled pNps. Following the apyrase treatment, the samples were treated with nuclease P1 (5 U/10 l reaction) in 75 mM ammonium acetate (pH 5.3) buffer to remove the 3′ phosphates. The mixture was then extracted with chloroform, ethyl ether and dried in a SpeedVac (Savant). The pellet was resuspended in water, and 20,000 c.p.m. were loaded onto a cellulose thin-layer chromatography plate (Merck) and analyzed by 2D-TLC. The nucleotides were separated using isobutyric acid, 25% ammonium hydroxide, water (50:1.1:28.9, by vol.) as the solvent system for the first dimension (solvent A in Figure 4). The solvent system for the second dimension is 0.1 M sodium phosphate pH 6.8, ammonium sulfate, n-propanol (100:60:2, v/w/v). After chromatography, the plates were dried at room temperature and subjected to PhosphorImager analysis (Molecular Dynamics). Nucleotide assignments were made using published maps (Grosjean et al., 2007; Grosjean et al., 2004).
Supplementary Material
HIGHLIGHTS.
First example of non-canonical editing in trypanosomes
First example of RNA editing necessary for splicing of a tRNA intron
Editing may occur in closely related organisms
Edited copies of the intron exist in the genome of closely related trypanosomes
Acknowledgments
We thank V. Gopalan, C. Rappleye, N. Ruiz and members of the Alfonzo and Trotta lab for comments and discussions. The research presented was supported in part by an NIH GM084065-05 to JDA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abelson J, Trotta CR, Li H. tRNA splicing. J Biol Chem. 1998;273:12685–12688. doi: 10.1074/jbc.273.21.12685. [DOI] [PubMed] [Google Scholar]
- Alfonzo JD, Blanc V, Estevez AM, Rubio MA, Simpson L. C to U editing of the anticodon of imported mitochondrial tRNA(Trp) allows decoding of the UGA stop codon in Leishmania tarentolae. Embo J. 1999;18:7056–7062. doi: 10.1093/emboj/18.24.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, Hertz-Fowler C, Horn D. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011;21:915–924. doi: 10.1101/gr.115089.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R, Aphasizheva I. Uridine insertion/deletion editing in trypanosomes: a playground for RNA-guided information transfer. Wiley Interdiscip Rev RNA. 2011;2:669–685. doi: 10.1002/wrna.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder S, Marchfelder A, Brennicke A. RNA editing of tRNA(Phe) and tRNA(Cys) in mitochondria of Oenothera berteriana is initiated in precursor molecules. Mol Gen Genet. 1994;244:67–74. doi: 10.1007/BF00280188. [DOI] [PubMed] [Google Scholar]
- Blanc V, Davidson NO. APOBEC-1-mediated RNA editing. Wiley Interdiscip Rev Syst Biol Med. 2010;2:594–602. doi: 10.1002/wsbm.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvin K, Li H. RNA-splicing endonuclease structure and function. Cell Mol Life Sci. 2008;65:1176–1185. doi: 10.1007/s00018-008-7393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castandet B, Choury D, Begu D, Jordana X, Araya A. Intron RNA editing is essential for splicing in plant mitochondria. Nucleic Acids Res. 2010;38:7112–7121. doi: 10.1093/nar/gkq591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chateigner-Boutin AL, Small I. Organellar RNA editing. Wiley Interdiscip Rev RNA. 2011;2:493–506. doi: 10.1002/wrna.72. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Crain PF, Alfonzo JD, Rozenski J, Kapushoc ST, McCloskey JA, Simpson L. Modification of the universally unmodified uridine-33 in a mitochondria-imported edited tRNA and the role of the anticodon arm structure on editing efficiency. Rna. 2002;8:752–761. doi: 10.1017/s1355838202022045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nicola Negri E, Fabbri S, Bufardeci E, Baldi MI, Gandini Attardi D, Mattoccia E, Tocchini-Valentini GP. The eucaryal tRNA splicing endonuclease recognizes a tripartite set of RNA elements. Cell. 1997;89:859–866. doi: 10.1016/s0092-8674(00)80271-8. [DOI] [PubMed] [Google Scholar]
- Fabbri S, Fruscoloni P, Bufardeci E, Di Nicola Negri E, Baldi MI, Attardi DG, Mattoccia E, Tocchini-Valentini GP. Conservation of substrate recognition mechanisms by tRNA splicing endonucleases. Science. 1998;280:284–286. doi: 10.1126/science.280.5361.284. [DOI] [PubMed] [Google Scholar]
- Fan H, Goodier JL, Chamberlain JR, Engelke DR, Maraia RJ. 5′ processing of tRNA precursors can Be modulated by the human La antigen phosphoprotein. Mol Cell Biol. 1998;18:3201–3211. doi: 10.1128/mcb.18.6.3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey J, Weil JH, Tomita K, Cosset A, Dietrich A, Small I, Marechal-Drouard L. Editing of plant mitochondrial transfer RNAs. Acta Biochim Pol. 2001;48:383–389. [PubMed] [Google Scholar]
- Gommans WM. A-to-I editing of microRNAs: regulating the regulators? Semin Cell Dev Biol. 2012;23:251–257. doi: 10.1016/j.semcdb.2011.09.018. [DOI] [PubMed] [Google Scholar]
- Goringer HU, Katari VS, Bohm C. The structural landscape of native editosomes in African trypanosomes. Wiley Interdiscip Rev RNA. 2011;2:395–407. doi: 10.1002/wrna.67. [DOI] [PubMed] [Google Scholar]
- Grosjean H, Droogmans L, Roovers M, Keith G. Detection of enzymatic activity of transfer RNA modification enzymes using radiolabeled tRNA substrates. Methods Enzymol. 2007;425:55–101. doi: 10.1016/S0076-6879(07)25003-7. [DOI] [PubMed] [Google Scholar]
- Grosjean H, Keith G, Droogmans L. Detection and quantification of modified nucleotides in RNA using thin-layer chromatography. Methods Mol Biol. 2004;265:357–391. doi: 10.1385/1-59259-775-0:357. [DOI] [PubMed] [Google Scholar]
- Grosjean H, Szweykowska-Kulinska Z, Motorin Y, Fasiolo F, Simos G. Intron-dependent enzymatic formation of modified nucleosides in eukaryotic tRNAs: a review. Biochimie. 1997;79:293–302. doi: 10.1016/s0300-9084(97)83517-1. [DOI] [PubMed] [Google Scholar]
- Hopper AK, Phizicky EM. tRNA transfers to the limelight. Genes Dev. 2003;17:162–180. doi: 10.1101/gad.1049103. [DOI] [PubMed] [Google Scholar]
- Jackman JE, Alfonzo JD. Transfer RNA modifications: nature’s combinatorial chemistry playground. Wiley Interdiscip Rev RNA. 2013;4:35–48. doi: 10.1002/wrna.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Gott JM, Gray MW. Doing it in reverse: 3′-to-5′ polymerization by the Thg1 superfamily. Rna. 2012;18:886–899. doi: 10.1261/rna.032300.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke A, Paabo S. Editing of a tRNA anticodon in marsupial mitochondria changes its codon recognition. Nucleic Acids Res. 1993;21:1523–1525. doi: 10.1093/nar/21.7.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PF, Abelson J. The yeast tRNATyr gene intron is essential for correct modification of its tRNA product. Nature. 1983;302:681–687. doi: 10.1038/302681a0. [DOI] [PubMed] [Google Scholar]
- Kolev NG, Tschudi C, Ullu E. RNA interference in protozoan parasites: achievements and challenges. Eukaryot Cell. 2011;10:1156–1163. doi: 10.1128/EC.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamattina L, Weil JH, Grienenberger JM. RNA editing at a splicing site of NADH dehydrogenase subunit IV gene transcript in wheat mitochondria. FEBS Lett. 1989;258:79–83. doi: 10.1016/0014-5793(89)81620-5. [DOI] [PubMed] [Google Scholar]
- Li M, Wang IX, Li Y, Bruzel A, Richards AL, Toung JM, Cheung VG. Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2011;333:53–58. doi: 10.1126/science.1207018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. MODOMICS: a database of RNA modification pathways--2013 update. Nucleic Acids Res. 2013;41:D262–267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallela A, Nishikura K. A-to-I editing of protein coding and noncoding RNAs. Crit Rev Biochem Mol Biol. 2012;47:493–501. doi: 10.3109/10409238.2012.714350. [DOI] [PubMed] [Google Scholar]
- Marechal-Drouard L, Ramamonjisoa D, Cosset A, Weil JH, Dietrich A. Editing corrects mispairing in the acceptor stem of bean and potato mitochondrial phenylalanine transfer RNAs. Nucleic Acids Res. 1993;21:4909–4914. doi: 10.1093/nar/21.21.4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M, Schiffer S, Marchfelder A. tRNA 3′ processing in plants: nuclear and mitochondrial activities differ. Biochemistry. 2000;39:2096–2105. doi: 10.1021/bi992253e. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Sharp PA. Site-specific modification of pre-mRNA: the 2′-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- Mottram JC, Bell SD, Nelson RG, Barry JD. tRNAs of Trypanosoma brucei. Unusual gene organization and mitochondrial importation. J Biol Chem. 1991;266:18313–18317. [PubMed] [Google Scholar]
- Paris Z, Fleming IM, Alfonzo JD. Determinants of tRNA editing and modification: avoiding conundrums, affecting function. Semin Cell Dev Biol. 2012;23:269–274. doi: 10.1016/j.semcdb.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin SV, Patel M, Furia BS, Peltz SW, Trotta CR. Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3′ end formation. Cell. 2004;117:311–321. doi: 10.1016/s0092-8674(04)00342-3. [DOI] [PubMed] [Google Scholar]
- Peebles CL, Gegenheimer P, Abelson J. Precise excision of intervening sequences from precursor tRNAs by a membrane-associated yeast endonuclease. Cell. 1983;32:525–536. doi: 10.1016/0092-8674(83)90472-5. [DOI] [PubMed] [Google Scholar]
- Petschek JP, Mermer MJ, Scheckelhoff MR, Simone AA, Vaughn JC. RNA editing in Drosophila 4f-rnp gene nuclear transcripts by multiple A-to-G conversions. J Mol Biol. 1996;259:885–890. doi: 10.1006/jmbi.1996.0365. [DOI] [PubMed] [Google Scholar]
- Phizicky EM. Have tRNA, will travel. Proc Natl Acad Sci U S A. 2005;102:11127–11128. doi: 10.1073/pnas.0504843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Greer CL. Pre-tRNA splicing: variation on a theme or exception to the rule? Trends Biochem Sci. 1993;18:31–34. doi: 10.1016/0968-0004(93)90085-2. [DOI] [PubMed] [Google Scholar]
- Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold-Hurek B, Shub DA. Self-splicing introns in tRNA genes of widely divergent bacteria. Nature. 1992;357:173–176. doi: 10.1038/357173a0. [DOI] [PubMed] [Google Scholar]
- Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3′ UTRs. Nat Struct Mol Biol. 2011;18:230–236. doi: 10.1038/nsmb.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio MA, Hopper AK. Transfer RNA travels from the cytoplasm to organelles. Wiley Interdiscip Rev RNA. 2011;2:802–817. doi: 10.1002/wrna.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- Sakurai M, Yano T, Kawabata H, Ueda H, Suzuki T. Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat Chem Biol. 2010;6:733–740. doi: 10.1038/nchembio.434. [DOI] [PubMed] [Google Scholar]
- Schneider A, McNally KP, Agabian N. Splicing and 3′-processing of the tyrosine tRNA of Trypanosoma brucei. J Biol Chem. 1993;268:21868–21874. [PubMed] [Google Scholar]
- Sie CP, Kuchka M. RNA editing adds flavor to complexity. Biochemistry (Mosc) 2012;76:869–881. doi: 10.1134/S0006297911080025. [DOI] [PubMed] [Google Scholar]
- Smith HC, Bennett RP, Kizilyer A, McDougall WM, Prohaska KM. Functions and regulation of the APOBEC family of proteins. Semin Cell Dev Biol. 2012;23:258–268. doi: 10.1016/j.semcdb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szweykowska-Kulinska Z, Senger B, Keith G, Fasiolo F, Grosjean H. Intron-dependent formation of pseudouridines in the anticodon of Saccharomyces cerevisiae minor tRNA(Ile) Embo J. 1994;13:4636–4644. doi: 10.1002/j.1460-2075.1994.tb06786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocchini-Valentini GP, Baldi MI, Gandini-Attardi D, Mattoccia E. Cleavage site recognition by the tRNA splicing endoribonuclease. Gene. 1993;135:93–97. doi: 10.1016/0378-1119(93)90053-6. [DOI] [PubMed] [Google Scholar]
- Trotta CR, Miao F, Arn EA, Stevens SW, Ho CK, Rauhut R, Abelson JN. The yeast tRNA splicing endonuclease: a tetrameric enzyme with two active site subunits homologous to the archaeal tRNA endonucleases. Cell. 1997;89:849–858. doi: 10.1016/s0092-8674(00)80270-6. [DOI] [PubMed] [Google Scholar]
- Ullu E, Tschudi C, Chakraborty T. RNA interference in protozoan parasites. Cell Microbiol. 2004;6:509–519. doi: 10.1111/j.1462-5822.2004.00399.x. [DOI] [PubMed] [Google Scholar]
- Wickstead B, Ersfeld K, Gull K. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol Biochem Parasitol. 2002;125:211–216. doi: 10.1016/s0166-6851(02)00238-4. [DOI] [PubMed] [Google Scholar]
- Wolin SL, Matera AG. The trials and travels of tRNA. Genes Dev. 1999;13:1–10. doi: 10.1101/gad.13.1.1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.