

Abstract
The epithelial Na+ channel (ENaC) plays a critical role in Na+ absorption, and mutations in this channel cause diseases of Na+ homeostasis, including a genetic form of hypertension (Liddle’s syndrome). To investigate cAMP-mediated stimulation of ENaC, α, β, and γENaC were coexpressed in Fischer rat thyroid epithelia to generate apical Na+ channels and transepithelial Na+ current. cAMP agonists stimulated Na+ current by 70%. Following covalent modification of cysteines introduced into ENaC, cAMP increased the rate of appearance of unmodified channels at the cell surface. In addition, cAMP increased the fluorescent labeling of ENaC at the apical cell surface. Inhibition of vesicle trafficking by incubating epithelia at 15°C prevented the cAMP-mediated stimulation of ENaC. These results suggest that cAMP stimulates Na+ absorption in part by increasing translocation of ENaC to the cell surface. Stimulation of ENaC by cAMP was dependent on a sequence (PPPXY) in the COOH terminus of each subunit. This sequence is the target for mutations that cause Liddle’s syndrome, suggesting that cAMP-mediated translocation of ENaC to the cell surface is defective in this genetic form of hypertension.
Introduction
The epithelial Na+ channel (ENaC) is a heteromultimer formed by 3 homologous subunits (αENaC, βENaC, and γENaC) (1–4). ENaC forms the pathway for Na+ absorption across epithelia, including the kidney, lung, and colon (5, 6). Na+ enters the cell through ENaC at the apical membrane and then exits at the basolateral membrane through the Na+, K+-ATPase, resulting in the transepithelial movement of Na+. As a rate-limiting step for Na+ absorption, ENaC plays a critical role in Na+ homeostasis and blood pressure control, and therefore the function of this channel is tightly regulated.
The cytoplasmic COOH-termini of the β and γ subunits play an important role in the control of Na+ absorption. Truncation of the COOH-terminus or mutations in a conserved COOH-terminal PY motif (PPPXY) cause Liddle’s syndrome, a genetic form of hypertension (7–9). These mutations abolish the interaction between ENaC and Nedd4, a ubiquitin protein-ligase. As a result, Na+ absorption is increased, at least in part, by an increase in the expression of ENaC at the cell surface (8, 10).
Na+ absorption through ENaC is stimulated by cAMP (6). However, it is not known whether the COOH-terminal PY motif of ENaC or Nedd4 are involved in cAMP-dependent regulation. In the kidney collecting duct, intracellular levels of cAMP are increased by vasopressin, which is released in the defense against hypovolemia, hypotension, and increased plasma osmolality. Vasopressin and cAMP not only increase Na+ absorption, but they also increase water absorption in the collecting duct by stimulating translocation of aquaporin-2 water channels to the apical cell surface (11). Whether cAMP stimulates ENaC by a similar mechanism is unclear (6). It is also possible that cAMP stimulates ENaC by increasing the open-state probability (PO) of the channel. Although a recent report supports such a mechanism (12), previous reports found no effect on PO (13, 14). Thus, the mechanisms by which cAMP stimulates ENaC are not certain.
The goal of this work was to investigate the mechanisms of cAMP-mediated ENaC stimulation. The small number of ENaC channels expressed at the apical surface of epithelia has presented a major technical hurdle to addressing this question with traditional biochemical techniques. Therefore, to investigate whether cAMP stimulates translocation of ENaC to the cell surface, I used a novel functional approach based on the covalent modification of ENaC at the cell surface. This strategy took advantage of the sensitivity of electrophysiological techniques to detect the activity of a small number of channels. Fluorescent labeling of channels at the cell surface was performed as an additional test of this hypothesis. Because of the critical role of the COOH-terminus of βENaC and γENaC in Na+ homeostasis, I also asked whether the COOH-terminus is involved in the regulation of ENaC by cAMP and whether this regulation is altered in Liddle’s syndrome.
Methods
Cell culture and transfection.
Fischer rat thyroid (FRT) cells were grown on permeable filter supports (Millicell HA, 0.4-μm pore size, 27 mm diameter; Millipore, Bedford, Massachusetts, USA) in Ham’s F-12 media (Sigma Chemical Co., St. Louis, Missouri, USA) with 5% FCS (Sigma), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C, as previously described (15). One to 3 days after seeding, cells were cotransfected with α, β, and γhENaC, each cloned into the pMT3 plasmid (3, 4). Mutations in the α, β, and γ subunits were generated by site-directed mutagenesis (Muta-Gene; Bio-Rad, Hercules, California, USA), and the accuracy of the sequence was confirmed by DNA sequencing in the University of Iowa Sequencing Core (Iowa City, Iowa, USA). The 3 plasmids (1 μg/monolayer each) were mixed with DMRIE/DOPE (Vical Inc., San Diego, California, USA; 15 μg/monolayer) in 360 μL/monolayer Opti-MEM (GIBCO BRL Baltimore, Maryland, USA) for 15 minutes and transferred to the apical surface of the monolayer. Six hours later, the apical media was replaced with Ham’s F-12 media containing 5% FCS and amiloride (10 μM). In most experiments α, β, and γhENaC were coexpressed. However, in some experiments (shown in Figure 1, a and c), epithelia were transfected with empty vector (pMT3), αhENaC alone, or α with either βhENaC or γhENaC.
Figure 1.

Expression of ENaC in FRT epithelia results in transepithelial Na+ absorption. Epithelia were transfected with (a) pMT3 (Control) (b) α, β, and γhENaC, or (c) pMT3 (C), or the indicated hENaC subunits. (a and b) Representative time courses of short-circuit current (ISC) with apical and basolateral membranes bathed in symmetrical NaCl solutions. Amiloride (10 μM) was added to the apical bathing solution (filled bar), and 0 current is indicated. (c) Plot of amiloride-sensitive ISC (mean ± SEM, n = 4–12).
Current measurements.
Na+ transport was measured 3–7 days after transfection. Monolayers were studied in modified Ussing chambers (Jim’s Instruments, Iowa City, Iowa, USA) with the apical and basolateral surfaces bathed in 135 mM NaCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 2.4 mM K2HPO4, 0.6 mM KH2PO4, 10 mM dextrose, and 10 mM HEPES (pH 7.4) at 37°C and bubbled with O2. Transepithelial current was measured under short-circuit conditions (ISC). Amiloride-sensitive ISC was determined as the difference in current with and without amiloride (10 μM) in the apical bathing solution. Because γG536C decreases the affinity of the channel for amiloride (16), benzamil (100 μM), a more potent blocker of ENaC, was used for experiments with this mutant. A mixture of cAMP agonists was added to the apical bathing solution: 200 μM 8-(4-chlorophenylthio)-cAMP sodium (CPT-cAMP; Sigma), 100 μM 3-isobutyl-1-methylxanthine (IBMX; Sigma), and 10 μM forskolin (Sigma). ENaC containing either γG536C (with wild-type α and βhENaC) or βS520C (with wild-type α and γhENaC) were covalently modified by adding 1 mM [2 (trimethylammonium)ethyl]methanethiosulfonate bromide (MTSET; Toronto Research Chemicals, North York, Ontario, Canada) to the apical bathing solution. MTSET had no effect on ISC in untransfected FRT epithelia or epithelia expressing wild-type ENaC. Following removal of MTSET from the bathing solution, the maximal rate of increase in ISC was determined from the slope of the time course of current.
Surface expression of ENaC.
FRT epithelia transfected with α, β, and γG536C ENaC were grown on permeable filter supports, as above. Three to 4 days after transfection, the apical and basolateral membranes were bathed in the NaCl solution described above, and 1 mM MTSET was added twice to the apical solution for 5 minutes at room temperature to modify cysteines at the cell surface. After washing off MTSET, the monolayers were treated (or not treated) with 200 μM CPT-cAMP, 100 μM IBMX, and 10 μM forskolin at 37°C for 15 minutes, placed on ice, and the cAMP agonists were removed. The remainder of the protocol was carried out with ice-cold solutions and with the monolayers on ice to prevent trafficking of membrane proteins. Ten millimoles sodium (2-sulfonatoethyl)methanethiosulfonate (MTSES; Toronto Research Chemicals) was added twice to the apical membrane for 5 minutes to block cysteines in endogenous proteins translocated to the cell surface. MTSES does not modify γG536C (17). After removal of MTSES, 200 μM N-Biotinylcaproylaminoethyl methanethiosulfonate (MTSEA-Biotincap; Toronto Research Chemicals) was added twice to the apical membrane for 10 minutes and then washed off. γG536C is accessible to modification by MTSEA-Biotincap; in Xenopus oocytes, MTSEA-Biotincap irreversibly blocks channels containing γG536C, but has no effect on wild-type ENaC (P.M. Snyder, unpublished observation). Channels modified with MTSEA-Biotincap were labeled with 10 μM NeutrAvidin Alexa Fluor 488 conjugate (Molecular Probes Inc., Eugene, Oregon, USA) for 30 minutes. The apical membrane was thoroughly washed, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 (Pierce Chemical Co., Rockford, Illinois, USA) for 30 minutes. F-actin was labeled with 5 U/mL Texas red-X phalloidin (Molecular Probes) for 30 minutes. Epifluorescence was detected at 488 nm (green) and 568 nm (red) using a confocal microscope (Bio-Rad MRC 1024ES, krypton/argon laser). Optical sections were obtained at 0.5-μm steps and superimposed. Mean pixel fluorescence at 488 nm (corresponding to Alexa 488) was quantitated after subtraction of background from an unstained region. Apical versus basolateral localization of labeling was determined by obtaining vertical sections. Note that in this transient expression system not all cells express ENaC.
Results
Expression of ENaC in FRT epithelia and stimulation by cAMP.
FRT epithelia had minimal transepithelial ISC when the apical and basolateral membranes were bathed in NaCl solutions, and amiloride (a blocker of epithelial Na+ channels) added to the apical bathing solution did not alter ISC (Figure 1, a and c). This is consistent with a previous report that FRT cells lack endogenous apical membrane Na+ channels (15). Transfection of epithelia with ENaC (α, β, and γ) increased ISC, and this current was completely blocked by addition of amiloride to the apical surface (Figure 1, b and c). Thus, transfection resulted in transepithelial Na+ absorption, with expression of ENaC at the apical surface providing a pathway for Na+ entry. Na+ absorption required the coexpression of all 3 subunits; expression of the α subunit alone, α with β, or α with γ, did not increase amiloride-sensitive ISC compared with untransfected or mock-transfected epithelia (Figure 1c). This contrasts with expression in Xenopus oocytes, where maximal Na+ current required coexpression of all 3 subunits, but expression of αENaC alone or in combination with either the β or γ subunits generated Na+ current (1–4). Thus, in epithelia, all 3 subunits may be required to form functional Na+ channels. This is consistent with the finding that mutations in any 1 of the 3 subunits can cause the Na+-wasting disorder pseudohypoaldosteronism type I (7).
To determine whether cAMP stimulates ENaC in FRT epithelia, cells expressing α, β, and γENaC were treated with cAMP agonists (200 μM CPT-cAMP, 100 μM IBMX, and 10 μM forskolin). cAMP increased amiloride-sensitive ISC approximately 70% and reached a plateau in 10 to 15 minutes (Figures 2, a and b). Treatment with 10 μM IBMX and 10 μM forskolin (without CPT-cAMP) increased current to a similar extent (82 ± 7%, n = 8); cAMP had no effect on ISC in untransfected epithelia (not shown).
Figure 2.

Na+ absorption is stimulated by cAMP. (a) Representative time course of current in epithelia expressing wild-type α, β, and γhENaC. cAMP agonists (200 μM CPT-cAMP, 100 μM IBMX, and 10 μM forskolin) and amiloride (10 μM) were added to the apical membrane (bars). (b) Amiloride-sensitive ISC (relative to without cAMP) in cells not treated or treated with cAMP agonists, as indicated (mean ± SEM, n = 32). *P < 0.0001 by Student’s t test.
The rate of translocation of ENaC to the cell surface is increased by cAMP.
cAMP could potentially stimulate Na+ current by increasing the rate of ENaC translocation to the cell surface. To determine the basal rate of translocation without cAMP, channels at the cell surface were covalently modified by MTSET, a cysteine-reactive reagent, and the rate of appearance of unmodified channels at the cell surface was determined. In a previous study, MTSET had no effect on wild-type ENaC current (18). In contrast, introduction of a cysteine into the pore (γG536C) (16, 17) resulted in irreversible block in response to covalent modification by MTSET, which decreased Na+ current by 90% (18). Because MTSET is impermeable to the cell membrane, only channels at the cell surface are modified when this reagent is added to the apical bathing solution (19). This strategy is illustrated in Figure 3a.
Figure 3.

Translocation of ENaC to the cell surface is stimulated by cAMP. (a) Model showing ENaC at plasma membrane and in an intracellular pool. Channels at the cell surface are irreversibly blocked by covalent modification of an introduced cysteine by MTSET. After washout of MTSET, unmodified channels are translocated to the cell surface, replacing modified channels that are internalized. (b) ISC recordings in cells expressing wild-type α and βhENaC with γG536C. MTSET (1 mM) was added to the apical membrane (filled bars). (c) Plot of initial rate of increase in ISC (μA/cm2 per minute; mean ± SEM, n = 9–13) after washout of MTSET, in the presence or absence of cAMP agonists, as indicated. *P < 0.0001. (d) ISC in cells expressing wild-type α and βhENaC with γG356C that were treated with cAMP agonists (open bars). (e) Plot of time course of increase in ISC (relative to peak increase) with or without cAMP, as indicated (mean ± SEM, n = 4–5).
In Figure 3b, γG536C was coexpressed with wild-type αENaC and βENaC in FRT epithelia, and the channels were blocked by addition of MTSET (1 mM) to the apical bathing solution. After removal of MTSET, there was a gradual increase in ISC (Figure 3b). Because MTSET irreversibly blocks channels at the cell surface, this time-dependent increase in current was the result of translocation of unmodified (unblocked) channels to the cell surface (illustrated in Figure 3a). Consistent with this conclusion, the current was blocked by MTSET (Figure 3b), indicating that the channels responsible for the increase in ISC had not been modified during the initial treatment with MTSET. The rate of increase in ISC is determined by the rate of insertion of new channels and by the rate of internalization of channels from the cell surface (Figure 3a). Because internalization of blocked channels would have a minimal effect on Na+ current, the initial rate of increase following washout of MTSET (0.065 μA/cm2 per minute) provides a measure of the rate of appearance of new, unblocked channels at the cell surface (Figure 3c). This value, together with the Na+ current before MTSET (1.87 μA/cm2) allows an estimate of 29 minutes as the time to replace the entire population of ENaC at the cell surface in the absence of cAMP. I cannot exclude the possibility that channel blockade by MTSET altered the rate of translocation of ENaC to the cell surface.
Addition of cAMP agonists following removal of MTSET caused a much more rapid increase in ISC (Figure 3d); the initial rate of increase was 4.6-fold higher than without cAMP (Figure 3c). Figure 3e plots the time course of the increase in ISC (relative to the peak increase) with or without cAMP; ISC peaked 4-fold faster with cAMP than without. This current resulted from the appearance of unmodified channels at the cell surface, because it was blocked by MTSET (Figure 3d). Together, these results indicate that cAMP increased the rate of translocation of ENaC to the cell surface. The data do not exclude an additional effect of cAMP on PO. However, because MTSET blocks channels at the cell surface, increased ISC resulting from an increase in PO would depend on translocation of unmodified (unblocked) channels to the cell surface. Thus, if it did not stimulate translocation of ENaC to the cell surface, cAMP would not have altered the rate of increase in ISC. It is also possible that cAMP increased the sensitivity of ENaC to modification by MTSET. However, a second treatment with cAMP agonists (after the second treatment with MTSET) increased amiloride-sensitive ISC, and this current was also blocked by MTSET (Figure 3d). If cAMP increased only the MTSET-sensitivity of channels resident at the cell surface, this second treatment with cAMP would not have been expected to increase MTSET-inhibitable current.
Na+ current is inhibited by cAMP after covalent modification of βS520C.
To further test whether cAMP stimulates translocation of ENaC to the cell surface, a cysteine was introduced at another site (βS520C). In contrast to γG536C, modification of this cysteine by MTSET stimulates ENaC current (17). When βS520C was coexpressed in FRT epithelia with wild-type αENaC and γENaC, cAMP increased amiloride-sensitive ISC similar to wild-type ENaC (not shown). Modification of βS520C with MTSET in the apical bathing solution irreversibly increased Na+ current (Figure 4a). More importantly, MTSET altered the response to cAMP; after removal of MTSET, cAMP decreased Na+ current (Figure 4, a left and b), in contrast to the increase in current with γG536C (Figure 4b). The rate of decrease was much greater with cAMP than in cells not treated with cAMP (Figure 4a, right). This decrease in current is not consistent with a cAMP-mediated increase in PO, which should have increased Na+ current. To determine if cAMP increased the appearance of unmodified channels at the cell surface, channels were treated a second time with MTSET (downward arrow in Figure 4a). MTSET increased ISC significantly more in cells that had been treated with cAMP agonists than in untreated cells (Figure 4c), suggesting that cAMP stimulated translocation of unmodified channels to the cell surface. If these unmodified channels were exchanged for modified (stimulated) channels at the cell surface, this would explain the cAMP-mediated decrease in Na+ current (after MTSET; Figure 4d). Such a finding suggests that cAMP also increased the rate of ENaC internalization, although to a lesser extent than the increase in translocation of ENaC to the cell surface.
Figure 4.

Na+ current is inhibited by cAMP after covalent modification of βS520C. (a) Representative time course of current in epithelia expressing wild-type α and γhENaC with βS520C. MTSET (1 mM, filled) and cAMP agonists (open bars) were added to the apical membrane (bars). Epithelia were treated with MTSET, washed, and then treated (left) or not treated (right) with cAMP agonists. ISC stimulated by a second treatment with MTSET (downward arrow) was then determined. (b) Plot of cAMP-induced change in ISC following removal of MTSET (relative to the current change by MTSET; mean ± SEM, n = 5–8) for epithelia expressing either γG536C or βS520C with the other 2 wild-type subunits. (c) Plot of ISC (mean ± SEM, n = 5–6) stimulated by second treatment with MTSET (downward arrow in a) in cells treated or not treated with cAMP agonists, as indicated. *P < 0.003. (d) Model showing ENaC at plasma membrane and in an intracellular pool. MTSET irreversibly stimulates ENaC at the cell surface (black). The predicted response if cAMP increases ENaC translocation to the cell surface is shown.
Block of vesicle trafficking prevents stimulation by cAMP.
Vesicle trafficking to the cell surface can be prevented by lowering the temperature to 15°C (20). Therefore, if translocation of ENaC to the cell surface is required for cAMP to increase Na+ current, this should be prevented by incubation of epithelia at 15°C. In Figure 5a, FRT epithelia expressing γG536C with wild-type αENaC and βENaC were incubated at 15°C and treated with MTSET. Following removal of MTSET, the increase in ISC was nearly abolished; with or without cAMP, the rate of increase in ISC was significantly diminished compared with epithelia at 37°C (Figure 5b). Incubation at 15°C did not cause irreversible damage to the epithelia; when the epithelia were returned to 37°C, cAMP rapidly increased MTSET-inhibitable ISC (Figure 5a). The reduction in temperature does not block cAMP-induced phosphorylation. cAMP stimulates another epithelial channel, CFTR, by cAMP-dependent protein kinase–mediated (PKA-mediated) phosphorylation, which increases the PO of channels located at the cell surface (21). In contrast to the result with ENaC, stimulation of CFTR was intact at 15°C (not shown) or even at 5°C (22). Thus, the data are consistent with a requirement for vesicle trafficking in the cAMP-mediated stimulation of ENaC.
Figure 5.

Block of vesicle trafficking prevents stimulation by cAMP. (a) Representative time course of current in epithelia expressing wild-type α and βhENaC with γG536C. MTSET (1 mM) and cAMP agonists were added to the apical membrane (bars). Epithelia were initially incubated in an Ussing chamber at 15°C and then transferred to a chamber at 37°C, as indicated. (b) Plot of initial rate of increase in ISC (μA/cm2 per minute; mean ± SEM, n = 5–13) after washout of MTSET, in the presence or absence of cAMP agonists, and at 15°C or 37°C, as indicated. *P < 0.0001 versus epithelia at 37°C.
Apical surface labeling of ENaC is increased by cAMP.
To provide additional evidence that cAMP increases translocation of the channel complex to the cell surface, ENaC at the apical membrane was fluorescently labeled by covalent modification of Cys536 introduced in γENaC. This strategy was designed to selectively detect channels that translocate to the cell surface in response to cAMP. FRT epithelia on permeable filter supports were transfected with α, β, and γG536C ENaC. Before treatment with cAMP, the cells were treated with a nonfluorescent reagent (MTSET) to block cysteines in endogenous membrane proteins and in γG536C ENaC already present at the cell surface. The cells were then treated (or not treated) with cAMP agonists. Following cAMP treatment, MTSES was used to block cysteines in non-ENaC proteins that trafficked to the cell surface. This negatively charged reagent does not modify γG536C (17). γG536 ENaC that translocated to the cell surface in response to cAMP was then modified with MTSEA-Biotincap and labeled with a fluorescent NeutrAvidin (green). Fluorescence was detected and quantitated by confocal microscopy. The cells were colabeled with Texas red-X phalloidin (red), which labels F-actin. In cells not treated with cAMP, there was a low level of background green fluorescence at the cell surface (Figure 6, a left and d), similar to untransfected controls (Figure 6, c left and d). cAMP produced a large increase in green fluorescence at the cell surface in cells expressing ENaC (173%; Figure 6, a right and d). Vertical sections revealed that the staining was exclusively at the apical membrane (Figure 6b), consistent with the apical localization of ENaC in epithelia. In untransfected epithelia, cAMP produced only a small increase in fluorescence (28%; Figure 6, c and d), likely arising from endogenous proteins translocated to the cell surface that were not completely blocked by MTSES. These results support the conclusion that cAMP increases translocation of ENaC from an intracellular store to the cell surface.
Figure 6.
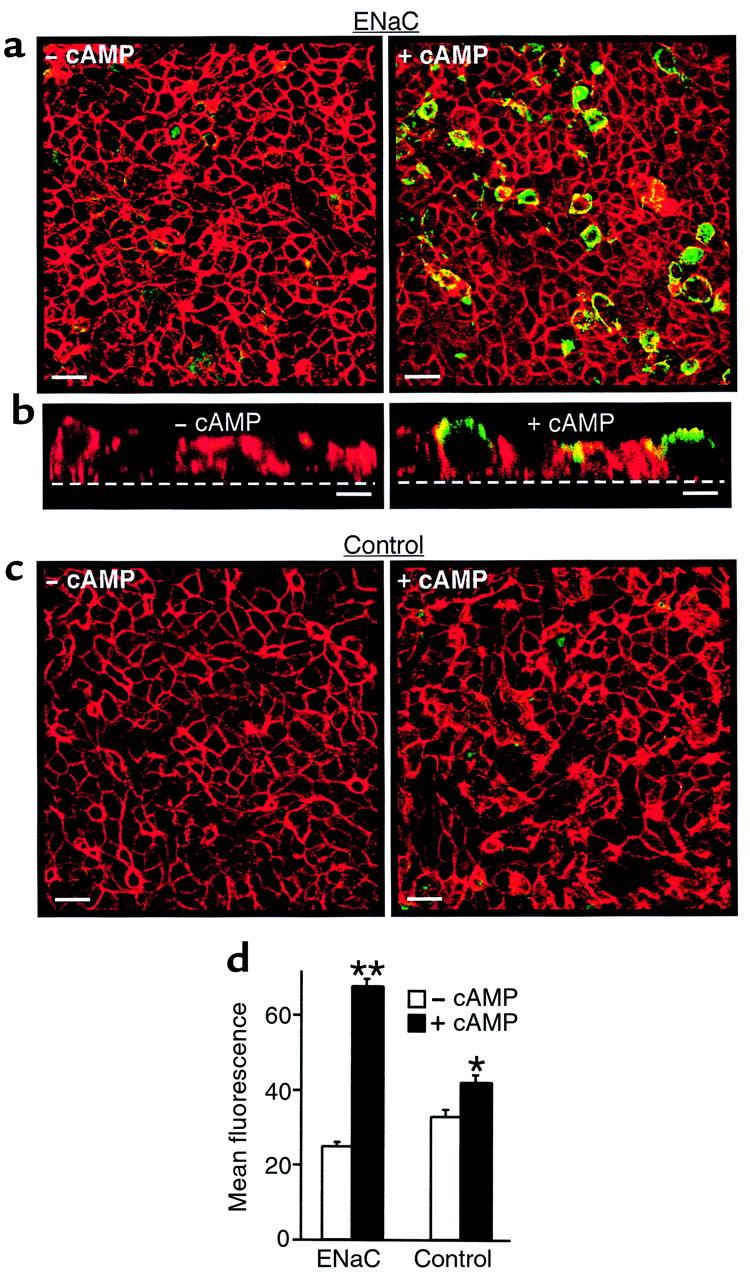
Apical surface labeling of ENaC is increased by cAMP. (a) Confocal images of FRT epithelia transfected with α, β, and γG536C ENaC with or without treatment with cAMP agonists (15 minutes), as indicated. ENaC at the cell surface is stained green, and F-actin is stained red. Scale bars: 20 μm. (b) Vertical sections through epithelia in a, with or without cAMP treatment, as indicated. The dashed line indicates the location of the permeable filter support. Scale bars: 5 μM. (c) Confocal images of untransfected FRT epithelia with or without treatment with cAMP agonists, as indicated. (d) Mean fluorescence (488 nm, green) for cells transfected (ENaC) or not transfected (Control) with α, β, and γG536C ENaC, and treated or not treated with cAMP agonists, as indicated (mean ± SEM, n = 9 fields). *P < 0.005, **P < 1.5 × 10–10 versus – cAMP.
Liddle’s syndrome mutations abolish cAMP-dependent stimulation of ENaC.
To investigate whether the cytoplasmic COOH-termini of ENaC subunits are required for cAMP-mediated stimulation, I tested the effect of mutations that delete most of the COOH-terminus beyond the second membrane-spanning segment (αS594X, βR566X, γK576X). In the β and γ subunits, such mutations cause Liddle’s syndrome (23, 24). When either the α or γ subunit was truncated (and coexpressed with the other 2 wild-type subunits), cAMP produced only a minimal increase in amiloride-sensitive ISC (Figure 7c), in contrast to its effect on wild-type ENaC (Figure 7, a and c). Even more dramatic, truncation of the β subunit abolished the cAMP-mediated stimulation (Figure 7, b and c). Instead, cAMP inhibited amiloride-sensitive ISC. Coexpression of all 3 truncated subunits had the same effect as truncation of the β subunit alone. Thus, deletion of the COOH-terminus of any of the 3 ENaC subunits is sufficient to disrupt cAMP-mediated stimulation of Na+ absorption.
Figure 7.

COOH-terminal truncations disrupt stimulation by cAMP. (a and b) Representative time courses of current in epithelia expressing wild-type α and γhENaC with either wild-type β (in a) or βR566X (in b). cAMP agonists and amiloride (10 μM) were added to the apical membrane (bars). The βR566X mutation increased amiloride-sensitive ISC 1.9-fold (wild-type, 3.6 μA/cm2; βR566X, 6.8 μA/cm2). (c) Percent change in amiloride-sensitive ISC by cAMP agonists in epithelia expressing the indicated combinations of hENaC subunits (mean ± SEM, n = 7–32). *P < 0.02 versus wild-type by Student’s t test.
To localize the COOH-terminal sequence in βENaC required for stimulation by cAMP, smaller deletions were tested. When the 9 COOH-terminal residues were deleted (βE632X), the cAMP-induced increase in Na+ absorption was similar to that in wild-type ENaC (Figure 8). In contrast, deletion of 19 residues (βS622X) abolished stimulation. This deletion overlaps with a tyrosine-based motif (PPPXY), which is conserved in the COOH-terminus of all 3 subunits and which is the site of mutations involved in Liddle’s syndrome (7–9). Mutation of the 3 prolines in this motif to alanine (βP616-618A) abolished stimulation by cAMP (Figure 8), similar to truncation of the COOH-terminus (βR566X). Mutation of the tyrosine (βY620A) had a similar effect (Figure 8). A serine (Ser622) is located nearby, although the surrounding residues do not fit the consensus for phosphorylation by cAMP-dependent protein kinase (Figure 8). When this serine was mutated to alanine, cAMP still stimulated amiloride-sensitive ISC, although less than the wild-type channel (Figure 8). Therefore, phosphorylation of Ser622 was not required for stimulation. These results indicate that the tyrosine-based COOH-terminal motif is required for stimulation by cAMP and suggest that regulation of Na+ absorption by cAMP is disrupted in Liddle’s syndrome.
Figure 8.

Deletions and mutations of PPPXY motif disrupt stimulation by cAMP. Percent change in amiloride-sensitive ISC by cAMP agonists in epithelia expressing wild-type α and γhENaC with the indicated β subunits (mean ± SEM, n = 8–32). *P < 0.0004 versus wild-type by Student’s t test. Schematics of the β COOH-terminus indicate the positions of deletions and site-directed mutations.
Discussion
In FRT epithelia expressing ENaC, cAMP agonists produced a large increase in Na+ current. Three potential mechanisms could explain this stimulation. First, cAMP could increase the single-channel conductance. However, there is no evidence to support this mechanism; previous studies of ENaC in native epithelia (13) or expressed in fibroblasts (12) found that cAMP had no effect on the single-channel conductance of ENaC.
Second, cAMP could increase PO, similar to the stimulation of CFTR (21). Here the data for ENaC are conflicting. In Xenopus kidney (A6) epithelia and rat cortical collecting duct, cAMP had no effect on the PO of ENaC (13, 14). In contrast, Stutts and coworkers recently reported that cAMP increased the PO of ENaC expressed in fibroblasts and suggested that the presence of CFTR might prevent this stimulation (12). The present data do not directly address whether cAMP increased PO. However, even if PO increased, it is not the only mechanism responsible for cAMP-mediated stimulation of ENaC.
The third potential mechanism is an increase in the number of channels at the cell surface, resulting from either a decrease in channel internalization or an increase in translocation of channels to the cell surface. Previous reports on this mechanism have also conflicted. Patch-clamp studies on A6 epithelia (13) or rat cortical collecting duct (14) suggested an increase in the density of conducting Na+ channels in response to cAMP. Immunoprecipitation of iodinated cell surface proteins with an anti-antiamiloride antibody in A6 epithelia suggested that vasopressin increased antigen expression at the cell surface (25). However, no increase in surface expression was found using an antibody against a biochemically purified Na+ channel (26). An important limitation of these studies is that the molecular identity of the protein(s) recognized by the antibodies and their relationship to ENaC are unknown. In this work I used a novel functional approach which provided a real-time assay of the translocation of ENaC to the cell surface and a fluorescence assay that selectively detected ENaC that translocated to the cell surface. Four findings strongly suggest that increased translocation was, at least in part, responsible for the cAMP-mediated increase in Na+ current. First, following modification of cell-surface ENaC (γG536C or βS520C) with MTSET, cAMP increased the rate of appearance of unmodified channels at the cell surface. This was confirmed both by functional and fluorescence assays. Second, the block of channels containing γG536C by MTSET did not prevent stimulation by cAMP. Third, cAMP inhibited ENaC following MTSET modification of channels containing βS520C. This finding also suggests that cAMP does not block ENaC internalization. Instead, cAMP appears to increase internalization, although to a lesser extent than the increase in insertion. Fourth, cAMP-mediated stimulation was abolished by an intervention that prevents vesicle trafficking (15°C incubation). Although not specific for vesicle trafficking, low temperature does not, in general, prevent cAMP-dependent phosphorylation and increased PO. For example, 15°C (and 5°C) incubation did not prevent stimulation of CFTR by cAMP.
The data have implications regarding the relative sizes of the cell surface and intracellular pools of ENaC. When channels at the cell surface were blocked by MTSET, cAMP produced a complete recovery in ISC. In addition, after this cAMP-stimulated current was blocked by MTSET, a second treatment with cAMP produced a further (although smaller) increase in ISC (Figure 3d). This suggests that the intracellular pool of ENaC is larger than the population of channels at the cell surface. The incomplete cAMP-mediated recovery from the second treatment with MTSET suggests a partial depletion of the intracellular pool. Alternatively, the incomplete recovery may have resulted from recycling of MTSET-blocked channels to the cell surface. Interestingly, the recovery in ISC to the block by MTSET was less complete in cells not treated with cAMP than in treated cells (compare Figure 3, b and d). Perhaps cAMP increased the size of the intracellular pool of ENaC available for translocation to the cell surface. It is also possible that different pools are responsible for cAMP-mediated and cAMP-independent translocation of ENaC to the cell surface. The finding that cAMP decreased ISC following modification of βS520C with MTSET suggests that modified channels at the cell surface were replaced by unmodified channels. This is also consistent with a relatively large intracellular pool of ENaC. It is interesting that when βS520C was modified by MTSET, cAMP inhibited ISC close to the initial baseline current (before MTSET). This suggests the possibility that MTSET and/or cAMP may have additional effects on the function or trafficking of βS520C ENaC.
To determine whether the COOH-termini of ENaC subunits play a role in the cAMP-mediated translocation of ENaC to the apical surface, I tested the effect of deletions and mutations in these cytoplasmic domains. Deletion of the COOH-terminus from βENaC abolished stimulation by cAMP, resulting instead in a small amount of inhibition. Identical mutations in the α and γ subunits also disrupted stimulation by cAMP. Surprisingly, these deletions did not disrupt cAMP-mediated stimulation by removing a consensus site for phosphorylation by PKA or a cyclic nucleotide-binding motif. Instead, progressive deletions and site-directed mutations identified the responsible sequence as a tyrosine-based motif (PPPXY). Mutation of conserved residues in this motif also prevented stimulation by cAMP. This sequence fits the consensus of the PY motif that forms a binding site for proteins, including Nedd4 (27, 28). This suggests that cAMP might stimulate ENaC indirectly through the interaction of the PY motif with a regulatory protein. Because Nedd4 binds to this motif, it is tempting to speculate that the interaction between Nedd4 and ENaC is involved in stimulation by cAMP. Perhaps phosphorylation of a site distinct from the PY motif alters the binding of Nedd4 to ENaC, resulting in increased translocation of the channel to the cell surface. Alternatively, interaction of ENaC with proteins other than Nedd4 might be involved. Such a protein might be required for the phosphorylation of a different site in ENaC, possibly acting as an A kinase anchoring protein (AKAP) (29). Recent work indicates that ENaC can be phosphorylated by PKA (30), although the specific residues that are phosphorylated and the functional relevance of phosphorylation are not yet known. Alternatively, phosphorylation of the interacting protein might be required for stimulation by cAMP. Such an interaction might be directly required for the translocation of ENaC to the cell surface. Conversely, interaction of a protein with the PY motif might inhibit channel translocation, and cAMP might increase translocation by preventing this interaction. Thus, Liddle’s syndrome mutations might disrupt cAMP-mediated stimulation either by preventing translocation or by increasing translocation in the absence of cAMP. It is also possible that defective internalization in Liddle’s syndrome depletes the intracellular pool of ENaC, disrupting the translocation of channels to the cell surface.
This potential requirement for an interacting protein might explain the differing responses to cAMP reported for different tissues. Although Na+ absorption is stimulated by cAMP in mammalian kidney collecting duct and amphibian epithelia (6), it is not stimulated in the distal colon (6), human airway epithelia (31), or Xenopus oocytes (32, 33). Differential expression of an interacting regulatory protein might underlie this tissue-specific regulation. Such a role has been proposed for CFTR; previous work reported that in the absence of CFTR, cAMP stimulated ENaC (34), but in the presence of CFTR, cAMP produced inhibition (33, 34). Although the mechanism of this functional interaction is unknown, it is interesting that CFTR expression and mutations in the PY motif of ENaC both prevent stimulation by cAMP. However, the presence or absence of CFTR cannot completely explain the tissue-specific regulation of ENaC by cAMP. cAMP stimulates Na+ current in some tissues that express both ENaC and CFTR, such as mammalian kidney cortical collecting duct (6, 35), canine airway (36), and A6 epithelia (37). Conversely, cAMP and cAMP-dependent protein kinase did not stimulate ENaC in some studies when CFTR was lacking, for example, in Xenopus oocytes (32, 33), or when ENaC was translated in vitro and reconstituted in planar lipid bilayers (32).
The current data have important implications for our understanding of Na+ homeostasis. To respond to states of Na+ excess or Na+ depletion, the rate of Na+ absorption through ENaC in the renal collecting duct must vary widely (38, 39). The data suggest that cAMP-mediated translocation of ENaC to the cell surface contributes to this regulation of Na+ absorption. Such a mechanism is similar to the cAMP-dependent regulation of aquaporin-2 water channels; in response to vasopressin, increased cellular levels of cAMP stimulate translocation of aquaporin-2 from an intracellular store to the cell surface (11). Interestingly, aquaporin-2 and ENaC are both expressed at the apical membrane of the kidney collecting duct, and both are stimulated by vasopressin. Perhaps this common mechanism of regulation by cAMP allows the coordinate control of Na+ and water homeostasis. The current data also have implications for our understanding of disease. The COOH-terminal mutations in ENaC that abolished stimulation by cAMP are the same mutations that cause Liddle’s syndrome. This suggests that cAMP-mediated regulation of ENaC is disrupted in Liddle’s syndrome and raises the possibility that dysfunctional regulation of the channel could also play a role in other forms of hypertension and disorders of Na+ homeostasis.
Acknowledgments
I thank Diane Olson, Phil Karp, and Pary Weber for technical support, and Michael Welsh, Christopher Adams, Heather Drummond, and my other colleagues for helpful discussions and critical review of this manuscript. The FRT cells were a generous gift from C. Zurzolo. P.M. Snyder was supported by the Roy J. Carver Charitable Trust and by the National Heart, Lung, and Blood Institute (HL-58812, HL-03575) and National Institute of Diabetes and Digestive and Kidney Diseases (DK-52617), National Institutes of Health.
References
- 1.Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. 1993;361:467–470. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- 2.Canessa CM, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 3.McDonald FJ, Snyder PM, McCray PB, Jr, Welsh MJ. Cloning, expression, and tissue distribution of a human amiloride–sensitive Na+ channel. Am J Physiol. 1994;266:L728–L734. doi: 10.1152/ajplung.1994.266.6.L728. [DOI] [PubMed] [Google Scholar]
- 4.McDonald FJ, Price MP, Snyder PM, Welsh MJ. Cloning and expression of the beta- and gamma-subunits of the human epithelial sodium channel. Am J Physiol. 1995;268:C1157–C1163. doi: 10.1152/ajpcell.1995.268.5.C1157. [DOI] [PubMed] [Google Scholar]
- 5.Benos DJ, Awayda MS, Ismailov II, Johnson JP. Structure and function of amiloride-sensitive Na+ channels. J Membr Biol. 1995;143:1–18. doi: 10.1007/BF00232519. [DOI] [PubMed] [Google Scholar]
- 6.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 7.Lifton RP. Molecular genetics of human blood pressure variation. Science. 1996;272:676–680. doi: 10.1126/science.272.5262.676. [DOI] [PubMed] [Google Scholar]
- 8.Snyder PM, et al. Mechanism by which Liddle’s syndrome mutations increase activity of a human epithelial Na+ channel. Cell. 1995;83:969–978. doi: 10.1016/0092-8674(95)90212-0. [DOI] [PubMed] [Google Scholar]
- 9.Schild L, et al. Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO J. 1996;15:2381–2387. [PMC free article] [PubMed] [Google Scholar]
- 10.Firsov D, et al. Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci USA. 1996;93:15370–15375. doi: 10.1073/pnas.93.26.15370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nielsen S, et al. Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proc Natl Acad Sci USA. 1995;92:1013–1017. doi: 10.1073/pnas.92.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stutts MJ, Rossier BC, Boucher RC. Cystic fibrosis transmembrane conductance regulator inverts protein kinase A-mediated regulation of epithelial sodium channel single channel kinetics. J Biol Chem. 1997;272:14037–14040. doi: 10.1074/jbc.272.22.14037. [DOI] [PubMed] [Google Scholar]
- 13.Marunaka Y, Eaton DC. Effects of vasopressin and cAMP on single amiloride-blockable Na channels. Am J Physiol. 1991;260:C1071–C1084. doi: 10.1152/ajpcell.1991.260.5.C1071. [DOI] [PubMed] [Google Scholar]
- 14.Frindt G, Silver RB, Windhager EE, Palmer LG. Feedback regulation of Na channels in rat CCT. III. Response to cAMP. Am J Physiol. 1995;268:F480–F489. doi: 10.1152/ajprenal.1995.268.3.F480. [DOI] [PubMed] [Google Scholar]
- 15.Sheppard DN, Carson MR, Ostedgaard LS, Denning GM, Welsh MJ. Expression of cystic fibrosis transmembrane conductance regulator in a model epithelium. Am J Physiol. 1994;266:L405–L413. doi: 10.1152/ajplung.1994.266.4.L405. [DOI] [PubMed] [Google Scholar]
- 16.Schild L, Schneeberger E, Gautschi I, Firsov D. Identification of amino acid residues in the alpha, beta, and gamma subunits of the epithelial sodium channel (ENaC) involved in amiloride block and ion permeation. J Gen Physiol. 1997;109:15–26. doi: 10.1085/jgp.109.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snyder PM, Olson DR, Bucher DB. A pore segment in DEG/ENaC Na+ channels. J Biol Chem. 1999;274:28484–28490. doi: 10.1074/jbc.274.40.28484. [DOI] [PubMed] [Google Scholar]
- 18.Snyder PM, Cheng C, Prince LS, Rogers JC, Welsh MJ. Electrophysiological and biochemical evidence that DEG/ENaC cation channels are composed of nine subunits. J Biol Chem. 1998;273:681–684. doi: 10.1074/jbc.273.2.681. [DOI] [PubMed] [Google Scholar]
- 19.Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 20.Kuismanen E, Saraste J. Low temperature-induced transport blocks as tools to manipulate membrane traffic. Methods Cell Biol. 1989;32:257–274. doi: 10.1016/s0091-679x(08)61174-7. [DOI] [PubMed] [Google Scholar]
- 21.Tabcharani JA, Chang XB, Riordan JR, Hanrahan JW. Phosphorylation-regulated Cl– channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991;352:628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- 22.Denning GM, Ostedgaard LS, Cheng SH, Smith AE, Welsh MJ. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J Clin Invest. 1992;89:339–349. doi: 10.1172/JCI115582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimkets RA, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the β subunit of the epithelial sodium channel. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 24.Hansson JH, et al. Hypertension caused by a truncated epithelial sodium channel γ subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11:76–82. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- 25.Kleyman TR, Ernst SA, Coupaye-Gerard B. Arginine vasopressin and forskolin regulate apical cell surface expression of epithelial Na+ channels in A6 cells. Am J Physiol. 1994;266:F506–F511. doi: 10.1152/ajprenal.1994.266.3.F506. [DOI] [PubMed] [Google Scholar]
- 26.Oh Y, Smith PR, Bradford AL, Keeton D, Benos DJ. Regulation by phosphorylation of purified epithelial Na+ channels in planar lipid bilayers. Am J Physiol. 1993;265:C85–C91. doi: 10.1152/ajpcell.1993.265.1.C85. [DOI] [PubMed] [Google Scholar]
- 27.Staub O, et al. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- 28.Goulet CC, et al. Inhibition of the epithelial Na+ channel by interaction of Nedd4 with a PY motif deleted in Liddle’s syndrome. J Biol Chem. 1998;273:30012–30017. doi: 10.1074/jbc.273.45.30012. [DOI] [PubMed] [Google Scholar]
- 29.Scott JD, McCartney S. Localization of A-kinase through anchoring proteins. Mol Endocrinol. 1994;8:5–11. doi: 10.1210/mend.8.1.8152430. [DOI] [PubMed] [Google Scholar]
- 30.Shimkets RA, Lifton R, Canessa CM. In vivo phosphorylation of the epithelial sodium channel. Proc Natl Acad Sci USA. 1998;95:3301–3305. doi: 10.1073/pnas.95.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mall M, Bleich M, Greger R, Schreiber R, Kunzelmann K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J Clin Invest. 1998;102:15–21. doi: 10.1172/JCI2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Awayda MS, Ismailov II, Berdiev BK, Fuller CM, Benos DJ. Protein kinase regulation of a cloned epithelial Na+ channel. J Gen Physiol. 1996;108:49–65. doi: 10.1085/jgp.108.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mall M, Hipper A, Greger R, Kunzelmann K. Wild type but not delta F508 CFTR inhibits Na+ conductance when coexpressed in Xenopus oocytes. FEBS Lett. 1996;381:47–52. doi: 10.1016/0014-5793(96)00079-8. [DOI] [PubMed] [Google Scholar]
- 34.Stutts MJ, et al. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- 35.Todd-Turla KM, Rusvai E, Naray-Fejes-Toth A, Fejes-Toth G. CFTR expression in cortical collecting duct cells. Am J Physiol. 1996;270:F237–F244. doi: 10.1152/ajprenal.1996.270.1.F237. [DOI] [PubMed] [Google Scholar]
- 36.Cullen JJ, Welsh MJ. Regulation of sodium absorption by canine tracheal epithelium. J Clin Invest. 1987;79:73–79. doi: 10.1172/JCI112811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marunaka Y, Tohda H. Effects of vasopressin on single Cl– channels in the apical membrane of distal nephron cells (A6) Biochim Biophys Acta. 1993;1153:105–110. doi: 10.1016/0005-2736(93)90281-4. [DOI] [PubMed] [Google Scholar]
- 38.Stokes JB. Potassium secretion by cortical collecting tubule: relation to sodium absorption, luminal sodium concentration, and transepithelial voltage. Am J Physiol. 1981;241:F395–F402. doi: 10.1152/ajprenal.1981.241.4.F395. [DOI] [PubMed] [Google Scholar]
- 39.Reif MC, Troutman SL, Schafer JA. Sodium transport by rat cortical collecting tubule. Effects of vasopressin and desoxycorticosterone. J Clin Invest. 1986;77:1291–1298. doi: 10.1172/JCI112433. [DOI] [PMC free article] [PubMed] [Google Scholar]