

Abstract
The pregnane X receptor (PXR) regulates the metabolism and excretion of xenobiotics and endobiotics by regulating the expression of drug-metabolizing enzymes and transporters. The unique structure of PXR allows the binding of many drugs and drug leads to it, possibly causing undesired drug-drug interactions. Therefore, it is crucial to evaluate whether lead compounds bind to PXR. Fluorescence-based assays are preferred because of their sensitivity and non-radioactive nature. One fluorescent PXR probe is currently commercially available; however, because its chemical structure is not publicly disclosed, it is not optimal for studying ligand-PXR interactions. Here we report the characterization of BODIPY FL Vinblastine, generated by labeling vinblastine with the fluorophore 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene (BODIPY FL), as a high-affinity ligand for human PXR with a Kd value of 673 nM. We provide evidence that BODIPY FL Vinblastine is a unique chemical entity different from either vinblastine or the fluorophore 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene in its function as a high-affinity human PXR ligand. We describe a BODIPY FL Vinblastine-based human PXR Time-Resolved Fluorescence Resonance Energy Transfer assay, which was used to successfully test a panel of human PXR ligands. The BODIPY FL Vinblastine–based biochemical assay is suitable for high-throughput screening to evaluate whether lead compounds bind to PXR.
Keywords: PXR, drug metabolism, xenobiotics, high-throughput screening, BODIPY FL Vinblastine, Time-Resolved Fluorescence Resonance Energy Transfer Assay
Introduction
The pregnane X receptor (PXR) is a key xenobiotic receptor that regulates the metabolism and excretion of xenobiotics and endobiotics by regulating the expression of drug-metabolizing enzymes and drug transporters [1,2]. Expression of a PXR target gene is regulated by the binding of PXR to the promoter region of the target gene, such as that of cytochrome P450 3A4 (CYP3A4), a key enzyme in the metabolism of more than 50% of all clinically prescribed drugs [3]. By affecting drug metabolism, changes in the expression of such mediators can influence the therapeutic and toxicologic response to drugs and cause adverse drug-drug interactions [4,5].
PXR activity is regulated mainly by direct ligand binding (and the unique structure of PXR allows the binding of many drugs and drug leads to it) [4], although it can also be regulated by various cell signaling pathways [5]. Radioisotope-based binding assays (scintillation proximity assays) were initially used to investigate direct binding of PXR to its ligands. They assessed the competitive binding of a group of tritium-labeled putative PXR ligands, such as 3,5-Bis(1,1-dimethylethyl)-4-hydr-oxyphenyl]ethenylidene]bis-phosphonic acid tetraethyl ester (SR12813) [6-8], N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]-benzenesulfonamide (TO901317) [9] and N-methyl-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl) ethyl]phenyl]benzenesulfonamide (NMTB) [10] to PXR. When Invitrogen offered the LanthaScreen® Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) Pregnane X Competitive Binding Assay kit, containing the first PXR fluorescent probe (Fluormone PXR [SXR] Green), it was quickly used in multiple studies [11-20]. However, this commercially available assay is not optimal for studying ligand-PXR interactions. First, the chemical structure of Fluormone PXR (SXR) Green is proprietary information and is not publicly disclosed. Second, Fluormone PXR (SXR) Green is available only as a component of the assay kit, at a pre-diluted concentration of 4 μM in 50% methanol/water [21].
To study ligand-PXR interactions, a PXR fluorescent probe that offers a publicly available structure and flexibility for various assay formats is highly valuable. Here we introduce BODIPY FL Vinblastine, vinblastine labeled with the fluorophore 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene (BODIPY fluorescein, or BODIPY FL) (Figure 1A), as the first PXR fluorescent probe with a publicly available structure. We report here that BODIPY FL Vinblastine has high binding affinity to the human PXR (hPXR) protein, with a Kd value of 673 nM in an hPXR TR-FRET binding assay. Furthermore, other known hPXR ligands could compete with BODIPY FL Vinblastine for binding to hPXR. We have developed and validated an hPXR TR-FRET assay that uses BODIPY FL Vinblastine as a fluorescent probe to evaluate the binding of a panel of hPXR ligands to hPXR in a high-throughput format.
Figure 1.
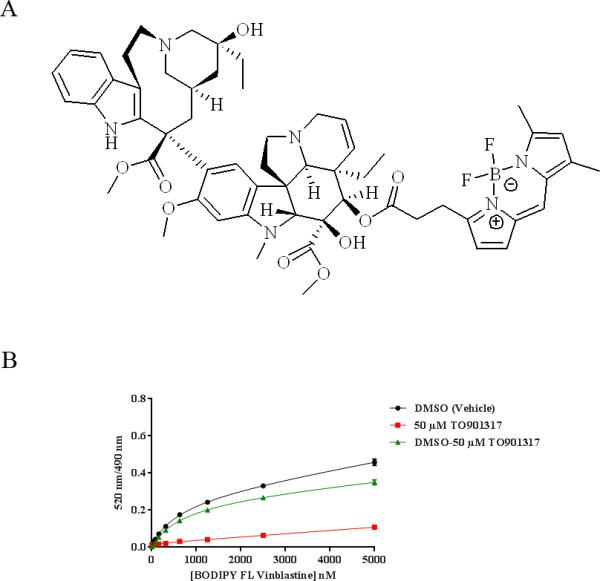
BODIPY FL Vinblastine and its interaction with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST. (A) Structure of BODIPY FL Vinblastine. (B) Binding curves of the indicated concentrations of BODIPY FL Vinblastine to 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST after 30 minutes of incubation in the presence of DMSO (total binding) or of 50 μM TO901317 (non-specific binding). The total binding curve and the non-specific binding curve were derived from titrated BODIPY FL Vinblastine (5 000 – 0.31 nM, with 1:2 dilution, at 15 concentration levels) treated with DMSO (vehicle, negative control) or 50 μM TO901317 (positive control), respectively. The specific binding curve was generated by subtracting non-specific binding from total binding. The specific equilibrium binding constant (Kd) derived from the specific binding curve was 673 ± 18 nM.
Materials and Methods
Materials
GST-hPXR-LBD, LanthaScreen Tb-anti-GST antibody, TR-FRET PXR (SXR) assay buffer, BODIPY FL Vinblastine (CAS: 1431846-82-0), BODIPY FL propionic acid (4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid; CAS: 126250-45-1), BODIPY FL hydrazide (4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid, hydrazide; CAS: 178388-71-1), BODIPY FL EDA (4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl ethylenediamine, hydrochloride; CAS: 209541-26-4), and 1 M DTT (dithiothreitol; CAS: 3483-12-3) were purchased from Invitrogen (Carlsbad, CA). Dimethyl sulfoxide (DMSO; CAS: 67-68-5) was purchased from Fisher Scientific (Pittsburgh, PA). TO901317 (N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]-benzenesulfonamide; CAS: 293754-55-9) was purchased from Cayman Chemical (Ann Arbor, MI). SR12813 ([[3,5-Bis(1,1-dimethylethyl)-4-hydr-oxyphenyl]ethenylidene]bis-phosphonic acid tetraethyl ester; CAS: 126411-39-0) was purchased from Enzo Life Sciences (Farmingdale, NY). Vinblastine (vincaleukoblastine; CAS: 143-67-9) and vincristine (22-oxovincaleukoblastine; CAS: 2068-78-2) were purchased from Tocris Bioscience (Minneapolis, MN). Clotrimazole (1-[(2-chlorophenyl)diphenylmethyl]-1H-imidazole; CAS: 23593-75-1) and rifampicin (3-(4-methylpiperazinyliminomethyl)rifamycin SV, CAS: 13292-46-1) were purchased from Sigma (St. Louis, MO). Hyperforin (CAS: 238074-03-8), ginkgolide A (CAS: 15291-75-5) and ginkgolide B (CAS: 15291-77-7) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). 384-well black polypropylene plates were purchased from Matrical Bioscience (Spokane WA). DMSO was used as solvent to dissolve all compounds used in this study.
Methods
All assays were carried out in 20 μL TR-FRET PXR (SXR) assay buffer with 5 nM GST-hPXR-LBD, 0.05 mM DTT, and 5 nM Tb-anti-GST at room temperature (approximately 25 °C) in 384-well black polypropylene plates, and all assays were performed in triplicate. All chemicals were solubilized in DMSO. The final DMSO concentration was 1.1% in all assays with the exception of the DMSO tolerance test, in which DMSO concentrations are specified. In a typical assay, chemical (solubilized in DMSO) diluted in TR-FRET PXR (SXR) assay buffer as 2 × working solution (containing 2% DMSO) was first dispensed (10 μL/well) into 384-well plate, followed by the dispensing of BODIPY FL Vinblastine solution [4 × working solution in TR-FRET PXR (SXR) assay buffer with 0.4 % DMSO] (5 μL/well), and a mixture of 20 nM GST-hPXR-LBD and 20 nM Tb-anti-GST solution (prepared in TR-FRET PXR (SXR) assay buffer containing 0.2 mM DTT (5 μL/well). After mixing of all assay components by shaking for 1 minute on a IKA® MTS 2/4 digital microtiter plate shaker (IKA® Works, Inc., Wilmington, NC), the plates were briefly centrifuged at 201 × g (1,000 rpm) for 30 second in an Eppendorf® 5810 centrifuge with the A-4-62 swing-bucket rotor (Eppendorf AG, Hamburg, Germany). The typical assay incubation time was 30 min, with the exception of the longitudinal signal stability assays, in which the incubation time is specified. All assay data were generated by using a PHERAstar plate reader from BMG Labtech (Durham, NC) to measure the fluorescent emission ratio (520 nm/490 nm) of each well, using a 340-nm excitation filter, 100-μs delay time, and 200-μs integration time. Raw data from the plate reader were directly used for analysis. The curve-fitting software GraphPad Prism 5.04 (GraphPad Software, La Jolla, CA) was used to generate graphs and curves and to determine Kd and IC50 values. The methods described here were applied to all the specific assays described below; additional information is included in the description of each specific assay where applicable.
Probe Kd Determination in the hPXR TR-FRET Assay
Serial dilutions of BODIPY FL Vinblastine (1:2 titration, 15 concentrations of 5000–0.31 nM) were incubated with 5 nM GST-hPXR-LBD, 0.05 mM DTT, and 5 nM Tb-anti-GST plus either 1.1% DMSO (vehicle) or 50 μM TO901317 (final DMSO concentration was 1.1%) for 30 min before TR-FRET signals were collected. The collected data were fit into a one-site total binding equation for the DMSO vehicle group and a linear non-specific binding equation for the 50 μM TO901317 group. The specific equilibrium binding constant (Kd) was derived from the specific binding curve, derived as curveDMSO-curveTO901317, by fitting the data into a one-site specific binding equation.
Optimization of Probe Concentration for hPXR TR-FRET Binding Assays
BODIPY FL Vinblastine (100, 250, or 500 nM) was incubated with 5 nM GST-hPXRLBD, 0.05 mM DTT, and 5 nM Tb-anti-GST plus either1.1 % DMSO (vehicle), 10 μM TO901317 (final DMSO concentration was 1.1%), or 50 μM TO901317 (final DMSO concentration was 1.1%) for 30 min, and then TR-FRET signals were collected.
Determination of Signal Stability in the BODIPY FL Vinblastine-based hPXR TR-FRET Assay
BODIPY FL Vinblastine (250 nM) was incubated with 5 nM GST-hPXR-LBD, 0.05 mM DTT, and 5 nM Tb-anti-GST plus either DMSO, 50 μM TO901317, or serial dilutions of TO901317 (50 μM to 0.28 nM with 1-to-3 titration for 12 concentration levels), and TR-FRET signals were collected at 0.5, 1, 2, 3, 4, and 5h. Final DMSO concentration in all samples was 1.1%. In the Z’-factor signal stability test, 16 data points were included in each high-signal (DMSO, negative vehicle control to determine total binding) or low-signal (50 μM TO901317, positive control to determine non-specific binding) group, and the Z’-factor value was calculated by using equation 1 [22].
| (1) |
where σ+ is the standard deviation of the negative control (DMSO) group; σ– is the standard deviation of the positive control (50 μM TO901317) group; Mean+ is the mean of the negative-control (DMSO) group; and Mean– is the mean of the positive-control (50 μM TO901317) group. The data from TO901317 titration were fit into a one-site competition binding equation to derive IC50 values.
DMSO Tolerance Test for the BODIPY FL Vinblastine-based hPXR TR-FRET Assay
BODIPY FL Vinblastine (250 nM) was incubated with 5 nM GST-hPXR-LBD, 0.05 mM DTT, and 5 nM Tb-anti-GST plus either DMSO, 50 μM TO901317, or serial dilutions of TO901317 (50 μM to 0.28 nM, with 1:3 titration at 12 concentration levels). The final DMSO concentration was 0.2%, 0.5%, 1%, 2%, 5% or 10% in each group. After 30-min incubation, the TR-FRET signals were collected. The data from titrated TO901317 were fit into a one-site competition binding equation to determine the IC50 values.
Binding Affinity of a Panel of hPXR Ligands, Vincristine, and Vinblastine with hPXR in the BODIPY FL Vinblastine-based TR-FRET Assay
Serial dilutions of TO901317, SR12813, clotrimazole, rifampicin, hyperforin, ginkgolide A, ginkgolide B, vincristine, vinblastine (100 μM to 0.56 nM, 1:3 titration at 12 concentration levels), DMSO, or 50 μM TO901317 were incubated with 250 nM BODIPY FL Vinblastine, 5 nM GST-hPXR-LBD, 0.05 mM DTT, and 5 nM Tb-anti-GST for 30 min, and then TR-FRET signals were collected. Where applicable, the data were fit into a one-site competitive binding equation in a dose-dependent manner to derive IC50 values. The inhibition constant ( K i) value was subsequently calculated by using equation 2 [23].
| (2) |
where IC50 is the concentration of inhibitor that inhibits 50% of binding, [L] is the concentration of BODIPY FL Vinblastine (250 nM), and KL is the Kd value of BODIPY FL Vinblastine in the assay (673 nM). The Ki values were used to compare the binding affinity of compounds to GST-hPXR-LBD.
Non-specific Binding of BODIPY Dyes in the hPXR TR-FRET Assay
BODIPY FL Vinblastine, BODIPY FL propionic acid, BODIPY FL hydrazide, or BODIPY FL EDA (250 nM) was incubated with 5 nM GST-hPXR-LBD, 0.05 mM DTT, and 5 nM Tb-anti-GST plus either DMSO or 50 μM TO901317 for 30 min, and then TR-FRET signals were collected. In a parallel control group, 250 nM BODIPY FL Vinblastine, BODIPY FL propionic acid, BODIPY FL hydrazide, or BODIPY FL EDA was incubated with only 5 nM GST-hPXR-LBD and 0.05 mM DTT plus either DMSO or 50 μM TO901317 for 30 min, and then TR-FRET signals were collected. Final DMSO concentration in all samples was 1.1%.
Statistical analysis
Results were expressed as the mean ± standard deviation of at least three independent experiments. Sample and control values were compared by using Student's t-test, and p ≤ 0.05 was considered to indicate a statistically significant difference.
Results and Discussion
BODIPY FL Vinblastine Binds to the hPXR Ligand-Binding Domain with High Affinity
We used a TR-FRET assay to evaluate the binding ability of a panel of fluorescent compounds to the ligand-binding domain (LBD) of hPXR and found that BODIPY FL vinblastine, a fluorescent analog of the anticancer drug vinblastine generated by linking it to the BODIPY FL fluorophore (Figure 1A), is a hPXR ligand (Figure 1B). The specific equilibrium binding constant (Kd) of BODIPY FL Vinblastine and hPXR-LBD in the TR-FRET assay system was determined by individually plotting the total binding curve (black curve in Figure 1B, in the presence of DMSO vehicle control) and non-specific binding curve (red curve in Figure 1B, in the presence of the high-affinity hPXR ligand TO901317) from experimental data, then generating the specific binding curve (green curve in Figure 1B) by subtraction of non-specific binding from total binding (DMSO – TO901317). The total binding curve and the non-specific binding curve were derived from titrated BODIPY FL Vinblastine (5000–0.31 nM, with 1:2 dilution, at 15 concentration levels) incubated with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST plus either DMSO or 50 μM TO901317, respectively.
The unique structure of PXR allows the binding of many drugs and drug leads to it [4]. TO901317, originally described as an LXR ligand, is a potent hPXR agonist [9] which at 50 μM inhibited all specific binding of BODIPY FL Vinblastine to hPXRLBD at the concentration range tested; any binding activity in the presence of 50 μM TO901317 was therefore considered non-specific. The Kd value of BODIPY FL Vinblastine, derived from the specific binding curve, was 673 nM ± 18 nM, indicating relatively high binding affinity. In the total binding experiments, the TR-FRET signal increased when the concentration of BODIPY FL Vinblastine increased from nanomolar to low micromolar, and it then increased slowly. In contrast, in the non-specific binding experiments the overall TR-FRET signal was relatively low and increased slowly in a linear manner over the concentration range tested. Because of the low non-specific binding, the specific binding curve was very similar to the total binding curve. These results indicate that BODIPY FL Vinblastine is a high-affinity hPXR fluorescent probe and that it has low non-specific activity in the assay system.
BODIPY FL Vinblastine is not the first high-affinity fluorescent hPXR probe. However, the undisclosed chemical structure of the Fluormone PXR (SXR) Green probe [21] makes it less valuable for studying ligand-PXR interactions. Here we describe the first hPXR fluorescent probe whose structure is publicly available; further, the compound is available in pure solid form, allowing the preparation of stock solutions of higher concentration in a preferred solvent. Therefore, BODIPY FL Vinblastine is a potentially useful fluorescent probe for studying regulation of PXR by ligand binding.
Determination of the Optimal BODIPY FL Vinblastine Concentration for the hPXR TR-FRET Assay
In a fluorescence-based assay, an optimal fluorescent probe concentration is crucial. To avoid deviating from the Cheng-Prusoff equation for subsequent calculation of the Ki value of a compound, a concentration at or somewhat below the Kd value of the probe should generally be tested first [23]. As the Kd value of BODIPY FL Vinblastine is 673 nM, we tested 3 different concentrations of BODIPY FL Vinblastine: 100, 250, and 500 nM. Each probe concentration was tested under 4 different treatment conditions to gain insight as to the total binding of BODIPY FL Vinblastine to hPXR (DMSO group), the non-specific binding of BODIPY FL Vinblastine under different concentrations of the competing ligand TO901317, and whether the non-specific binding is PXR-mediated (DMSO in the absence of hPXR protein) (Figure 2A). Consistent with the data shown in Figure 1B, in Figure 2A, both total binding and non-specific binding increased when the concentration of BODIPY FL Vinblastine increased. TO901317 at 10 μM and 50 μM inhibited the TR-FRET signal equally effectively at a lower concentration of BODIPY FL Vinblastine (100 nM), with fluorescent emission ratios of 0.019 and 0.018, respectively; however, 50 μM TO901317 inhibited the signal more effectively than 10 μM TO901317 at higher BODIPY FL Vinblastine concentration (500 nM), with fluorescent emission ratios of 0.031 and 0.050, respectively. The positive control (TO901317) was tested at concentrations as high as 100 μM, but no additional inhibition of BODIPY FL Vinblastine binding was observed beyond that achieved by 50 μM of TO901317 (data not shown; see the dose-responsive curves in Figure 5). Therefore, 50 μM TO901317 was used as the positive control in subsequent experiments to determine the assay background and noise signal levels.
Figure 2.
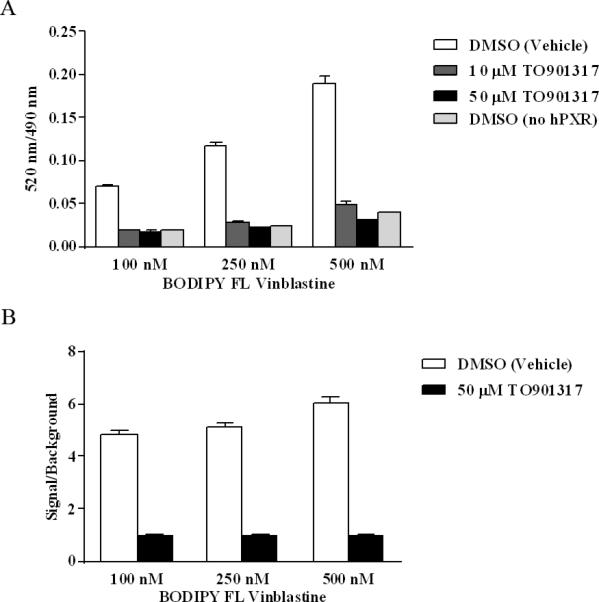
Interaction between the indicated concentrations of BODIPY FL Vinblastine, 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST after 30 minutes of incubation. (A) BODIPY FL Vinblastine interaction with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST in the presence of DMSO, 10 μM TO901317, or 50 μM TO901317, and interaction of BODIPY FL Vinblastine and 5 nM Tb-anti-GST (without GST-hPXR-LBD) with DMSO. (B) Signal-to-background ratio (signal/background) of BODIPY FL Vinblastine interaction with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST, where signal and background are the 520 nm/490 nm ratio obtained from DMSO and 50 μM TO901317, respectively. For each BODIPY FL Vinblastine concentration tested in both (A) and (B), the difference between DMSO and either 10 μM, or 50 μM TO901317was statistically significant (p < 0.0001).
Figure 5.
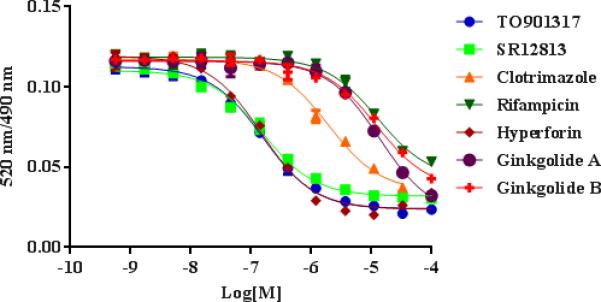
Dose-response curves of a panel of hPXR ligands in the presence of 250 nM BODIPY FL Vinblastine, 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST after 30 minutes of incubation.
To determine whether non-specific BODIPY FL Vinblastine binding is mediated by hPXR, we omitted hPXR protein from the assay. In the absence of GST-hPXR-LBD, increasing BODIPY FL Vinblastine concentration (from 100, 250, to 500 nM) correspondingly increased the fluorescent emission ratio (0.019, 0.025, and 0.04, respectively), suggesting that BODIPY FL Vinblastine can bind weakly to other components of the assay system, such as the Tb-anti-GST antibody (Figure 2A). BODIPY FL Vinblastine binding in the absence of hPXR protein (fluorescent emission ratio 0.019, 0.025, and 0.04, corresponding to 100, 250, and 500 nM of BODIPY FL Vinblastine) was comparable to the non-specific BODIPY FL Vinblastine binding in the presence of GST-PXR-LBD protein and either 50 μM TO901317 (0.018, 0.023, and 0.031) or 10 μM TO901317 (0.019, 0.028, and 0.050). These results indicate that while BODIPY FL Vinblastine can bind weakly and non-specifically to components of the assay system in an hPXR-independent manner, its binding to hPXR is specific and can be abolished by a higher concentration of the hPXR ligand TO901317. The ratio of total binding signal (DMSO negative control group) to non-specific binding signal (50 μM TO901317 positive control group) is shown in Figure 2B as signal/background: 4.8, 5.1 and 6.0 for BODIPY FL Vinblastine concentrations of 100, 250, and 500 nM, respectively. As the TR-FRET assay is robust and radiometric, all three signal/background ratios are suitable for a high-throughput screening (HTS) assay. In fact, with the Invitrogen PXR TR-FRET kit, HTS of 8280 chemicals was successfully accomplished, with signal/background ratios ranging from 2.5 to only 3.5 and Z’-factor > 0.5) [13]. We chose 250 nM of BODIPY FL Vinblastine for further experiments, considering both the signal/background ratio (the higher he better) and non-specific binding (the lower the better). However, 100 nM and 500 nM of BODIPY FL Vinblastine probe concentrations can both be used as well to achieve acceptable assay performance.
Signal from the BODIPY FL Vinblastine-based hPXR TR-FRET Assay Is Stable
Signal stability is an important parameter in an HTS assay. To assess the signal stability, we measured the binding activity of 250 nM BODIPY FL Vinblastine with or without various concentrations of TO901317 at 0.5, 1, 2, 3, 4, and 5 hours in reactions containing 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST. Both the total binding (with DMSO vehicle control) and the non-specific binding (with 50 μM TO901317) were relatively stable (Figure 3A). Correspondingly, the signal/background ratios were relatively stable as well, with a slightly decreasing trend over time: 5.02, 5.05, 4.99, 4.77, 4.66, and 4.36 for 0.5, 1, 2, 3, 4, and 5 h, respectively (Figure 3B). The Z’-factor remained constant over the entire testing period: 0.840, 0.834, 0.831, 0.834, 0.836 and 8.835 at 0.5, 1, 2, 3, 4, and 5 hours, respectively (Figure 3C). The IC50 values for TO901317 increased slightly during the first 2 h (162 nM, 170 nM, and 185 nM at 0.5, 1, and 2h, respectively) and then progressively increased to 231 nM, 266 nM and 299 nM after 3, 4 and 5 hours respectively, of incubation (Figure 3D). The consistent Z’-factor values over 5 hours demonstrated that the assay is very stable and is suitable for HTS. While all incubation times can be used for HTS, the signal/background ratio and IC50 values suggest an optimal incubation time of 2 h or less for the BODIPY FL Vinblastine-based hPXR TR-FRET assay; an HTS assay using the Invitrogen PXR TR-FRET kit also demonstrated an optimal incubation time of approximately 2 hours [13]. Because a shorter incubation time contributes to higher throughput, a 0.5 h incubation time was selected for further experiments.
Figure 3.

Longitudinal signal stability of the interaction of 250 nM BODIPY FL Vinblastine with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST. (A) Interaction of 250 nM BODIPY FL Vinblastine with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST at the indicated time points in the presence of DMSO or 50 μM TO901317. (B) Signal-to-background ratio of the interaction of 250 nM BODIPY FL Vinblastine with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST at the indicated time points. (C) Z’-factor values of the interaction of 250 nM BODIPY FL Vinblastine with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST at the indicated time points. The Z’-factor was calculated from the total binding signal (DMSO) and background signal (50 μM TO901317) by using equation 1 (see Methods). (D) The TO901317 dose-response curves in the presence of 250 nM BODIPY FL Vinblastine, 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST at the indicated time points. For each time point tested in both (A) and (B), the difference between DMSO and 50 μM TO901317 was statistically significant (p < 0.0001).
BODIPY FL Vinblastine-based hPXR TR-FRET Assay Tolerates a Wide Range of DMSO Concentrations
DMSO tolerance is another important assay parameter, as DMSO is a solvent commonly used for compounds in drug discovery. We used DMSO to dissolve all the compounds used in this study. In the DMSO tolerance test, the TR-FRET signals from BODIPY FL Vinblastine binding were collected after 30 minutes of incubation with either DMSO vehicle control or various concentrations of TO901317 in 0.2%, 0.5%, 1%, 2%, 5% and 10% final DMSO concentrations, plus 250 nM BODIPY FL Vinblastine, 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST. The total binding signal (at 520 nm/490 nm ratio) remained stable at 0.106, 0.109, 0.106, 0.104, and 0.100 when the DMSO concentration increased (0.2%, 0.5%, 1%, 2%, and 5%), and then decreased to 0.088 when the DMSO concentration increased to 10% (Figure 4A). The non-specific binding signal (in the presence of 50 μM of TO901317) remained constant at 0.0199, 0.0210, 0.0200, 0.0199, 0.0196, and 0.0201, corresponding to DMSO concentrations of 0.2%, 0.5%, 1%, 2%, 5%, and 10% (Figure 4A). The signal/background ratio was also relatively stable, ranging from 5.35 to 5.09 at DMSO concentrations below 5%, and falling to 4.36 when the DMSO concentration was 10% (Figure 4B). In the TO901317 competition assay, the IC50 values of TO901317 remained constant throughout the DMSO concentration range tested: 158, 160, 154, 164, 152, and 160 nM, corresponding to DMSO concentrations of 0.2%, 0.5%, 1%, 2%, 5%, and 10% (Figure 4C). These results indicate that the BODIPY FL Vinblastine-based TR-FRET assay can tolerate a wide range of DMSO concentrations up to 5%, comparable to the Invitrogen PXR TR-FRET kits [21]. DMSO concentrations up to 1.1% were used in this report with the exception of the DMSO tolerance test.
Figure 4.

DMSO tolerance in the interaction of 250 nM BODIPY FL Vinblastine with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST after 30 minutes of incubation. (A) Interaction of 250 nM BODIPY FL Vinblastine with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST in the presence of DMSO control or 50 μM TO901317 at the indicated final DMSO concentrations. (B) Signal-to-background ratio in the interaction of 250 nM BODIPY FL Vinblastine with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST in the presence of the indicated DMSO concentrations. (C) TO901317 dose-response curves in the presence of 250 nM BODIPY FL Vinblastine, 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST at the indicated DMSO concentrations. For each DMSO concentration tested in both (A) and (B), the difference between DMSO and 50 μM TO901317 was statistically significant (p < 0.0001).
Binding Affinity of a Panel of hPXR Ligands in the BODIPY FL Vinblastine-based hPXR TR-FRET Assay
To further validate and evaluate the BODIPY FL Vinblastine-based hPXR TR-FRET assay, we selected a panel of seven hPXR ligands: TO901317, SR12813, clotrimazole, rifampicin, hyperforin, ginkgolide A and ginkgolide B. The hPXR binding activity values of all seven compounds have previously been derived by using the Invitrogen hPXR TR-FRET binding kit [11-16,24]. In addition, TO901317 [25], SR12813 [26,27], rifampicin [28] and hyperforin [29] ligand-hPXR co-crystal structures have been reported. In the BODIPY FL Vinblastine-based TR-FRET assay, TR-FRET signals were collected after 30-minute incubation with serial dilutions of the seven hPXR ligands, plus 250 nM BODIPY FL Vinblastine, 5 nM GST-hPXR-LBD, and 5 nM Tb-anti-GST. The data were fit into a one-site competitive binding equation to derive the dose response curves (Figure 5). Because of incomplete solubility, data points for clotrimazole 100 μM were not included. TO901317, SR12813, clotrimazole, rifampicin, hyperforin, ginkgolide A, and ginkgolide B had IC50 values of 159 nM, 157 nM, 1.94 μM, 12.7 μM, 147 nM, 13.7 μM, and 12.1 μM, respectively (Ki values of 116 nM, 114 nM, 1.41 μM, 9.26 μM, 107 nM, 9.99 μM, and 8.82 μM, respectively) (Table 1).
Table 1.
Summary of the Activity of a Panel of hPXR Ligands
| Activity in This Study, Using BODIPY FL vinblastine as Probe | Activity Reported, Using Fluormone PXR (SXR) Green as Probe | |||
|---|---|---|---|---|
| Chemical | IC50 | K i | IC50a | K i b |
| TO901317 | 159 nM | 116 nM | 44 nM [12], 52 nM [24], 90 nM[13] | 22 nM, 26 nM, 45 nM |
| SR12813 | 157 nM | 114 nM | 42 nM [15], 49 nM [11], 69 nM [24], 140 nM [16], 710 nM[13] | 21 nM, 24.5 nM, 34.5 nM 70 nM, 355 nM |
| Clotrimazole | 1.94 μM | 1.41 μM | 2.0 μM [24] | 1.0 μM |
| Rifampicin | 12.7 μM | 9.26 μM | 14.13 μM [24] | 7.06 μM |
| Hyperforin | 147 nM | 107 nM | 140 nM [24], 710 nM [13] | 70 nM, 355 nM |
| Ginkgolide A | 13.7 μM | 9.99 μM | 54% (1000 μM) [14] | NAc |
| Ginkgolide B | 12.1 μM | 8.82 μM | 48% (1000 μM) [14] | NA |
Values obtained from published reports (see References)
Values calculated from published data by using equation 2 (see Methods)
NA: Not applicable
Of the ligands with previously reported IC50 values (TO901317, SR12813, clotrimazole, rifampicin, and hyperforin), the BODIPY FL Vinblastine-based TR-FRET assay yielded IC50 or Ki values in the order (low to high) of hyperforin ≈ SR12813 ≈ TO901317 < clotrimazole < rifampicin, in general agreement with the order reported using the Invitrogen hPXR binding kit(SR12813 ≈ TO901317 < hyperforin < clotrimazole < rifampicin), although the values measured with the BODIPY FL Vinblastine-based TR-FRET assay were somewhat higher (Table 1). The Ki value of hyperforin was similar to those of TO901317 and SR12813 in the BODIPY FL Vinblastine-based assay but higher than those of TO901317 and SR12813 obtained by using the Invitrogen kit (Table 1). The reported activity of SR12813 and hyperforin varies, possibly reflecting different assay conditions. We used the lowest reported IC50 values for comparison to our data. Both ginkgolide A and ginkgolide B are reported to be hPXR ligands [14], but their IC50 or Ki values had not been reported. In the BODIPY FL Vinblastine-based TR-FRET assay, both ginkgolide A and ginkgolide B competed with BODIPY FL Vinblastine for binding to hPXR in a dose-dependent manner (Figure 5); similar curves were reported using Fluormone PXR (SXR) Green as the fluorescent probe [14]. When comparing the binding affinity of a compound to PXR from different assays, it is critical to carry out the comparison in the context of specific assay conditions and assay formats used. Xiao et al. highlighted such importance in their evaluation of drug-PXR interactions by using different in vitro assays [30]. By using a circular dichroism assay and a PXR-SRC (steroid receptor coactivator) tethered protein, they determined hypothetical Kd values of clotrimazole, rifampicin and hyperforin to be 0.29, 0.54 and 0.05 μM, respectively. By using an affinity selection-mass spectrometry assay and a PXR protein (without SRC), they determined the Kd values of clotrimazole and rifampicin to be 0.33 and 10 μM, respectively. When they compared the PXR without SRC to the PXR-SRC tethered protein, they found significant differences in the shape and the volume of the corresponding ligand binding sites, and suggested that Kd values determined by the circular dichroism assay be treated as a ranking order instead of an estimation of the binding affinity. In our BODIPY FL Vinblastine-based TR-FRET assay, the Kd values of clotrimazole, rifampicin and hyperforin are 1.41, 9.26 and 0.107 μM, respectively (Table 1). Consistent with the statement made by Xiao et al, the ranking order, but not the exact value of the Kd, is consistent among different assays.
Taken together, our results indicate that BODIPY FL Vinblastine binds specifically to hPXR and that the BODIPY FL Vinblastine-based hPXR TR-FRET assay can be used to measure the binding affinity of compounds to hPXR. The rank order of affinity of hyperforin, SR12813, and TO901317 determined by our assay was slightly different from that determined by the Fluormone PXR (SXR) Green-based assay (Table 1). This discrepancy may reflect different binding modes of BODIPY FL Vinblastine and Fluormone PXR (SXR) Green to hPXR. It is well known that PXR has a relatively large and flexible ligand-binding pocket that can accommodate ligands of different sizes [25-29,31-33] and even a single ligand with different conformations [26]. It would be of interest to use a co-crystal structural approach to compare the binding modes of BODIPY FL Vinblastine-hPXR and Invitrogen's Fluormone PXR (SXR) Green-hPXR.
BODIPY FL Vinblastine is a Unique Chemical Entity with High Binding Affinity to hPXR
Because BODIPY FL Vinblastine binds to the LBD of hPXR with high affinity (Kd= 673 nM), we investigated whether this affinity is due to BODIPY FL or to vinblastine. Vinblastine has been shown to activate hPXR in cell-based assays [34,35]. However, there is no evidence of direct interaction between vinblastine and hPXR. We first tested the effect of DMSO (vehicle control), 50 μM TO901317, and various concentrations of vinblastine or its close analog, vincristine, on 250 nM BODIPY FL Vinblastine, incubated with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST for 30 minutes. As expected, 50 μM TO901317, when compared to DMSO, significantly (p < 0.0001) decreased the binding of BODIPY FL Vinblastine to hPXR (Figure 6A). Surprisingly, vinblastine and vincristine at 11.1 and 33.3 μM failed to compete with and inhibit the binding of BODIPY FL Vinblastine to hPXR (p > 0.05; compared to DMSO). At 100 μM, vinblastine and vincristine only marginally but statistically significantly (p=0.0004 and 0.009, respectively; compared to DMSO vehicle control) inhibited the binding of BODIPY FL Vinblastine to hPXR (25.4% and 12.1%, respectively) (Figure 6A). The slightly higher binding affinity of vinblastine than that of vincristine is consistent with cell-based assays in which vinblastine is a stronger activator of hPXR than vincristine [34].
Figure 6.

Specific and non-specific interaction of vincristine, vinblastine, BODIPY FL Vinblastine, BODIPY FL propionic acid, BODIPY FL hydrazide or BODIPY FL EDA with 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST after 30 minutes of incubation. (A) Competition of TO901317, vincristine, or vinblastine with 250 nM BODIPY FL Vinblastine in the presence of 5 nM GST-hPXR-LBD and 5 nM Tb-anti-GST. (B) Structures of BODIPY FL propionic acid, BODIPY FL hydrazide, and BODIPY FL EDA. (C) Interaction of 250 nM BODIPY FL Vinblastine, BODIPY FL propionic acid, BODIPY FL hydrazide, or BODIPY FL EDA with 5 nM Tb-anti-GST with or without 5 nM GST-hPXR-LBD in the presence of DMSO or 50 μM TO901317. The inset shows an enlargement of the BODIPY FL Vinblastine bar graph.
The unexpected failure of vinblastine to efficiently compete with and inhibit the binding of BODIPY FL–labeled vinblastine to hPXR prompted us to investigate whether the BODIPY FL fluorophore mediates the binding of BODIPY FL Vinblastine to hPXR. We compared the binding affinity of 250 nM of BODIPY FL Vinblastine, BODIPY FL propionic acid, BODIPY FL hydrazide and BODIPY FL EDA (Figure 6B), in the presence of either DMSO vehicle control (total binding) or 50 μM TO901317 (non-specific binding), after incubation for 30 minutes with 5 nM Tb-anti-GST and 5 nM GST-hPXR-LBD (for hPXR mediated binding) or no GST-hPXR-LBD (for non-hPXR-mediated binding).
As shown in Figure 6C, binding of BODIPY FL Vinblastine to hPXR was specifically inhibited by 50 μM of TO901317. In contrast, BODIPY FL propionic acid, BODIPY FL hydrazide and BODIPY FL EDA generated high non-specific, hPXR-independent TR-FRET signals (1.72, 1.75 and 3.27 for BODIPY FL propionic acid, BODIPY FL hydrazide, and BODIPY FL EDA, respectively), possibly by interacting with the Tb-anti-GST antibody. In the presence of GST-hPXR-LBD, the TR-FRET signals decreased to 0.65, 0.65, and 1.16 for BODIPY FL propionic acid, BODIPY FL hydrazide and BODIPY FL EDA, respectively. The TR-FRET signals generated by BODIPY FL propionic acid, BODIPY FL hydrazide and BODIPY FL EDA were not inhibited by the hPXR-specific ligand TO901317 in either the presence or absence of GST-hPXR-LBD. These results demonstrate that the BODIPY fluorophore, in either its acid form (BODIPY FL propionic acid) or its basic form (BODIPY FL hydrazide and BODIPY FL EDA), can generate strong TR-FRET assay signals that are independent of hPXR, possibly by interacting non-specifically with the Tb-anti-GST antibody. Therefore, the BODIPY fluorophore does not bind to hPXR. Interestingly, BODIPY is reported to enhance ligand binding affinity to certain target proteins [36]. Experiments investigating the effect of BODIPY on a broad range of protein-based biochemical assays will further our understanding of the non-target–specific bioactivity of BODIPY.
In summary, we have found that vinblastine labeled with the BODIPY FL fluorophore (BODIPY FL Vinblastine) can bind to GST-hPXR-LBD in a TR-FRET assay containing Tb-anti-GST antibody. Binding can be inhibited by either the potent hPXR ligand TO901317 (Figure 1) or by omitting GST-hPXR-LBD from the assay (Figure 2A), indicating that BODIPY FL Vinblastine binds specifically to the LBD of hPXR. We further confirmed the specific binding of BODIPY FL Vinblastine to hPXR LBD by showing that a panel of hPXR ligands competed with and inhibited the binding of BODIPY FL Vinblastine to hPXR LBD (Figure 5 and Table 1). Importantly, we have demonstrated that BODIPY FL Vinblastine is a unique chemical entity: a high-affinity (Kd = 673 nM) ligand that binds to the LBD of hPXR and that differs from either vinblastine or the fluorophore BODIPY FL. Here, we present BODIPY FL Vinblastine as a new and high-affinity hPXR ligand. The BODIPY FL Vinblastine-based hPXR TR-FRET assay has a high signal/background ratio (Figure 2B) and high signal stability, both of which contribute to high and consistent Z’ values (Figure 3); it also has a wide range of DMSO tolerance (Figure 4). In testing a panel of hPXR ligands, this validated assay produced results comparable to those of Invitrogen's hPXR TR-FRET assay, the probe of which [Fluormone PXR (SXR) Green] has an undisclosed structure. While BODIPY FL Vinblastine is a high affinity hPXR fluorescent probe and a potentially useful biochemical tool to study drug-hPXR interactions, vinblastine does not bind to hPXR significantly. Therefore, BODIPY FL Vinblastine and vinblastine should be treated as two different chemical entities with distinctly different binding affinity to hPXR.
Acknowledgments
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant GM086415], National Institutes of Health National Cancer Institute [Grant P30-CA21765], the American Lebanese Syrian Associated Charities (ALSAC) and St. Jude Children's Research Hospital. The authors thank Sharon Naron for editing the manuscript.
Abbreviations
- BODIPY FL
4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene
- BODIPY FL propionic acid
4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid
- BODIPY FL hydrazide
4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid, hydrazide
- BODIPY FL EDA
4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl ethylenediamine, hydrochloride
- CYP3A4
cytochrome P450 3A4
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- GST
glutathione-S-transferase
- HTS
high-throughput screening
- hPXR
human pregnane X receptor
- LBD
ligand binding domain
- NMTB
N-methyl-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl) ethyl]phenyl]benzenesulfonamide
- PXR
pregnane X receptor
- SR12813
Tetraethyl 2-(3,5-di-tert-butyl-4-hydroxyphenyl)ethenyl-1,1-bisphosphonate
- TO901317
N-[4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)phenyl]-N-(2,2,2-trifluoroethyl)benzenesulfonamide
- TR-FRET
time-resolved fluorescence resonance energy transfer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no competing financial interest.
References
- 1.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 2.Gong H, Sinz MW, Feng Y, Chen T, Venkataramanan R, Xie W. Animal models of xenobiotic receptors in drug metabolism and diseases. Methods Enzymol. 2005;400:598–618. doi: 10.1016/S0076-6879(05)00034-0. [DOI] [PubMed] [Google Scholar]
- 3.Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr.Drug Metab. 2008;9:310–322. doi: 10.2174/138920008784220664. [DOI] [PubMed] [Google Scholar]
- 4.Timsit YE, Negishi M. CAR and PXR: the xenobiotic-sensing receptors. Steroids. 2007;72:231–246. doi: 10.1016/j.steroids.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang YM, Ong SS, Chai SC, Chen T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert.Opin.Drug Metab Toxicol. 2012;8:803–817. doi: 10.1517/17425255.2012.685237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NC, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol.Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- 7.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, Goodwin B, Liddle C, Blanchard SG, Willson TM, Collins JL, Kliewer SA. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J.Biol.Chem. 2000;275:15122–15127. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Z, Kim S, Chen T, Lin JH, Bell A, Bryson J, Dubaquie Y, Yan N, Yanchunas J, Xie D, Stoffel R, Sinz M, Dickinson K. Correlation of high-throughput pregnane X receptor (PXR) transactivation and binding assays. J.Biomol.Screen. 2004;9:533–540. doi: 10.1177/1087057104264902. [DOI] [PubMed] [Google Scholar]
- 9.Mitro N, Vargas L, Romeo R, Koder A, Saez E. T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett. 2007;581:1721–1726. doi: 10.1016/j.febslet.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Li H, Moore LB, Johnson MD, Maglich JM, Goodwin B, Ittoop OR, Wisely B, Creech K, Parks DJ, Collins JL, Willson TM, Kalpana GV, Venkatesh M, Xie W, Cho SY, Roboz J, Redinbo M, Moore JT, Mani S. The phytoestrogen coumestrol is a naturally occurring antagonist of the human pregnane X receptor. Mol.Endocrinol. 2008;22:838–857. doi: 10.1210/me.2007-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin W, Wu J, Dong H, Bouck D, Zeng FY, Chen T. Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. J.Biol.Chem. 2008;283:30650–30657. doi: 10.1074/jbc.M806132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duniec-Dmuchowski Z, Fang HL, Strom SC, Ellis E, Runge-Morris M, Kocarek TA. Human pregnane X receptor activation and CYP3A4/CYP2B6 induction by 2,3-oxidosqualene:lanosterol cyclase inhibition. Drug Metab Dispos. 2009;37:900–908. doi: 10.1124/dmd.108.025130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shukla SJ, Nguyen DT, Macarthur R, Simeonov A, Frazee WJ, Hallis TM, Marks BD, Singh U, Eliason HC, Printen J, Austin CP, Inglese J, Auld DS. Identification of pregnane X receptor ligands using time-resolved fluorescence resonance energy transfer and quantitative high-throughput screening. Assay.Drug Dev.Technol. 2009;7:143–169. doi: 10.1089/adt.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lau AJ, Yang G, Rajaraman G, Baucom CC, Chang TK. Human pregnane X receptor agonism by Ginkgo biloba extract: assessment of the role of individual ginkgolides. J.Pharmacol.Exp.Ther. 2010;335:771–780. doi: 10.1124/jpet.110.172338. [DOI] [PubMed] [Google Scholar]
- 15.Dong H, Lin W, Wu J, Chen T. Flavonoids activate pregnane x receptor-mediated CYP3A4 gene expression by inhibiting cyclin-dependent kinases in HepG2 liver carcinoma cells. BMC.Biochem. 2010;11:23. doi: 10.1186/1471-2091-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Tang Y, Robbins GT, Nie D. Camptothecin attenuates cytochrome P450 3A4 induction by blocking the activation of human pregnane X receptor. J.Pharmacol.Exp.Ther. 2010;334:999–1008. doi: 10.1124/jpet.110.168294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rulcova A, Prokopova I, Krausova L, Bitman M, Vrzal R, Dvorak Z, Blahos J, Pavek P. Stereoselective interactions of warfarin enantiomers with the pregnane X nuclear receptor in gene regulation of major drug-metabolizing cytochrome P450 enzymes. J.Thromb.Haemost. 2010;8:2708–2717. doi: 10.1111/j.1538-7836.2010.04036.x. [DOI] [PubMed] [Google Scholar]
- 18.Venkatesh M, Wang H, Cayer J, Leroux M, Salvail D, Das B, Wrobel JE, Mani S. In vivo and in vitro characterization of a first-in-class novel azole analog that targets pregnane X receptor activation. Mol.Pharmacol. 2011;80:124–135. doi: 10.1124/mol.111.071787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lau AJ, Yang G, Rajaraman G, Baucom CC, Chang TK. Differential effect of meclizine on the activity of human pregnane X receptor and constitutive androstane receptor. J.Pharmacol.Exp.Ther. 2011;336:816–826. doi: 10.1124/jpet.110.175927. [DOI] [PubMed] [Google Scholar]
- 20.Lau AJ, Yang G, Yap CW, Chang TK. Selective agonism of human pregnane x receptor by individual ginkgolides. Drug Metab Dispos. 2012;40:1113–1121. doi: 10.1124/dmd.112.045013. [DOI] [PubMed] [Google Scholar]
- 21. [September 2013];LanthaScreen TR-FRET PXR (SXR) Competitive Binding Assay Kit. http://tools.invitrogen.com/content/sfs/manuals/lanthascreen_PXR_man.pdf.
- 22.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J.Biomol.Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 23.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem.Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 24.Hereley SB, Marks BD, Stafslien DK, Singh U, Eliason HC, Wolken JK, Frazee WJ. [September 2013];Fluorescence-based Biochemical Assays for the Study of Pregnane X Receptor and Constitutive Androstane Receptor. http://tools.invitrogen.com/content/sfs/posters/1007-ISSX-PXR-CAR-poster-.pdf.
- 25.Xue Y, Chao E, Zuercher WJ, Willson TM, Collins JL, Redinbo MR. Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg.Med.Chem. 2007;15:2156–2166. doi: 10.1016/j.bmc.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 27.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J.Mol.Biol. 2003;331:815–828. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 28.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol.Endocrinol. 2005;19:1125–1134. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 29.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. 2.1 A crystal structure of human PXR in complex with the St. John's wort compound hyperforin. Biochemistry. 2003;42:1430–1438. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 30.Xiao L, Nickbarg E, Wang W, Thomas A, Ziebell M, Prosise WW, Lesburg CA, Taremi SS, Gerlach VL, Le HV, Cheng KC. Evaluation of in vitro PXR-based assays and in silico modeling approaches for understanding the binding of a structurally diverse set of drugs to PXR. Biochem.Pharmacol. 2011;81:669–679. doi: 10.1016/j.bcp.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 31.Teotico DG, Bischof JJ, Peng L, Kliewer SA, Redinbo MR. Structural basis of human pregnane X receptor activation by the hops constituent colupulone. Mol.Pharmacol. 2008;74:1512–1520. doi: 10.1124/mol.108.050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng Y, Redinbo MR. Activation of the human nuclear xenobiotic receptor PXR by the reverse transcriptase-targeted anti-HIV drug PNU-142721. Protein Sci. 2011;20:1713–1719. doi: 10.1002/pro.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xue Y, Moore LB, Orans J, Peng L, Bencharit S, Kliewer SA, Redinbo MR. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol.Endocrinol. 2007;21:1028–1038. doi: 10.1210/me.2006-0323. [DOI] [PubMed] [Google Scholar]
- 34.Harmsen S, Meijerman I, Febus CL, Maas-Bakker RF, Beijnen JH, Schellens JH. PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemother.Pharmacol. 2010;66:765–771. doi: 10.1007/s00280-009-1221-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith NF, Mani S, Schuetz EG, Yasuda K, Sissung TM, Bates SE, Figg WD, Sparreboom A. Induction of CYP3A4 by vinblastine: Role of the nuclear receptor NR1I2. Ann.Pharmacother. 2010;44:1709–1717. doi: 10.1345/aph.1P354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J, Felts S, Llauger L, He H, Huezo H, Rosen N, Chiosis G. Development of a fluorescence polarization assay for the molecular chaperone Hsp90. J.Biomol.Screen. 2004;9:375–381. doi: 10.1177/1087057104265995. [DOI] [PubMed] [Google Scholar]