

Abstract
Although mononuclear cell infiltration is a hallmark of cellular rejection of a vascularized allograft, efforts to inhibit rejection by blocking leukocyte-endothelial cell adhesion have proved largely unsuccessful, perhaps in part because of persistent generation of chemokines within rejecting grafts. We now provide, to our knowledge, the first evidence that in vivo blockade of specific chemokine receptors is of therapeutic significance in organ transplantation. Inbred mice with a targeted deletion of the chemokine receptor CCR1 showed significant prolongation of allograft survival in 4 models. First, cardiac allografts across a class II mismatch were rejected by CCR1+/+ recipients but were accepted permanently by CCR1–/– recipients. Second, CCR1–/– mice rejected completely class I– and class II–mismatched BALB/c cardiac allografts more slowly than control mice. Third, levels of cyclosporin A that had marginal effects in CCR1+/+ mice resulted in permanent allograft acceptance in CCR1–/– recipients. These latter allografts showed no sign of chronic rejection 50–200 days after transplantation, and transfer of CD4+ splenic T cells from these mice to naive allograft recipients significantly prolonged allograft survival, whereas cells from CCR1+/+ mice conferred no such benefit. Finally, both CCR1+/+ and CCR1–/– allograft recipients, when treated with a mAb to CD4, showed permanent engraftment, but these allografts showed florid chronic rejection in the former strain and were normal in CCR1–/– mice. We conclude that therapies to block CCR1/ligand interactions may prove useful in preventing acute and chronic rejection clinically.
Introduction
Mononuclear cell recruitment to an allograft is a classic hallmark of cellular rejection. At least in broad terms, such leukocyte recruitment from the vascular pool across activated endothelial cells and into tissues is now reasonably well understood (1). Thus, leukocytes roll along selectin-expressing endothelium adjacent to a chemoattractant source, attach more firmly, change shape, migrate between adjacent endothelial cells as a result of integrin and other adhesion molecule binding, and migrate through extravascular tissues along chemotactic gradients to reach their destination. The latter chemokine/chemokine receptor phase is the least understood, with little in vivo data available. However, given the burgeoning field of chemokine biology, dissecting which molecules are generated in a given inflammatory setting, and especially the nature of chemokine receptors responsible for leukocyte recruitment, might well prove key to developing better therapeutic strategies for the prevention and treatment of allograft rejection. The current literature on chemokine receptor expression in organ transplants is limited to 2 papers noting expression of CXCR4 (ref. 2) and CCR5 (ref. 3) by mononuclear cells infiltrating rejecting human renal allografts. No mechanistic or interventional studies involving targeting of chemokine receptors in transplantation have yet been published.
The current studies involve serial analysis of intragraft chemokine and chemokine receptor expression within completely MHC-mismatched mouse cardiac allografts. On the basis of our initial data, in which several chemokine receptors and their ligands were associated with host mononuclear cell infiltration, we undertook a detailed analysis of the significance of 1 of the more highly expressed chemokine receptors, CCR1 (4), which binds RANTES, macrophage inflammatory protein 1-alpha (MIP-1α), and various monocyte chemoattractant proteins (MCPs). Our studies demonstrate that compared with control CCR1+/+ mice, CCR1–/– mice show significantly delayed, or in some cases an absence of, acute or chronic rejection, such that targeting of CCR1 may eventually prove of therapeutic significance clinically.
Methods
Mice.
Generation of mice with a targeted disruption of the CCR1 gene (CCR1–/–) were described previously (5); mice used as allograft recipients were of the same genetic background (B6/129, H-2b, intercrossed 10–20 generations) as CCR1+/+ mice. Additional control inbred C57BL/6, 129, and B6/129 mice, plus MHC class I– and class II–disparate BALB/c (H-2d), and MHC class II–disparate C57BL/6.CH-2bm12 (bm12) mice, were obtained from The Jackson Laboratory (Bar Harbor, Maine, USA). All mice were housed under specific pathogen–free conditions.
Transplantation.
Heterotopic cardiac allografting (BALB/c→B6/129, bm12→B6/129) in male 8- to 10-week-old mice (CCR1–/– or CCR1+/+) was performed with anastomoses to the abdominal aorta and vena cava (6). In additional studies, use of inbred B6 or 129 mice as allograft recipients gave identical survival times (n ≥ 6/group; data not shown) to those of the B6/129 recipients of BALB/c allografts detailed in Results. In each experiment (n = 6 to 10/group), events within the allograft or isograft plus the paired recipient heart, a reference tissue exposed to the same circulation, were analyzed. At harvest at day 100 after transplant or the times indicated for the respective protocol, midventricular samples were fixed in formalin for light microscopy or were snap-frozen in liquid nitrogen for immunohistology and RNA extraction.
Immunosuppression.
Cyclosporin A (CsA) (Novartis, Basel, Switzerland) was dissolved in olive oil and administered daily (10 mg/kg intraperitoneally) for 14 days beginning at transplantation. Rat mAb to mouse CD4 (GK1.5, IgG2b) was prepared from culture supernatant using protein G-Sepharose (Sigma Chemical Co., St. Louis, Missouri, USA). Recipients were injected with CD4 mAb daily (1 mg/kg intraperitoneally) for 7 days beginning at engraftment (6).
Adoptive transfer studies.
Given the potent effects of CsA on allograft survival in CCR1–/– mice, detailed in Results, transfer of unfractionated spleen cells (40 × 107) or purified CD4+ splenic T cells (8 × 106) from primary allograft recipients to naive allograft recipients was tested. Spleens were harvested at rejection in CsA-treated CCR1+/+ mice or at day 50 or 200 in CsA-treated CCR1–/– mice. Spleens were teased apart, red cells lysed, and unfractionated washed spleen cells used, or CD4+ T cells were isolated by positive selection using Dynal beads (Dynal, Oslo, Norway), according to manufacturer directions. Cell purity was assessed by flow cytometric analysis with CD4, CD8, and CD19 mAb’s, and CD4+ populations were more than 95% pure. Spleen cells and CD4+ T cells were infused intravenously into naive mice of the same strain 1 week before cardiac allografting using BALB/c donors (7). No additional immunosuppression was used, and animals were observed until rejection.
In vitro T-cell proliferative responses.
Mixed lymphocyte responses (MLRs) were assessed by culturing responder and mitomycin C–inactivated stimulator splenocytes in RPMI-1640 medium containing 5% FBS, 1% penicillin/streptomycin, and 5 × 10-5 M 2-mercaptoethanol in 96 flat-bottom wells (8). Cultures were incubated at 37°C in 5% CO2 for 3–5 days and were pulsed with [3H]thymidine for 6 hours before harvesting; mean counts per minute and SD were calculated using 12 wells per group. Concanavalin-A (Con-A) mitogenic responses were measured as for MLR studies, except that mitogen (1.25–10 μg/mL; Sigma Chemical Co.) was substituted for stimulator cells in 72-hour cultures (8).
In situ hybridization.
A 708-bp CCR1 probe template was amplified from a pool of cDNAs derived from mouse spleen, thymus, and lymph node using primers CCR1-312 (catgtgcaagcttctctct) and CCR1-1019 (tggtcctttctagttggtcc). Further PCR amplification, to introduce RNA polymerase sites, was performed using the primers CCR1-T3 (aattaaccctcactaaagggcatgtgcaagcttctctctg) and CCR1-T7 (taatacgactcactatagggtggtcctttctagttggtcc). Sense and antisense digoxygenin-labeled mRNA probes were synthesized using the DIG RNA Labeling Kit/Genius 4 Kit (Roche Molecular Biochemicals, Indianapolis, Indiana, USA). Cryostat sections of cardiac allografts were air dried at room temperature for 1–2 hours, followed by pretreatment and Proteinase K (0.1 μg/mL for 5 minutes at room temperature) incubations as described elsewhere (9). Sections were hybridized for 16–18 hours at 60°C in hybridization buffer of 200 ng/mL of digoxygenin-labeled probe, 50% formamide (GIBCO BRL, Rockville, Maryland, USA) 5× SSC, 5× Denhardt’s solution (Sigma Chemical Co.), 0.5 mg/mL salmon sperm DNA (GIBCO), and 25 μg/mL yeast RNA (Sigma Chemical Co.). Sections were washed in 0.2× SSC for 1 hour at 60°C and in 0.2× SSC for 5 minutes at room temperature. Bound digoxygenin-labeled probe was detected using the DIG Nucleic Acid Detection Kit/Genius 3 (Roche Molecular Biochemicals) according to manufacturer’s directions, except that the incubation with alkaline phosphatase–labeled antidigoxygenin antibody (1:100) was carried out at 4°C overnight and 10% 70–100 kDa polyvinyl alcohol (Sigma Chemical Co.) was added to the alkaline phosphatase reaction buffer.
Northern blot analyses of T cells and T-cell clones.
Spleens and lymph nodes were harvested from C57BL/6 mice, and equal numbers of viable splenic and lymph node mononuclear cells were combined and cultured on CD3 mAb–precoated plates (clone 2C11; PharMingen, San Diego, California, USA) in DMEM medium containing 10% FBS, 50 μM 2-mercaptoethanol, 1% glutamine, and 1% penicillin/streptomycin (all reagents from GIBCO). Cultures for Th1 cell differentiation were supplemented with IL-12 (10 ng/mL) plus anti–IL-4 mAb (10 μg/mL), whereas cultures for Th2 cell differentiation received IL-4 (10 ng/mL) plus anti–IL-12 mAb (3 μg/mL) and anti–IFN-γ mAb (10 μg/mL); cytokines and neutralizing mAb’s were obtained from PharMingen. On day 2, human IL-2 (20 U/mL, Boehringer Mannheim, Indianapolis, Indiana, USA) was added to all cell cultures. Established T-cell clones (AE7/Th1, D10/Th2, CDC35/Th2), a gift of S.J. Szabo (Harvard School of Public Health, Boston, Massachusetts, USA), received biweekly stimulation with the appropriate antigen and irradiated spleen-derived antigen presenting cells. Clones were harvested on day 7 after antigen stimulation or after stimulation on CD3 mAb–precoated plates. At the times indicated, RNA was isolated using Rneasy Kits (QIAGEN Inc., Valencia, California, USA) and Northern blots were prepared using 10 μg of RNA per lane (10); blots were analyzed using the CCR1 probe described in the in situ hybridization section.
Ribonuclease protection assay of cytokine, chemokine, and chemokine receptor expression.
Cardiac RNA was isolated in guanidine-thiocyanate, with 2 rounds of acid phenol/chloroform extraction and alcohol precipitation (11). RNA integrity was confirmed by agarose gel electrophoresis and quantitated by optical density measurement (260 nm). RNA from each mouse was evaluated using the Riboquant system (PharMingen); mouse template sets mCK5 and mCK3b were used for detection of chemokines and cytokines, respectively; and template sets mCR5 and mCR6 were used to detect CC and CXC chemokine receptors. In addition, a riboprobe for mouse CXCR3 was prepared in house. In vitro transcription was carried out in transcription buffer supplemented with [α32P]UTP (3,000 Ci/mmol; Amersham Life Science, Arlington Heights, Illinois, USA) and T7 RNA polymerase. After DNaseI treatment, the riboprobe was isolated by phenol/chloroform extraction and ammonium acetate/ethanol precipitation, and labeling efficiency was determined by measuring Cherenkov activity in a scintillation counter. Each riboprobe set was diluted to the optimal activity defined by the manufacturer, added to 20 μg of kidney RNA, heated to 90°C, allowed to cool to 56°C, and annealed overnight. After RNase and proteinase K treatment, protected RNA hybrids were purified by phenol/chloroform extraction and ammonium acetate/ethanol precipitation and separated by electrophoresis on 5% polyacrylamide/8 M urea gels. Gels were dried, subjected to autoradiography using Kodak Biomax MS2 film (Eastman Kodak, Rochester, New York, USA), and autoradiographs were scanned into Adobe Photoshop (Adobe Systems Inc., San Jose, California, USA). RNA bands were quantitated by densitometric analysis with NIH Image (National Institutes of Health, Bethesda, Maryland, USA), and results were normalized for L32 and GAPDH gene expression.
Immunopathology.
Hearts were fixed in formalin, paraffin-embedded, and stained with hematoxylin and eosin for routine histology, Masson’s trichrome for evaluation of interstitial fibrosis, and elastin stain for assessment of transplant arteriosclerosis. Elastin-stained arteries were scored (>10 sections per graft; n = 6/time point) (ref. 6), in grafts harvested at rejection or at 50, 100, or 200 days after transplant, as: 0: less than 5% occlusion; 1: less than 5–20%; 2: greater than 20–40%; 3: greater than 40–60%; 4: greater than 60–80%; 5: greater than 80%. Cryostat sections fixed in paraformaldehyde-lysine-periodate were stained by peroxidase-antiperoxidase (PAP) technique (8), using goat anti-CCR1 or control antibody (Santa Cruz Biotech, Santa Cruz, California, USA), mAb’s to mouse leukocytes, and isotype-matched control mAb’s (PharMingen, San Diego, California, USA). mAb’s were directed against all leukocytes (CD45, 30F11.1), T cells (CD4, GK1.5 and CD8, 53-6.7), monocytes (CD11b, M1/70), granulocytes (GR-1, RB6-8C5), natural killer cells (NK-1.1, PK136), and IL-2R+ cells (CD25, PC61). Leukocytes were counted by digital scanning of 20 high-power fields per graft and analysis using IPLab software (Scanalytics, Fairfax, Virginia, USA); results were expressed as immunostained cells per field. Intragraft deposits of humoral reactants was assessed using rat mAb’s to mouse IgG, IgM, and C3 (Zymed, San Francisco, California, USA) (6).
Statistical analyses.
Data were compared by nonparametric analysis (Instat software; GraphPad, San Diego, California, USA), using the log-rank test for graft survival data and Mann-Whitney test for data from proliferation assays and immunohistological studies.
Results
CCR1+ mononuclear cells infiltrate rejecting mouse cardiac allografts.
The murine homologue of human CCR1 was characterized and cloned based on its expression by murine macrophages and eosinophils (12). Because, numerically, macrophages constitute the most prominent component of graft infiltrating cells (6, 8), we investigated whether CCR1 was expressed by leukocytes in rejecting mouse cardiac allografts. As shown by in situ hybridization studies (Figure 1, a and b), CCR1 mRNA was indeed expressed by graft infiltrating cells. Immunohistological studies confirmed CCR1 protein expression by more than 50% of intragraft mononuclear cells (Figure 1c), whereas no expression was seen in cardiac allografts within CCR1–/– recipients (Figure 1d).
Figure 1.

Detection of CCR1 expression after mouse cardiac allografting. (a) In situ hybridization studies of a day 5 allograft in a CCR1+/+ mouse, using a digoxygenin-labeled antisense probe, shows CCR1 mRNA expression by diffusely distributed leukocytes; (b) specificity of labeling was shown by lack of blue/violet staining using a corresponding sense probe. (c) Higher magnification of immunoperoxidase-stained cardiac allografts shows dense CCR1 expression by infiltrating mononuclear cells (brown reaction product) in a CCR1+/+ mouse, whereas (d) no CCR1 protein was detected in allografts within CCR1–/– recipients. (Cryostat sections, in situ hybridization, a and b, have no counterstain. ×100. Immunoperoxidase, c and d, have a hematoxylin counterstain. ×400. Data are representative of 4 grafts per group.)
CCR1–/– mice mount normal T-cell proliferative responses.
Before in vivo studies, the capacity of CCR1–/– mice to elicit T-cell responses was assessed. CCR1–/– mice showed identical mitogen-induced T-cell proliferation to that of CCR1+/+ mice (Figure 2a). In vitro assays of alloresponses showed that CCR1–/– mice also developed vigorous MLRs (P < 0.001) (Figure 2b), although the overall magnitude in repeated 5-day MLR assays was consistently 20–25% less in cultures from CCR1–/– versus CCR1+/+ mice (P < 0.01).
Figure 2.

T-cell proliferative responses in CCR1+/+ versus CCR1–/– mice. (a) Both groups showed comparable mitogen responses (mean ± SD, 6 wells per group, 48 hours); representative of 4 experiments. (b) Both groups displayed robust MLRs (**P < 0.001 versus respective responder cells alone) to mitomycin C–treated BALB/c stimulator cells, although CCR1 KO proliferated somewhat less than WT controls (*P < 0.01). Data represent mean ± SD of 6 wells per group in a 5-day MLR and are representative of 4 experiments.
CCR1 and MHC-mismatched cardiac graft survival.
Given expression of CCR1 by graft infiltrating leukocytes, we tested the effects of genetic deletion of CCR1 on cardiac allograft survival using completely MHC-mismatched strains (H2d→H2b). Compared with cardiac allografts, which were rejected by 7 days in CCR1+/+ recipients, whether B6, B6/129, or 129 strain (all H2b), and consistent with the mildly reduced alloresponses noted in vitro, CCR1–/– mice showed a doubling of survival time (Table 1; P < 0.001). However, a profound effect of CCR1 expression on allograft survival was revealed by brief therapy with CsA. Mice are CsA resistant, such that in CCR1+/+ mice given a 14-day course of CsA at 10 mg/kg per day intraperitoneally, allograft survival was only marginally prolonged by 2–3 days. By comparison, CCR1–/– recipients receiving the same course of therapy, remarkably, accepted their allografts permanently (Table 1; P < 0.001). Moreover, transfer of unfractionated spleen cells, harvested from CCR1–/– recipients of cardiac allografts at day 200 after transplant, to naive CCR1 recipients of BALB/c cardiac allografts resulted in more than 30 days of survival, and comparable results were seen by transfer of just the corresponding CD4+ T-cell population as present within unfractionated spleen cells (Table 1; each P < 0.01 versus controls).
Table 1.
Survival of cardiac allografts in CCR1–/– vs. CCR1+/+ recipients

CCR1 and intragraft cytokine, chemokine, and chemokine receptor mRNA expression.
Comparison of intragraft cytokine, chemokine, and chemokine receptor mRNA expression in untreated CCR1–/– versus CCR1+/+ recipients of cardiac allografts showed similar results for all but 2 genes (Figure 3a).
Figure 3.

Ribonuclease protection assay of intragraft cytokine, chemokine, and chemokine receptor expression in CCR1+/+ versus CCR1–/– cardiac allograft recipients at day 5 after transplant. Data show results from (a) untreated recipients and (b) mice receiving a 14-day course of a subtherapeutic regimen of CsA (10 mg/kg per day). Data shown are representative of 3 separate experiments.
First, CCR1 mRNA expression was markedly decreased, although not completely absent, in CCR1–/– recipients. We have found that normal hearts from CCR1+/+ mice show very low level expression by dendritic cells and wandering macrophages, such that some residual CCR1 mRNA expression could be donor derived. However, more importantly, the targeting construct used to inactivate the CCR1 gene deletes most, but not all, of the coding region such that some CCR1 mRNA production still occurs, although no functional protein is produced (5). The residual part of the gene encodes an mRNA fragment that is also the part of the gene detected by CCR1 probe in the commercial RPA kit, such that grafts in CCR1–/– mice show low-level mRNA expression.
Second, allografts in CCR1–/– recipients showed somewhat less IL-6 than in wild-type controls, but the significance of this is unclear. In contrast to the modest differences between CCR1–/– and control mice when used as unmodified allograft recipients, major effects on intragraft mRNA expression were seen when low-dose CsA was added (Figure 3b). Thus, in CCR1–/– mice, low-dose CsA reduced to baseline induction of the Th1 cytokines, IL-2, and IFN-γ and also suppressed (IL-4, IL-10, IL-13) or diminished (IL-6, IL-15) expression of several other cytokines. In addition, in CCR1–/– versus control recipients, low-dose CsA decreased or completely abrogated expression of all the chemokines tested, except MIP-2, and diminished intragraft expression of the chemokine receptors CCR2, CCR5, and CXCR3 (Figure 3b). Densitometric quantitative data for each group are shown in Figure 4.
Figure 4.

Densitometric quantitation of ribonuclease protection assay data derived from Figure 3; data are representative of 3 separate experiments. Use of CsA in CCR1–/– allograft recipients markedly suppressed or abolished intragraft expression of chemokines and chemokine receptors.
Effects of low-dose CsA therapy on intragraft infiltrates.
To assess the extent to which alterations in intragraft chemokine and chemokine receptor mRNA expression were accompanied by effects on graft cellularity, we undertook histological and immunohistological analyses of sequentially harvested cardiac allografts; representative data from day 5 after transplant are shown in Figure 5. Overall graft cellularity in CCR1–/– recipients, and in CCR1+/+ mice given low-dose CsA, was somewhat reduced compared with grafts in CCR1+/+ untreated mice, whereas allografts in CCR1–/– mice given low-dose CsA appeared normal and completely lacked infiltration by IL-2R+ mononuclear cells (Figure 5a). Quantitative analysis of leukocyte subsets in day 5 allografts showed that, compared with untreated controls, CCR1–/– mice had significantly decreased infiltration of T cells (TCR-α/β+) and CD4+ and CD8+ T-cell subsets (all P < 0.01), as well as macrophages (P < 0.05); NK and B cells were lacking in both groups. Again, CCR1–/– mice were comparable with respect to the composition and extent of cell infiltration seen in CCR1+/+ mice given low-dose CsA, whereas use of low-dose CsA in CCR1–/– recipients markedly suppressed all cell infiltration (P < 0.001) and immune activation (P < 0.001) (Figure 5b). No intragraft immunoglobulin or complement deposition was detected in any group.
Figure 5.

Immunopathology of cardiac allografts at day 5 after transplant. (a) Histology and intragraft immune activation, as assessed by detection of IL-2R+ mononuclear cells. Overall leukocyte infiltration was greatest in CCR1+/+, decreased in both CCR1–/– and CCR1+/+ plus low CsA groups, and least in the CCR1–/– plus low CsA samples, which appeared largely normal. Levels of immune activation were comparable in the first 3 groups and contrasted markedly with the CCR1–/– plus low CsA group. (b) Quantitative assessment, representative of 4 grafts per group, of intragraft infiltration, as assessed using cell lineage-specific mAb’s and ≥20 fields/graft; *P < 0.01 and **P < 0.001.
CCR1 expression by T cells and T-cell clones.
CCR1 expression by cells of the mouse mononuclear phagocyte lineage, as well as mouse eosinophils and neutrophils, has been documented previously, but CCR1 expression by mouse T cells has not been shown. Such knowledge is important given the reduction in T-cell recruitment to allografts in CCR1–/– mice (Figure 4), as well as the ability to prolong allograft survival in naive recipients by transfer of spleen cells from CCR1–/– mice given low-dose CsA (Table 1). Accordingly, we undertook Northern analyses of CCR1 expression by primary T cells and T-cell clones in culture. Freshly isolated mouse lymphocytes lacked CCR1 expression, but on activation under conditions that promote Th1 or Th2 differentiation, Th1 cells expressed CCR1 mRNA (Figure 6). Established Th1 clones (OF6 and AE7) also expressed CCR1, whereas Th2 clones were variable in their CCR1 expression; CDC35 cells showed little CCR1 mRNA, whereas D10 cells expressed as much CCR1 as Th1 clones (Figure 6).
Figure 6.

Northern analysis of CCR1 mRNA expression by T cells. (a) Primary murine lymphocytes were stimulated with plate-bound anti-CD3 mAb under conditions that polarize toward Th1 (IL-12 and anti–IL-4 mAb) or Th2 (IL-4 plus anti–IL-12 and anti–IFN-γ mAb’s) differentiation and harvested on the days indicated. (b) Established long-term T-cell clones were stimulated with antigen, rested in IL-2 for 7 days, and harvested, and Northern blots were prepared.
CCR1–/– mice do not develop chronic rejection.
We have previously shown that a brief course of CD4 mAb therapy induces donor-specific cardiac allograft tolerance in the completely MHC-mismatched BALB/c→B6/129 model but does not prevent development of florid transplant arteriosclerosis or other pathognomonic features of chronic allograft rejection (6, 8). We therefore tested whether administration of CD4 mAb in CCR1–/– versus CCR1+/+ mice would affect the extent of chronic rejection in this model. CD4 mAb therapy induced permanent engraftment in both groups, but CCR1–/– mice showed protection against development of chronic rejection. Whereas control mice showed focal infarcts, diffuse mononuclear cell infiltration, interstitial fibrosis, and uniform intimal proliferation (intimal score of 2.4 ± 1.1; n = 42 elastin-stained arteries observed), CCR1–/– mice lacked significant cell infiltration or interstitial fibrosis, showed well-preserved myocardial structure, and had normal vessels (intimal score of 0.3 ± 0.2; n = 35 elastin-stained arteries observed; P < 0.001) (Figure 7).
Figure 7.

CD4 mAb therapy (4 doses of GK1.5 mAb) induced permanent survival of BALB/c cardiac allografts in both CCR1+/+ (left) and CCR1–/– (right) recipients, but analysis of allografts harvested at day 50 showed significant differences in the development of chronic rejection (grafts shown are representative of 4 per group). In a typical CCR1+/+ recipient, (a) H&E staining shows widespread mononuclear cell infiltration, healed infarcts, and significant vascular pathology (arrows). A serial section (c) of the same graft stained by elastin shows advanced intimal proliferation and vascular occlusion of the arteries. In contrast, a typical allograft in a CCR1–/– recipient showed (b) only minor mononuclear cell infiltrates with normal myocardium, and, in a serial elastin-stained section, (d) normal vessels. (Representative of 4 animals per group, paraffin sections. ×250.)
CCR1–/– mice permanently accept MHC class II–mismatched cardiac allografts.
Last, as class II mismatches are key determinants of clinical allograft success for various organs, including the heart (13), we tested the requirement for CCR1 expression for rejection of MHC class II–mismatched cardiac allografts, using the bm12→B6/129 strain combination. CCR1+/+ mice rejected bm12 hearts by 33 days, whereas CCR1–/– mice accepted permanently bm12 allografts (>100 d; P < 0.01) (Table 1). Examination of well-functioning bm12 allografts harvested at 100 days or later from CCR1 KO mice showed excellent preservation of cardiac morphology, with no evidence of chronic rejection, including freedom from development of transplant arteriosclerosis (intimal score of 0.2 ± 0.1) (Figure 8).
Figure 8.
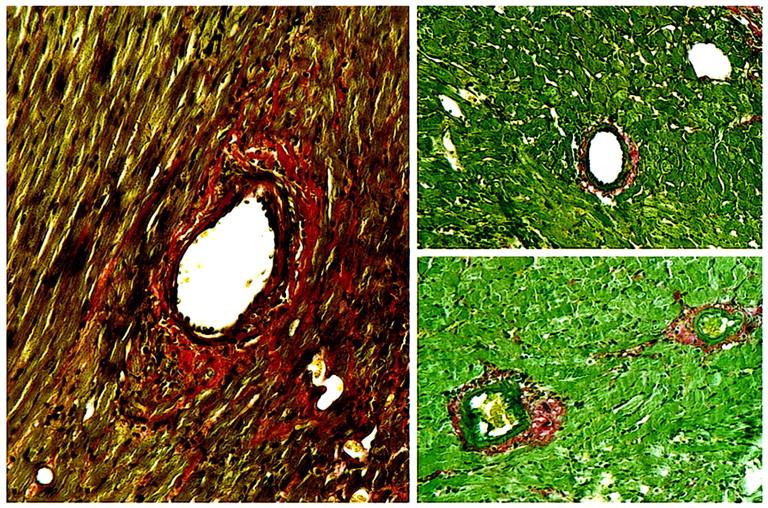
Examination of 3 consecutive bm12 cardiac allografts harvested at day 100 after transplant from untreated CCR1–/– recipients showed normal architecture without evidence of chronic rejection, including an absence of transplant arteriosclerosis. (Paraffin sections, Verhoeff’s elastin. ×250.)
Discussion
With the premise that chemokines likely mediate leukocyte recruitment to tissues during pathological responses, we have begun to dissect which chemokine and chemokine receptor pathways are present during alloresponses and which, if any, of these pathways are of functional significance. The current study sets out the first results of this approach and, using mice genetically deficient in the chemokine receptor, CCR1, identifies a key role for CCR1 in the recruitment of host macrophages and T cells and development of cardiac allograft rejection.
Our initial serial analysis showed that development of a cellular infiltrate in unmodified recipients of completely MHC-mismatched allografts was associated with intragraft expression of mRNA for CCR1, CCR2, CCR5, and CXCR3. At least some of their corresponding chemokine ligands were also expressed in this context, including MIP-1α (CCR1, CCR5), MCP-1 (CCR2), RANTES (CCR5), MIP-1β (CCR5), and IP-10 (CXCR3). There are no reported mAb’s to any of these mouse chemokine receptors, such that mAb-induced depletion of chemokine receptor–bearing cells in a rodent allograft recipient was not feasible, but we were able to demonstrate by in situ hybridization, and immunohistology with a commercially available polyclonal antibody, expression of CCR1 by infiltrating mononuclear cells. This led us to test CCR1–/– mice as allograft recipients.
CCR1–/– mice generated comparable chemokine responses to those seen in CCR1+/+ upon allografting but showed an approximate doubling in survival times of completely MHC-mismatched allografts (Table 1). Prolongation of graft survival was associated with a modest decrease in macrophage and T-cell recruitment, comparable to that seen with use of CsA in wild-type mice. These data were consistent with our in vitro evidence that CCR1–/– mice are immunocompetent even though their allogeneic proliferative responses were slightly less than in control mice (Figure 2). The latter difference in proliferative responses suggests a role for CCR1 other than that of purely chemokine-dependent cell recruitment. In the absence of blocking mAb’s to mouse CCR1, testing of this in murine MLR assays is not possible, but some clues are available. Using mouse CCR1 cell transfectants, we have recently shown that the main chemokine ligand for CCR1 in the mouse is MIP-1α; murine RANTES and MCP-3 did not show significant binding or induce chemotactic or other functional responses when used at physiological concentrations. Interestingly, MIP-1α is known to potentiate the activating effects of IFN-γ on mouse macrophage cytokine production (14), whereas Th2 cytokines including IL-4 (15), IL-10 (16), and TGF-β (17) each suppress MIP-1α production. Moreover, in humans, MIP-1α and RANTES can costimulate T-cell and T-cell clone proliferation and IL-2 production in the presence of CD3 mAb (18).
In contrast to wild-type controls, unmodified CCR1–/– mice accepted MHC class II–mismatched allografts permanently. Rejection in this model is dependent on IFN-γ production by alloresponsive CD4+ T cells (19, 20). Because we found that activated mouse Th1 cells express CCR1 (Figure 5), the failure of CCR1–/– recipients to reject bm12 cardiac allografts suggests a key role of CCR1 in initiating recruitment and/or activation of alloreactive IFN-γ–producing Th1 cells. Additional mechanisms serving to decrease alloresponse in CCR1–/– recipients could relate to the expression of CCR1 on macrophages and dendritic cells (21). However, the potent effect of CCR1–/– on host responses to allografts disparate for class II and possibly some non–H-2 genes points to a T cell–dependent event as critical. These data, reflecting that in the murine system CCR1 deletion affects more than macrophage functions alone, are likely relevant to the 2 additional key models investigated that explored use of the immunosuppressive agents, CsA and CD4 mAb, in CCR1–/– recipients of cardiac allografts.
A dosage of CsA of 10 mg/kg is well within the therapeutic range in patients and is typically decreased after transplant in order to achieve circulating trough levels of about 200 ng/mL. However, mice are quite resistant to CsA. The current experiments involved initial titering of CsA regimens (1.5, 2.5, 5, or 10 mg/kg per day) until any increase in cardiac allograft survival was achieved, and the lowest dose (10 mg/kg) at which this was achieved (2–3 days of prolongation in CCR1+/+ mice) (Table 1) was used thereafter. This regimen resulted in a remarkable synergism, with permanent engraftment and transferability of unresponsiveness, when using CsA in allografted CCR1–/– mice, and contrasts with our combinations in which CsA administration, a mainstay of clinical transplantation, can abrogate the beneficial effects of an otherwise effective therapeutic approach (22, 23). In CCR1–/– mice, CsA shut down or markedly suppressed intragraft production of almost all cytokines, chemokines, and their receptors (Figures 3b and 4) and led to essentially pristine allografts lacking cell infiltration (Figure 5) or deposition of humoral reactants. Our working hypothesis is that the alloantigen-induced activation of T cells from CCR1–/– mice is already somewhat compromised, and this is fully revealed in the presence of CsA, but the molecular basis for this, as well as the ability to transfer hyporesponsiveness to allografts in naive mice, remain to be elucidated.
Only fragmentary data from other systems are known as to the effects of CsA on chemokine production, and there are none involving effects of CsA on chemokine receptor expression. CsA was shown to block mitogen-induced production of the chemokines, IL-8 and IP-10, by human T cells (24), but had no effect on LPS-induced IL-8 production by human monocytes (25) and only modestly decreased production of IL-8 by LPS-treated alveolar macrophages (26). In contrast, CsA enhanced IL-1β–induced fibroblast production of IL-8 but decreased associated production of MCP-1 (27). The only data on the effects of CsA on chemokine production by cardiac cells arose from a study of cardiac myxoma cell in vitro in which their high level of constitutive production of IL-8 was completely inhibited by CsA (28). CsA is also known to inhibit production of lymphotactin (29), as was noted in the current study, although the significance of this chemokine in vivo remains unknown. Last, the immunosuppressive effects of CsA may be mediated, at least in part, via induction of TGF-β (30). Preliminary studies at day 5 did not show evidence of CsA-induced TGF-β production in our system (data not shown), although TGF-β expression beyond the initial period after transplant was not determined.
In direct contrast to control mice, which developed florid chronic rejection, CCR1–/– mice, in which long-term cardiac allograft survival was achieved by peritransplant therapy with a CD4 mAb, proved resistant to development of transplant arteriosclerosis or other stigmata of chronic allograft rejection (Figure 6). Rapid, vigorous recruitment of macrophages and T cells and thereafter, smooth muscle cells, to the intima of intracardiac vessels is a consistent feature of long-surviving allografts in this model (6). We have recently noted that rodent vascular smooth muscle cells can be induced by IFN-γ, TNF-α, and other stimuli to express CCR1 and display dose-dependent chemotactic responses to the CCR1 ligand, MIP-1α (31). Thus, the inability of CCR1 KO mice to develop transplant arteriosclerosis could potentially relate to failure of expression of CCR1 by host macrophages, T cells, or even vascular smooth muscle cells, to the extent that recruited smooth muscle cells have been shown, in some studies (32, 33), to include recipient-derived cells. In the clinical context, in which both recipient and donor express CCR1, CCR1 may be expressed by leukocytes and parenchymal cells. Indeed, this shared expression may represent a prime indication for use of CCR1 antagonists, as transplant arteriosclerosis is a hallmark of chronic rejection, and in the clinical setting, chronic rejection is the key limitation to long-term allograft survival, regardless of the use of various combinations of potent immunosuppressive agents, including CsA (34, 35).
In summary, a survey of the chemokines and chemokine receptors associated with development of cardiac allograft rejection in a well-established mouse model showed that intragraft induction of CCR1 ligands and CCR1 expression by host mononuclear cells were linked with acute rejection. Subsequent evaluation using CCR1–/– mice showed that targeting of CCR1 can delay rejection of completely MHC-disparate allografts and prevent rejection of class II–disparate grafts. Moreover, use of CCR1 targeting in conjunction with brief CsA or CD4 mAb therapy results in permanent engraftment and protection against development of chronic rejection. Questions remain as to which cell(s) expressing CCR1 is critical to development of allograft rejection and whether comparable effects to that seen using CCR1–/– mice can be achieved using anti-CCR1 mAb’s or small molecules that block ligand binding. However, we conclude from the current studies that targeting of CCR1 may have clinical application as a novel approach to clinical management of allograft recipients, including the prevention of chronic rejection.
Acknowledgments
This research was supported in part by a grant to W.W. Hancock from the National Institutes of Health (PO1-AI40152).
References
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Eitner F, Cui Y, Hudkins KL, Alpers CE. Chemokine receptor (CXCR4) mRNA-expressing leukocytes are increased in human renal allograft rejection. Transplantation. 1998;66:1551–1557. doi: 10.1097/00007890-199812150-00021. [DOI] [PubMed] [Google Scholar]
- 3.Eitner F, et al. Chemokine receptor (CCR5) expression in human kidneys and in the HIV infected macaque. Kidney Int. 1998;54:1945–1954. doi: 10.1046/j.1523-1755.1998.00211.x. [DOI] [PubMed] [Google Scholar]
- 4.Neote K, DiGregorio D, Mak JY, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell. 1993;72:415–425. doi: 10.1016/0092-8674(93)90118-a. [DOI] [PubMed] [Google Scholar]
- 5.Gerard C, et al. Targeted disruption of the beta-chemokine receptor CCR1 protects against pancreatitis-associated lung injury. J Clin Invest. 1997;100:2022–2027. doi: 10.1172/JCI119734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hancock WW, Buelow R, Sayegh MH, Turka LA. Antibody-induced transplant arteriosclerosis is prevented by graft expression of anti-oxidant and anti-apoptotic genes. Nat Med. 1998;4:1392–1396. doi: 10.1038/3982. [DOI] [PubMed] [Google Scholar]
- 7.Gao ZH, et al. Adoptively transferable tolerance induced by CD45RB monoclonal antibody. J Am Soc Nephrol. 1999;10:374–381. doi: 10.1681/ASN.V102374. [DOI] [PubMed] [Google Scholar]
- 8.Mottram PL, Han WR, Purcell LJ, McKenzie IFC, Hancock WW. Increased expression of IL-4 and IL-10 and decreased expression of IL-2 and interferon-g in long-surviving mouse heart allografts after brief CD4-monoclonal antibody therapy. Transplantation. 1995;59:559–565. [PubMed] [Google Scholar]
- 9.Breitschopf, H., and Suchanek, G. 1996. Detection of mRNA on paraffin embedded material of the central nervous system with DIG-labeled RNA probes. In Nonradioactive in situ hybridization application manual. Boehringer Mannheim Biochemicals. Vienna, Austria. 1–52.
- 10.Smiley ST, Boyer SN, Heeb MJ, Griffin JH, Grusby MJ. Protein S is inducible by interleukin 4 in T cells and inhibits lymphoid cell procoagulant activity. Proc Natl Acad Sci USA. 1997;94:11484–11489. doi: 10.1073/pnas.94.21.11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 12.Post TW, et al. Molecular characterization of two murine eosinophil beta chemokine receptors. J Immunol. 1995;155:5299–5305. [PubMed] [Google Scholar]
- 13.Smith JD, Rose ML, Pomerance A, Burke M, Yacoub MH. Reduction of cellular rejection and increase in longer-term survival after heart transplantation after HLA-DR matching. Lancet. 1995;346:1318–1322. doi: 10.1016/s0140-6736(95)92341-1. [DOI] [PubMed] [Google Scholar]
- 14.Fahey TJ, et al. Macrophage inflammatory protein 1 modulates macrophage function. J Immunol. 1992;148:2764–2769. [PubMed] [Google Scholar]
- 15.Berkman N, et al. Inhibition of macrophage inflammatory protein-1 alpha expression by IL-10: differential sensitivities in human blood monocytes and alveolar macrophages. J Immunol. 1995;155:4412–4418. [PubMed] [Google Scholar]
- 16.Standiford TJ, et al. Macrophage inflammatory protein-1 alpha from human blood monocytes and alveolar macrophages is inhibited by interleukin-4. Am J Respir Cell Mol Biol. 1993;9:192–198. doi: 10.1165/ajrcmb/9.2.192. [DOI] [PubMed] [Google Scholar]
- 17.Maltman J, Pragnell IB, Graham GJ. Specificity and reciprocity in the interactions between TGF-beta and macrophage inflammatory protein-1 alpha. J Immunol. 1996;156:1566–1571. [PubMed] [Google Scholar]
- 18.Taub DD, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. Chemokines and T lymphocyte activation. I. beta chemokines costimulate human T lymphocyte activation in vitro. J Immunol. 1996;156:2095–2103. [PubMed] [Google Scholar]
- 19.Tomura M, et al. Suppression of allograft responses induced by interleukin-6, which selectively modulates interferon-gamma but not interleukin-2 production. Transplantation. 1997;64:757–763. doi: 10.1097/00007890-199709150-00017. [DOI] [PubMed] [Google Scholar]
- 20.Ring GH, et al. Interferon-gamma is necessary for initiating the acute rejection of major histocompatibility complex class II-disparate skin allografts. Transplantation. 1999;67:1362–1365. doi: 10.1097/00007890-199905270-00012. [DOI] [PubMed] [Google Scholar]
- 21.Foti M, et al. Upon dendritic cell (DC) activation chemokines and chemokine receptor expression are rapidly regulated for recruitment and maintenance of DC at the inflammatory site. Int Immunol. 1999;11:979–986. doi: 10.1093/intimm/11.6.979. [DOI] [PubMed] [Google Scholar]
- 22.Larsen CP, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996;381:434–438. doi: 10.1038/381434a0. [DOI] [PubMed] [Google Scholar]
- 23.Kirk AD, et al. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in non-human primates. Nat Med. 1999;5:686–693. doi: 10.1038/9536. [DOI] [PubMed] [Google Scholar]
- 24.Zipfel PF, Bialonski A, Skerka C. Induction of members of the IL-8/NAP-1 gene family in human lymphocytes-T is suppressed by Cyclosporin-A. Biochem Biophys Res Commun. 1991;181:179–183. doi: 10.1016/s0006-291x(05)81398-1. [DOI] [PubMed] [Google Scholar]
- 25.Mrowietz U, Sticherling M, Mielke V, Schroder JM, Christophers E. Neutrophil-activating peptide 1/interleukin 8 mRNA expression and protein secretion by human monocytes: effect of cyclosporin A. Cytokine. 1991;3:322–326. doi: 10.1016/1043-4666(91)90500-d. [DOI] [PubMed] [Google Scholar]
- 26.Losa Garcia JE, et al. Effect of cyclosporin A on inflammatory cytokine production by human alveolar macrophages. Respir Med. 1998;92:722–728. doi: 10.1016/s0954-6111(98)90002-6. [DOI] [PubMed] [Google Scholar]
- 27.Burnstine MA, Elner SG, Elner VM. Orbital fibroblast chemokine modulation: effects of dexamethasone and cyclosporin A. Br J Ophthalmol. 1998;82:318–322. doi: 10.1136/bjo.82.3.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakamoto H, et al. Immunosuppressive drugs inhibit the production of interleukin-6 and interleukin-8 in cultured cardiac myxoma cells. Res Commun Mol Pathol Pharmacol. 1997;97:60–66. [PubMed] [Google Scholar]
- 29.Wang JD, et al. Lymphotactin: a key regulator of lymphocyte trafficking during acute graft rejection. Immunology. 1998;95:56–61. doi: 10.1046/j.1365-2567.1998.00570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khanna A, Li BG, Stenzel KH, Suthanthiran M. Regulation of new DNA synthesis in mammalian cells by cyclosporine: demonstration of a transforming growth factor beta-dependent mechanism of inhibition of cell growth. Transplantation. 1994;57:577–582. [PubMed] [Google Scholar]
- 31.Hancock WW, Kusaka M, Mackenzie HS, Tilney NL. Hypertension induces transplant arteriosclerosis in rat renal allografts via endothelin and chemokine receptor induction by vascular smooth muscle cells. Transplantation. 1999;67:S574. (Abstr.) [Google Scholar]
- 32.Kennedy LJ, Weissman IL. Dual origin of intimal cells in cardiac-allograft arteriosclerosis. N Engl J Med. 1971;285:884–887. doi: 10.1056/NEJM197110142851603. [DOI] [PubMed] [Google Scholar]
- 33.Vischjager M, Van Gulik TM, Van Marle J, Pfaffendorf M, Jacobs MJ. Function of cryopreserved arterial allografts under immunosuppressive protection with cyclosporine A. J Vasc Surg. 1996;24:876–882. doi: 10.1016/s0741-5214(96)70026-5. [DOI] [PubMed] [Google Scholar]
- 34.Tilney NL. Chronic rejection and its risk factors. Transplant Proc. 1999;31:41S–44S. doi: 10.1016/s0041-1345(98)02080-6. [DOI] [PubMed] [Google Scholar]
- 35.Hancock WW. Molecular basis of chronic rejection. Curr Opin Organ Transplant. 1999;4:3–10. [Google Scholar]