

Abstract
Objective:
To find out whether linarin can be used as a potential natural inhibitor to target CDK4 in retinoblastoma using virtual screening studies.
Materials and Methods:
In this study, molecular modeling and protein structure optimization was performed for crystal structure of CDK4 (PDB id: 3G33), and was subjected to Molecular Dynamics (MD) simulation for 10 nanoseconds, as a preparatory process for docking. Furthermore, the stable conformation obtained in the MD simulation was utilized for virtual screening against the library of natural compounds in Indian Plant Anticancer Compounds Database (InPACdb) using AutoDock Vina. Finally, best docked ligands were revalidated individually through semi-flexible docking by AutoDock 4.0.
Results:
The CDK4 structure was stereochemically optimized to fix clashes and bad angles, which placed 96.4% residues in the core region of Ramachandran plot. The final structure of CDK4 that emerged after MD simulation was proven to be highly stable as per different validation tools. Virtual screening and docking was carried out for CDK4 against optimized ligands from InPACdb through AutoDock Vina. This inferred Linarin (Inpacdb AC.NO. acd0073) as a potential therapeutic agent with binding energy of -8.9 kJ/mol. Furthermore, it was also found to be valid as per AutoDock 4.0 semi-flexible docking procedure, with the binding energy of -8.18 kJ/mol and Ki value of 1.01 μM.
Conclusion:
The docking results indicate linarin, a flavonoid plant compound, as a potential inhibitor of CDK4 compared to some of the currently practiced anticancer drugs for retinoblastoma. This finding can be extended to experimental validation to assess the in vivo efficacy of the identified compound.
Keywords: Cyclin-dependent kinase 4, InPACdb, molecular docking, molecular dynamics, retinoblastoma, virtual screening
INTRODUCTION
The cyclin-dependent kinases (CDKs) are a conserved family of proline-directed serine/threonine kinases that play a key role in regulation of cell cycle in eukaryotic cells.[1] The activity of CDKs is regulated most significantly through interaction with cyclins. CDK family is known to have a high level of structural homology, especially CDK2 shares 45% sequence identity with CDK4.[2] The resemblance has helped spearhead research involving design of inhibitors for CDK4, mimicking the CDK2 inhibitors. CDK4 associates with D-type cyclins (D1, D2, and D3), phosphorylates and inactivates the retinoblastoma protein family members (p107, p130, pRb).[1] In human beings, pRb is encoded by the RB1 gene located on 13q14.1-q14.2. Phosphorylation of pRb by CDK4 inactivates pRb, causing the activation of E2F target genes, which facilitates progression through the G1 phase of the cell cycle, eventually leading to the condition of retinoblastoma.[1]
Developing and identifying small molecule inhibitors targeting CDK4 has been a prime area of research since past two decades. Some of the drugs that have been proven as desirable targets for cancer are flavopiridol, butyrolactone, the purine-based olomoucine and its analogues, arcyriaflavin A, and isoquinolinedione analogues.[2] Flavopiridol, the first CDK inhibitor for human beings derived from an indigenous plant from India, demonstrated potent and specific in vitro inhibition of all CDKs tested (CDKs 1, 2, 4, and 7), clearly blocking the cell cycle progression at the G1/S and G2/M boundaries.[3] However, flavopiridol is not selective for CDK4. It inhibits other CDKs to almost the same extent as CDK4.[4] In similar lines, most CDK inhibitors suffer from low selectivity. The low selectivity of these chemical compounds may cause adverse side effects.[5,6] This brings up the need for more potent and selective CDK4 inhibitors with better efficiency for the condition of retinoblastoma. Natural compounds have their advantage over chemical inhibitors by being a safer substitute and ensure lesser/no side effects. A library of 125 natural compounds was taken from Indian Plant Anticancer Compounds Database (InPACdb) (http://www.inpacdb.org/)[7] to test them for their efficiency as therapeutic agent for CDK4.
This study aims to find out whether linarin can be used as a potential natural inhibitor to target CDK4 in retinoblastoma using virtual screening studies
MATERIALS AND METHODS
Molecular optimization was carried out for crystal structure of CDK4 as a preparative process for docking. The structure was fixed for stereochemical clashes and bumps. The modeled protein was then subjected to Molecular Dynamics (MD) simulation. The decoy having lowest potential energy was chosen from MD simulation, and was found to be valid as per many validation tools. Furthermore, the optimal structure was subjected to Virtual screening using the compounds from InPACdb, which were optimized using LigPrep, then, docking simulation was performed. On stringent scrutinization, one of the compounds emerged to show reliable inhibitory activity as expected for a CDK4 inhibitor.
The coordinates of CDK4 was downloaded from Protein Data Bank (PDBid: 3G33).[8] The sequence for the same was retrieved from Uniprot[9]; UniprotID: P11802. The unphosphorylated model of CDK4 (3G33:Chain A) was modeled using Modeller 9v8[10] and was refined to fix steric clashes and bumps using WHAT IF server.[11] To draw the initial starting models closer to their native state in terms of hydrogen bonds, side-chain positioning, and backbone topology, Modrefiner was used.[12] The output of the same was taken for loop refinement to obtain a structure which when evaluated using PROCHECK[13] placed 0.0% residues in the disallowed regions of Ramachandran plot. The refined protein was further subjected to MD simulation using GROMACS4.3.5[14] with GROMOS96 43a2 force field.[15] The system was solvated in a cubical box with SPCE waters and counter ions (2 Na+) were added to neutralize the charge of each system. The system was energy minimized by steepest decent for 1 000 steps. NVT and NPT steps were processed as a part of equilibration phase for 100ps each, after which MD simulation was initiated to be run for 10ns.[15]
The stabilized protein was validated using various tools such as Charmm-gui,[16] PatchFinderPlus,[17] Protein Quality Predictor (ProQ),[18] ProSA,[19,20] Predyflexy,[21] Psipred,[22] and Qsitefinder[23] for analyzing electrostatic property, protein flexibility, and binding site analysis. CDK4 is known to have ATP-binding region between 93 and 102 residues. Various groups have reported drug designs targeting the same pocket.[2]
KinDOCK server was used to provide both potential ligands and their putative binding orientation for our optimized protein.[24] The top ten ligands binding favorably to the ATP-binding region of CDK4 were taken, and the favorable interactions made at the binding site were noted and analyzed with the data acquired from the previous works, as listed in Table 1. Virtual screening was carried out with the 125 natural compounds from InPACdb.[7] The compounds were optimized using LigPrep[25] module of Schrodinger, then were docked to our protein of interest using AutoDock Vina module through Pyrx interface.[26] Based on binding energy as the parameter, top 10 ligands having lower binding energy values were chosen. Purine-based analogues emerged as the basic structure to mimic in our drug, on analyzing the structure of flavopiridol, isoquinolinedione analogues.[6,27,28] This gave us five ligands out of ten which mimicked purine-like analogues, for which semi-flexible docking was performed with CDK4 using AutoDock 4.0.[29] Ten conformers were generated for each of these best five CDK4 inhibitors. Binding energy, Ki value, and Hydrogen bonding interactions with the protein were used as the criteria for filtering the best inhibitor. Pymol (http://www.pymol.org/) and Accelrys Discovery Visualizer[30] were used to view and interpret the Protein-Ligand interactions.
Table 1.
List of CDK4 active site residues
RESULTS
Protein structure refinement
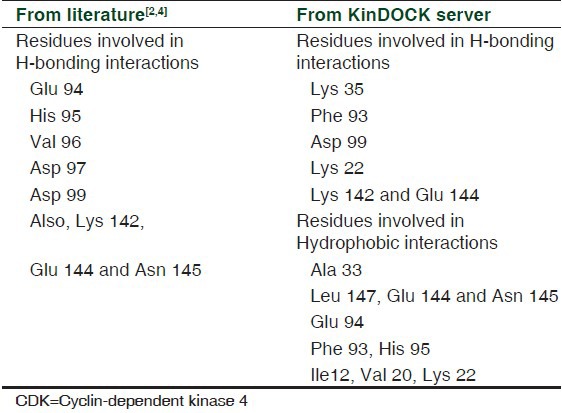
3G33: Chain A was taken for homology modeling using Modeller 9v8. Single template modeling was performed on 3G33: A as the template, with CDK4 sequence as the target. The protein was then fixed for bumps and clashes using WHAT IF server. Modrefiner was used to generate significant improvement in physical quality of local structures by drawing the initial model closer to native state in terms of hydrogen bonds, side-chain positioning, and backbone topology. The structure that was produced after refinement had Cys135 in disallowed region. This brought in the need for loop refinement to be carried out. The loop-refined model placed 96.4% of all residues in the allowed regions with no residues in the disallowed region of Ramachandran Plot as shown in Figure 1. The model after being fixed of stereochemical clashes was taken to be stabilized by running MD simulation.
Figure 1.
(a) Refined structure of the CDK4 protein, where helix is shown in red, yellow indicates beta sheets, and coil is shown in green color. (b) Ramachandran Plot of the protein after structural refinement
Molecular dynamics simulation
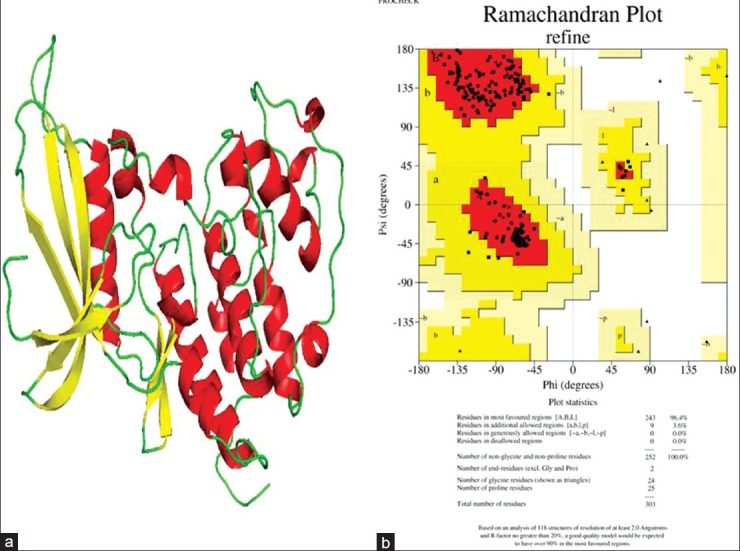
The refined protein was subjected to MD simulation using GROMACS 4.3.5 with GROMOS96 43a2 force field. The system was solvated in a cubical box with SPCE waters containing 23260 molecules and 2 Na+ ions were added to neutralize the charge of each system. The system was energy minimized by steepest decent method for 1000 steps, after which the potential energy of the system dropped from -1.25970e + 06 to -9.28789e + 05. Position restraint was performed using NVT and NPT steps that were processed as a part of equilibration phase for 100ps each, after which MD simulation was initiated to be run for 10ns. For analyzing simulation results, Xmgrace program (http://plasma-gate.weizmann.ac.il/Grace/.) was used to generate root mean square deviation (RMSD), potential energy, radius of gyration, and solvent-accessible surface graphs. RMSD graph shows a steep incline in the deviation starting from 1Å to 3.9Å, which stabilizes at around 3.5Å, and it drops to a minimum of around 3Å at 6800ps. The deviation pattern then retraces 3.5Å level till 10ns, as shown in Figure 2a. Potential energy traces a reliable drop in value over 10ns of MD simulation, with the lowest potential energy structure observed at 6820ps as shown in Figure 2b. Radius of gyration is indicative of compactness of the protein. There is an observable drop in its value from 2.7 to 2 nm, as shown in Figure 2c. The lowest value of radius of gyration at around 6820ps is indicative of the high degree of compactness observed in the protein in its lowest energy state. Overall decrease in radius of gyration from 2.065 to 1.99 nm shows the increase in compactness of the protein after 10ns of simulation. Solvent-accessible surface area indicates the residue's exposure to solvent. The protein in its lowest potential energy shows a dip in hydrophobicity levels (increase in hydrophilicity), clearly indicating that the protein converges in shape over time during simulation, as shown in Figure 2d. Pymol was used to visualize the 3D structure of the simulated protein. Starting with the structure taken for simulation, the binding pocket cavity change in the structure was analyzed for 10ns with an interval time of 1ns. ATP-binding site's flexibility increased with time, as shown in Figures 3a and b. The protein with the lowest potential energy (at 6 820ps) was taken as the final CDK4 structure for further studies, as shown in Figure 3b.
Figure 2.
(a) The RMSD graph, with deviation stabilizing close to 3.25Å at the end of 10ns. Overall deviation in rmsd over 10ns was observed close to 2.25Å. X-axis shows time in ps and Y-axis indicates RMSD in nm. (b) potential energy graph over 10ns. The lowest potential energy was noted at 6820ps, marked with a red circle. X-axis shows time in ps and Y-axis indicates potential energy value in kJ/mol. (c) Solvent-accessible surface graph, showing the drop in hydrophobicity near the structure with lowest potential energy. X-axis shows time in ps and Y-axis indicates Area in nm. (d) Radius of gyration graph, which drops in value by a good margin indicating the structure converging in shape acquiring compactness with time. X-axis shows time in ps and Y-axis indicates Radius of gyration in nm
Figure 3.
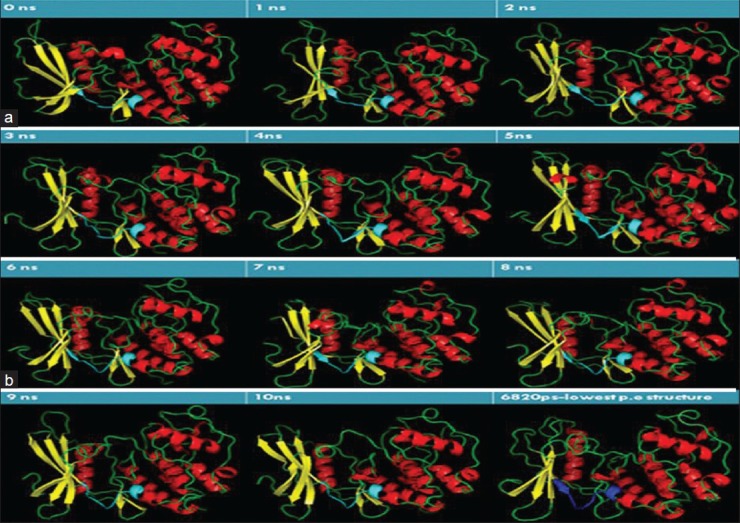
(a) ATP-binding site cavity change observed over 5ns, with interval of 1ns. The binding site cavity (shown in Cyan) converges in shape over time. Helix is shown in red, yellow indicates sheets, and coil is shown in green color. (b) ATP-binding site cavity change observed 6-10ns, with interval of 1ns. The binding site cavity (shown in Cyan) converges in shape over time. The structure with lowest potential energy assumes a compact figure as shown. The ATP-binding site in the structure with lowest potential energy is shown in blue. Helix is shown in red, yellow indicates sheets, and coil is shown in green color
Validating structure of simulated protein
To validate the structure of the simulated protein, various tools were used to check the electrostatic properties, binding site flexibility, and interactions involved in the active site.
Protein electrostatics
The PBEQ Solver of Charmm-gui solver helped interactively to visualize the calculated electrostatic potential on the solvent-accessible surface as well as isoelectrostatic potential contours of the stabilized protein molecule by solving the Poisson-Boltzmann (PB) equation. The positive charge on the ligand was important for improving selectivity for CDK4, because of the electronegative nature of the active site.[16] The active site was confirmed to be electronegative in nature with the red patch spread over the residues in the ATP-binding region, as shown in the Figure 4a.
Figure 4.
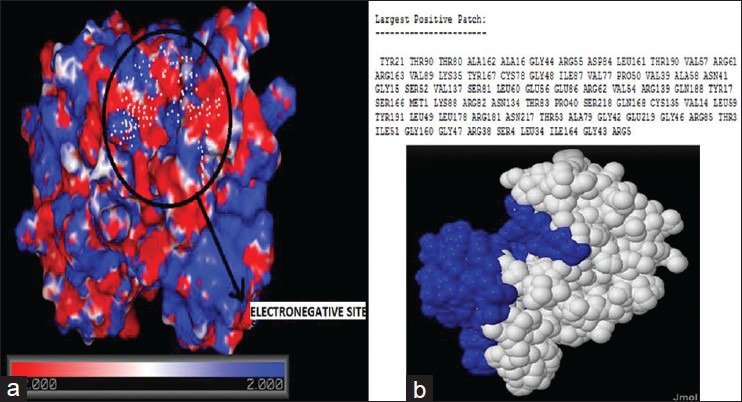
(a) The electrostatic potential map generated using charmm force field using charm-gui. The region encircled in black shows the electronegative binding pocket. Different shades of red shows varying levels of electronegativity of the residues, white color indicates residues with neutral charge, and different shades of red shows varying levels of electropositivity of the residues. (b) the results of patch finder plus that displays the largest positive patch (indicated in blue) in the protein. The largest positive patch does not include the residues in the binding site
The Pathfinder algorithm calculates the PB electrostatic potential of the protein and constructs the largest continuous positive patch on the protein surface. Clarity of result was established by the absence of positively charged residues in the protein's binding site, as shown in the Figure 4b.
Quality prediction using protein quality predictor and ProSA
ProQ is a neural network-based predictor of protein's structure quality based on a number of structural features. LGscore and MaxSub are the quality measures that are used. LG score attempts to minimize P value for distribution, while Maxsub attempts to maximize Levitt Gerstein score. As predicted for a very good model, the value of LG score was 5.380 and that of MaxSub was 0.612. Proteins that contain long target sequences are more likely to get good LG score, while the sequences that are short by length are more likely to get a good MaxSub score, which is well correlated with our result.
ProSA calculates the overall quality score for a specific input structure by displaying in a plot, the score of a specific model that relates to the scores computed from all experimental structures deposited in PDB. Z-score with higher negative value is indicative of a reliable result. Z-score of our model showed a value of -8.24 post-simulation, with the structure appearing close to X-ray region on the plot.
Residue-wise stability prediction
Predyflexy Server examines flexibility according to two different descriptors, the B-factor and root mean square fluctuations, from MD simulations. It defines three flexibility classes and proposes a method based on the LSP (Line Spectrum Pair) prediction method for predicting flexibility along the sequence. Predyflexy Server produced the protein with good flexibility in the active site region. Predicted flexibility class is indicated by 0, 1, and 2, with 0 being the least flexible and 2, the most flexible. Most of the residues in the ATP-binding site shared moderately and highly flexible residues with high confidence index, as shown in the Figure 5a.
Figure 5.
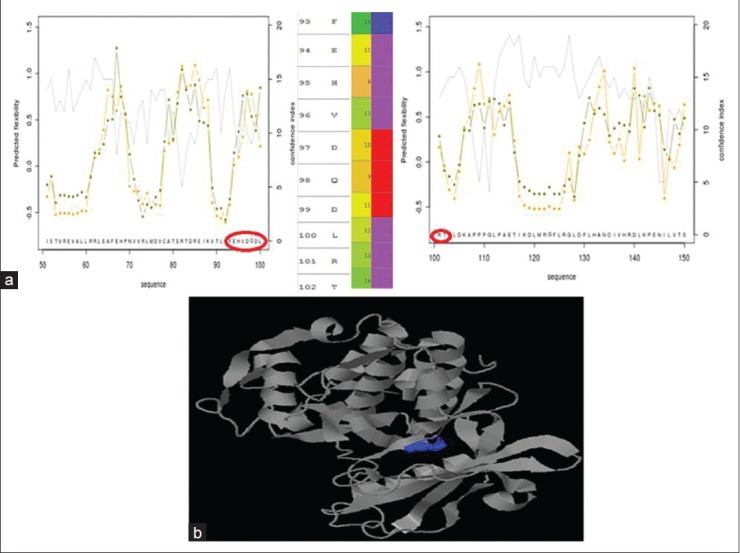
(a) Predicted flexibility score as calculated by Predyflexy server (shown in Y-axis) for residues in the protein (shown in Y-axis). ATP-binding site residues show good flexibility, which is shown in the zoomed image. First column indicates the residue number and the corresponding residue. Second column shows confidence index of prediction and the third column, indicated by values 0, 1, and 2, is indicated by degree of flexibility of the residue in the increasing order of magnitude. (b) the output of Q-SiteFinder, with various plausible binding sites indicated in different colors. The toggled surface marked in blue is the required binding site. The structure shown in grey is the protein structure
Structure reliability check using PSIPRED
PSIPRED, a highly accurate method for protein secondary structure prediction strategy, works by observing that most conserved regions of protein sequence are the ones which are functionally important and those that are buried in protein core, and maximum variability is assumed to be in the residues forming the surface of protein. PSIPRED results showed several stretches of residues that showed deviation from the model pre-simulation; and the change in the model, post-simulation, was predicted as being positive in nature with high confidence. The structure predicted by PSIPRED well correlated with the active site region with the helix patch-LRT showing high confidence prediction by PSIPRED.
Binding site analysis
Q-SiteFinder is a tool that helps us locate the energetically favorable binding sites by calculating the energy between the protein molecule and simple van der Waals probe. This tool predicts various plausible sites where the protein can favorably bind, considering protein electrostatics and spatial proximity. The sites are displayed with information of site volume, volume of the protein, precision of prediction, and the residues involved in interaction. The key residues involved in interaction were noted to be Glu94, His95, Val96, Asp97, and Asp99. Also, the residues Lys142, Glu 144, and Asn 145 were known to play important roles by contributing to the stability of the protein-ligand complex.[27,29] Q site finder predicted site no: 4, with site volume of 223Å as the desired pocket to target the ligand on. The residues Ile12, Val20, Lys22, Ala33, Lys35, Phe93, His95, Asp99, Glu144, Asn145, Ile146, Leu147, Ala157, and Ala170 made the binding pocket, displayed in dark blue color, as shown in Figure 5b. There was a heavy dip in the active site volume when compared to that of observed pre-simulation. However, the key residues required for interaction in the active site remained conserved.
KinDOCK server extracts a structural library of protein kinase-ligand complexes that provide both potential ligands and their putative binding orientation for a given protein kinase. Protein–protein structural superposition is performed, following which the ligands from the template complexes are taken as a potential ligand. SCORE software was used as the basis of evaluating the best complexes for computing theoretical affinity. Top ten best hits were taken based on the program score, and their binding interactions with the ligand were analyzed and noted. There was a pattern observed in the type of inhibitor designed for CDK4; the best hits were largely composed of adenine-based inhibitors. Lys22, Lys35, Phe93, Asp99, Lys142, and Glu144 were found to show good H-bonding interactions, while Ile12, Val20, Lys22, Ala33, Phe93, Glu94, His95, Glu144, Asn145, and Leu147 were shown to be key residues involved in hydrophobic interactions, as mentioned in Table 1.
Purine-based analogues largely dominated as the potential analogues for CDK4. Flavopiridol, the first CDK4 inhibitor and substituted isoquinoline-1,3-(2H,4H)-diones, a recently discovered CDK4 inhibitor were among the few reliable inhibitors for CDK4 that had its structure analogous to adenine moiety containing a purine ring.
Virtual screening and docking studies
After validating the model of the simulated protein, it was taken to be docked against a library of natural compounds. InPACdb containing 125 natural compounds were optimized using Ligprep module of Schrodinger (Version 9.0), before it was docked with the simulated structure of CDK4.
AutoDock Vina was used through PyRX interface to perform molecular docking and virtual screening, offering partial receptor flexibility providing high performance and accuracy of results. The ATP-binding site (residues-93-102) was set as flexible and Vina search space was centered around (9.8395, 46.9036, 40.9514) and the dimensions of the grid box were set to (27.2071, 28.7208, 25.0000). The docking results comprised binding energy value given in kJ/mol, as shown in Figure 6a. Top ten ligands with respect to binding energy score were taken for analysis. Five of these top 10 ligands were seen to compose purine-mimicking structure. The compound that emerged as the best among these natural inhibitors showed a binding energy value of -8.9 kJ/mol.
Figure 6.
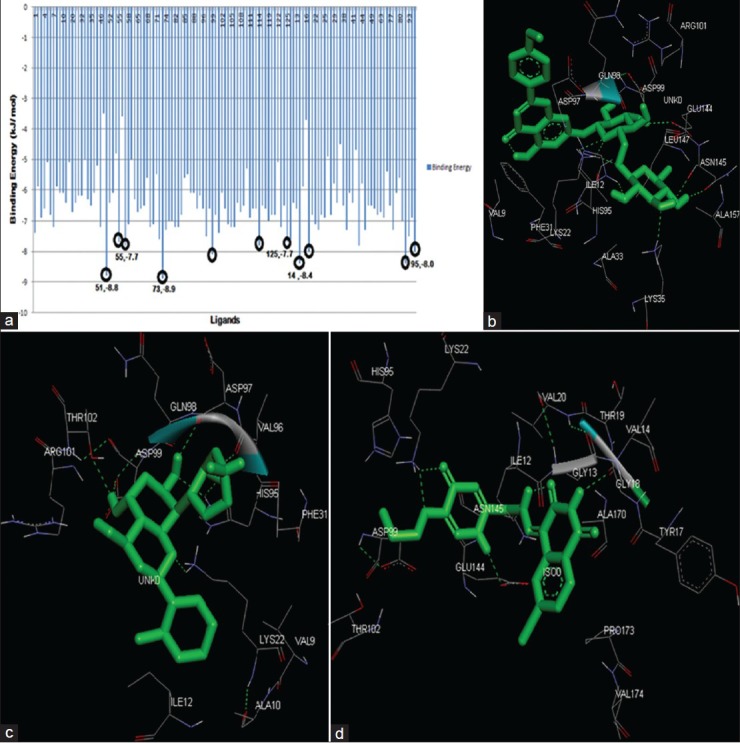
(a) The output of AutoDock Vina. The Lines encircled in black show the top ten ligands (X-axis) from the results in terms of binding energy value given in kJ/mol (Y-axis). Five ligands mimicking the structure purine analogues is numbered as (ligand number, binding energy). (b) Interactions at the binding site between CDK4 protein and Linarin. (c) Interactions at the binding site between CDK4 protein and Flavopiridol. (d) Interactions at the binding site between CDK4 protein, an Isoquinolinedione. The structure in green is the ligand molecule. The green dotted lines indicate H-bond interactions between the residue and ligand. The residues surrounding the ligands are the ones involved in hydrophobic interaction with it
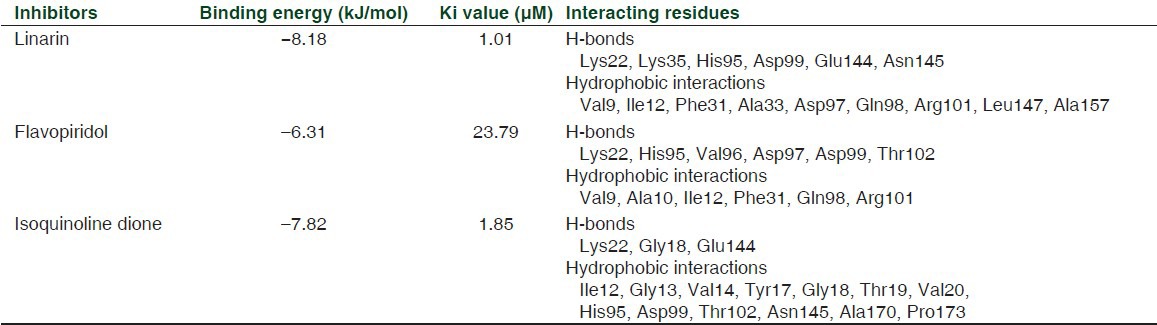
The five purine-mimicking inhibitors were revalidated individually using AutoDock 4.0 together with Flavopiridol and substituted Isoquinolinedione. Each of the top five natural inhibitors were docked using semi-flexible docking following Lamarckian G.A algorithm, and generated 10 favorable conformations each at the end of every docking run. Grid box was set to dimension- 62 × 62 × 62 with coordinates of 41.734, 28.76, and 66.937. The analysis of docking results showed protein-Linarin complex to have the best binding energy and Inhibitor constant of -8.18 kJ/mol and 1.01 μM, respectively (as tabulated), correlating with AutoDock Vina result. The highly flexible and long side chain of Linarin contributed to its 13 rotatable bonds and high rotatable torsions. This complex was studied for interactions in the binding pocket. The residues Lys22, Lys35, His95, Asp99, Glu144, and Asn145 were involved in H-bonding interaction and Val9, Ile12, Phe31, Ala33, Asp97, Gln98, Arg101, Leu147, and Ala157 where shown to be involved in hydrophobic interactions. A comparative study was done between Linarin, Flavopiridol, and Isoquinolinedione after docking was performed, the results of which are tabulated in Table 2. The interactions involved in the active site region between the protein and these three inhibitors are individually displayed in [Figure 6b–d].
Table 2.
Final list of shortlisted compounds as per AutoDock 4.0 validation
DISCUSSION
A detailed perspective from a molecular viewpoint has been provided regarding the suitability of Linarinas, the effective inhibitor of the protein kinase, CDK4. Complying to the stringent parameters of the docking programs, a reliable binding energy of -8.9 kJ/mol was obtained on using AutoDock Vina and further validation of the result using AutoDock 4.0 gave binding energy value of -8.18 kJ/mol. Inhibitory constant value was 1.01 μM, better than the values shown by Flavopiridol and substituted isoquinolinedione. Furthermore, CDK4 docking interaction with Flavopiridol, substituted isoquinolinedione and Linarin at the ATP-binding were compared, wherein, the stability of interaction was observed to be better in Linarin in comparison with its counterparts. Moreover, several potential CDK4 inhibitors were observed to be Purine-based analogues and so was Linarin. Also, acd0073 (Linarin) incidentally happened to fall under the class of Flavonoids.
Flavonoids, apart from being well known for their antioxidant activity in vitro, has long been investigated for their potential as viable anti-cancer agents.[31] Flavopiridol, the first CDK inhibitor in human beings, belonging to the family of flavonoids, was found to show activity for renal, prostate, colon, and gastric carcinomas.[3] Linarin sharing the same family characteristics was shown to have potency to serve as an inhibitor for human prostate cancer. Linarin has shown good sensitivity to cell cycle arrest in G1 phase.[31] The acquired results could extend its effectiveness as a reliable CDK4 inhibitor for the condition of retinoblastoma.
ACKNOWLEDGMENT
The authors sincerely thank SASTRA UNIVERSITY for granting access to use LigPrep module of Schrodinger.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Day PJ, Cleasby A, Tickle IJ, O’Reilly M, Coyle JE, Holding FP, et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc Natl Acad Sci U S A. 2009;106:4166–70. doi: 10.1073/pnas.0809645106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ikuta M, Kamata K, Fukasawa K, Honma T, Machida T, Hirai H, et al. Crystallographic approach to identification of cyclin-dependent kinase 4 (CDK4)-specific inhibitors by using CDK4 mimic CDK2 protein. J Biol Chem. 2001;276:27548–54. doi: 10.1074/jbc.M102060200. [DOI] [PubMed] [Google Scholar]
- 3.Senderowicz AM. Flavopiridol: The first cyclin-dependent kinase inhibitor in human clinical trials. Invest New Drugs. 1999;17:313–20. doi: 10.1023/a:1006353008903. [DOI] [PubMed] [Google Scholar]
- 4.Ishimaru T, Lau J, Jackson AL, Modiano JF, Weiss RH. pharmacological inhibition of cyclin dependent kinases causes p53 dependent apoptosis in renal cell carcinoma. J Urol. 2010;184:2143–9. doi: 10.1016/j.juro.2010.06.088. [DOI] [PubMed] [Google Scholar]
- 5.Shchemelinin I, Sefc L, Necas E. protein kinase inhibitors. Folia Biol (Praha) 2006;52:137–48. [PubMed] [Google Scholar]
- 6.Lu XY, Chen YD, Sun NY, Jiang YJ, You QD. Molecular-docking-guided 3D-QSAR studies of substituted isoquinoline-1,3-(2 H, 4 H)-diones as cyclin-dependent kinase 4 (CDK4) inhibitors. J Mol Model. 2010;16:163–73. doi: 10.1007/s00894-009-0529-7. [DOI] [PubMed] [Google Scholar]
- 7.Umashankar V, Nanditha Subramaniam, Kalabharath Indian Plant Anticancer Compound Database (InPACdb) Bioinformation. 2009:71–4. doi: 10.6026/97320630004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takaki T, Echalier A, Brown NR, Hunt T, Endicott JA, Noble ME. The structure of CDK4/cyclin D3 has implications for models of CDK activation. Proc Natl Acad Sci U S A. 2009;106:4171–6. doi: 10.1073/pnas.0809674106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. [Last accessed on 2012 Jan 17]. Available from: http://www.uniprot.org/uniprot/P11802 .
- 10.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen M.-y, Pieper U, Sali A. Comparative Protein Structure Modeling Using MODELLER. Current Protocols in Protein Science. 2007;50:2.9.1–2.9.31. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 11.Vriend G. WHAT IF: A molecular modeling and drug design program. J Mol Graph. 1990;8:52–6. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 12.Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J. 2011;101:2525–34. doi: 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–67. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 14.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS4: Algorithms for highly efficient, Load-Balanced, and scalable Molecular Simulation. J Chem Theory Comp. 2008;4:435–47. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 15.Mascarenhas NM, Bhattacharyya D, Ghoshal N. Why pyridine containing pyrido[2,3-d] pyrimidin-7-ones selectively inhibit CDK4 than CDK2: Insights from molecular dynamics simulation. J Mol Graph Model. 2010;28:695–706. doi: 10.1016/j.jmgm.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 16.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J ComputChem. 2008;29:1859–65. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 17.Shazman S, Celniker G, Haber O, Glaser F, Mandel-Gutfreund Y. Patch Finder Plus (PFplus): A web server for extracting and displaying positive electrostatic patches on protein surfaces. Nucleic Acids Res. 2007;35:W526–30. doi: 10.1093/nar/gkm401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallner B, Elofsson A. Can correct protein models be identified? Protein Sci. 2003;12:1073–86. doi: 10.1110/ps.0236803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiederstein M, Sippl MJ. Pro SA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–10. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–62. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 21.Bornot A, Etchebest C, de Brevern AG. Predicting protein flexibility through the prediction of local structures. Proteins. 2011;79:839–52. doi: 10.1002/prot.22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–5. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 23.Laurie AT, Jackson RM. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–16. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 24.Martin L, Catherinot V, Labesse G. KinDOCK: A tool for comparative docking of protein kinase ligands. Nucleic Acids Res. 2006;34:W325–9. doi: 10.1093/nar/gkl211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.New York, NY: Schrodinger, LLC; 2011. Lig Prep, version 2.5. [Google Scholar]
- 26.Trott O, Olson AJ. Auto Dock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J ComputChem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veselý J, Havlicek L, Strnad M, Blow JJ, Donella-Deana A, Pinna L, et al. Inhibition of cyclin-dependent kinases by purine analogues. Eur J Biochem. 1994;224:771–86. doi: 10.1111/j.1432-1033.1994.00771.x. [DOI] [PubMed] [Google Scholar]
- 28.Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ. Flavopiridol induces G1 arrest with inhibition of cyclin-dependent kinase (CDK) 2 and CDK4 in human breast carcinoma cells. Cancer Res. 1996;56:2973–8. [PubMed] [Google Scholar]
- 29.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28:1145–52. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 30.San Diego: Accelrys Software Inc; 2011. Accelrys Software Inc. Discovery Studio Modeling Environment, Release 3.1. [Google Scholar]
- 31.Singh RP, Agrawal P, Yim D, Agarwal C, Agarwal R. Acacetin inhibits cell growth and cell cycle progression, and induces apoptosis in human prostate cancer cells: Structure-activity relationship with linarin and linarin acetate. Carcinogenesis. 2005;26:845–54. doi: 10.1093/carcin/bgi014. [DOI] [PubMed] [Google Scholar]