

Abstract
Known single-nucleotide polymorphisms (SNPs) explain <2% of the variation in body mass index (BMI) despite the evidence of >50% heritability from twin and family studies, a phenomenon termed ‘missing heritability'. Using DNA alone for unrelated individuals, a novel method (in a software package called Genome-wide Complex Trait Analysis, GCTA) estimates the total additive genetic influence due to common SNPs on whole-genome arrays. GCTA has made major inroads into explaining the ‘missing heritability' of BMI in adults. This study provides the first GCTA estimate of genetic influence on adiposity in children. Participants were from the Twins Early Development Study (TEDS), a British twin birth cohort. BMI s.d. scores (BMI-SDS) were obtained from validated parent-reported anthropometric measures when children were about 10 years old (mean=9.9; s.d.=0.84). Selecting one child per family (n=2269), GCTA results from 1.7 million DNA markers were used to quantify the additive genetic influence of common SNPs. For direct comparison, a standard twin analysis in the same families estimated the additive genetic influence as 82% (95% CI: 0.74–0.88, P<0.001). GCTA explained 30% of the variance in BMI-SDS (95% CI: 0.02–0.59; P=0.02). These results indicate that 37% of the twin-estimated heritability (30/82%) can be explained by additive effects of multiple common SNPs, and provide compelling evidence for strong genetic influence on adiposity in childhood.
Keywords: Genome-wide Complex Trait Analysis (GCTA), missing heritability, children, twins, genetics
Introduction
Family history has long been recognised as an important risk factor for obesity.1, 2 Quantitative genetic analyses using twin, family and adoption designs have demonstrated that familial resemblance in body mass index (BMI) is largely due to genetic similarity, with high heritability estimates reported from twin studies (47–90%), and moderate-to-high estimates from family (24–81%) and adoption studies (20–60%).3, 4 A recent meta-analysis of twin studies showed that heritability estimates were on average 0.07 higher in children than in adults.3
Genome-wide Association Studies (GWAS) have made significant headway in identifying single-nucleotide polymorphisms (SNPs) that are related to the relative body weight, indexed using BMI.5 A large meta-analysis of 123 865 adults from 46 studies with follow-up in another 125,931 participants conducted by the Genetic Investigation of Anthropometric Traits (GIANT) consortium identified 32 SNPs robustly associated with adult BMI.5 The majority of these SNPs (23–28) demonstrated directionally consistent effects in age- and sex-adjusted BMI in children and adolescents.5, 6 A subsequent meta-analysis of 14 studies with 5530 cases of obesity and 8318 controls identified another two SNPs associated with childhood and adolescent obesity that also showed directionally consistent effects in the previous meta-analysis of adult BMI.5, 7
However, even in combination, the 32 established SNPs explain <2% of the variation in BMI in either adults or children,5 although there are some suggestions that the size of the association between combined genetic obesity risk and adiposity may vary over the lifespan, peaking during late childhood (age 11) and early adulthood (age 20)8 in line with heritability estimates. The mismatch between the high heritability estimates from quantitative genetic analyses and the small proportion of variation explained through GWAS findings across many complex traits have come to be known as the problem of ‘missing heritability'.9 Part of the missing heritability is likely to be due to rare genetic variants and some non-additive genetic effects. These contribute to the estimated genetic effect in quantitative genetic studies, but are not detected in GWAS analyses that only capture additive effects of common SNPs with minor allele frequencies of ⩾5%. A second possibility is that there are multiple additional common genetic variants that contribute to the genetic effect observed in quantitative genetic studies, but have such small effect sizes that they cannot be detected even in the huge data sets used in contemporary GWAS analyses. However, until there is direct molecular genetic evidence for these additional sources of genetic influence, missing heritability is not clarified, and questions will remain about whether the heritability of obesity has been overestimated by quantitative genetic studies.
A novel approach called Genome-wide Complex Trait Analysis (GCTA) takes advantage of the fact that the degree of genetic resemblance for common SNPs at the whole-genome level is normally distributed among unrelated individuals. This can be used to quantify the proportion of the variation in a particular phenotype that is explained by the total common SNP similarity, effectively a molecular genetic estimate of heritability.10 The purpose of GCTA is not to identify specific SNPs related to the target phenotype, but rather to estimate the total additive genetic effect of the common SNPs used on currently available DNA arrays. Its value relative to GWAS comes from the fact that the GCTA estimate includes the effect of SNPs well below the current GWAS threshold.
GCTA has made major inroads into explaining the missing heritability of adiposity in adults. The first report found a genetic effect due to additive effects of common SNPs of 16.5%11 a remarkable order-of-magnitude increase compared with the effect of known genetic variants, and not far off the lower limit for additive genetic influence as estimated from family studies (e.g.12). A second study produced very similar results, with GCTA estimates of 14 and 10% in two independent adult samples.13 In this study, we provide the first pediatric GCTA estimate of additive genetic effects on adiposity in a sample of unrelated children. We also include the twin-based estimate of heritability in the same sample for direct comparison with the GCTA estimate by including data from the co-twin in the same families.
Subjects and methods
Sample
Data were from the Twins Early Development Study (TEDS), a population-based cohort of monozygotic and dizygotic twins (>11 000 pairs) born between 1994 and 1996 in England and Wales.14 Twins and parents provided informed consent for each part of the study prior to data collection. King's College London's Ethics Committee provided ethical approval.
Genotyping
Genome-wide genotyping was completed in 2010 for one randomly selected child in each of 3665 families; of these, 3152 (1446 male and 1706 female subjects) survived quality control criteria for ancestry, heterozygosity, relatedness and hybridisation intensity outliers (for details see15). Genotyping and quality control was done using the Affymetrix 6.0 GeneChip SNP genotyping array (Affymetrix Inc, Santa Clara, CA, USA) using standard experimental protocols as part of the WTCCC2 project.16 SNPs were selected on their minor allele frequency (>0.01), genotype call-rate (>0.80), Hardy–Weinberg Equilibrium (>10−20) and plate effect P-value (>10−6), which resulted in ∼700 000 quality-controlled genotyped SNPs. In addition, there were ∼2.5 million SNPs imputed from HapMap 2 and 3, and WTCCC controls, using the programme IMPUTE v.2 software.17 Imputed SNPs were screened using much more stringent quality control that resulted in reduction to ∼1 000 000 SNPs, giving a total of 1.7 million (quality controled) SNPs (for details see15). To control for ancestral stratification, we performed principal component analysis using EIGENSTRAT from EIGENSOFT package18 and identified significant axes using the Tracy–Widom Test.19 This resulted in eight axes with P<0.05 that were used as covariates in GCTA analyses.
Measurement of adiposity
Height and weight data were obtained in 2005 when the children were 8–11 years old, as part of a study of the heritability of adiposity.20 Parents were sent detailed instructions and asked to record each child's weight to the nearest pound or tenth of a kg, and height to the nearest cm, as well as the date of measurement. Parent- and researcher-measured heights and weights were correlated 0.90 and 0.83 in a subsample of 228 families.20 BMI was calculated as weight (kg)/height (m)2. BMI values were converted to s.d. scores (BMI-SDS) that take into account the child's age and sex, using 1990 UK growth reference data21 and computed with the programme ImsGrowth.22 Reference values21 were used to exclude implausible heights (<1.05 or >1.80 m), weights (<12 or >80 kg) and BMIs (<11 or >32).
Statistical analyses
All analyses were conducted on BMI-SDS that had been residualised for age and sex effects using a regression procedure. We used the GCTA package23 to quantify the proportion of variance in BMI-SDS explained by 1.7 million SNPs for the unrelated children with genotype and BMI-SDS data. All possible pairs from a sample of 2269 individuals yields nearly 2.6 million pairwise comparisons (2 573 046). No pairs exceeded the GCTA standard cutoff coefficient of 0.025 for genetic relatedness, confirming that no two children in the analysed sample appeared to be genetically related in the traditional sense. We performed standard ACE model-fitting analyses using OpenMx24 to estimate the heritability of BMI-SDS for the same sample of children by including anthropometric data from their co-twin to provide a direct comparison for the estimate derived from GCTA. The fit of the model was not of primary interest in this study; however, to assure a ‘good fit', we used the full ACE Cholesky Decomposition Model (including additive genetic (A), shared environmental (C) and unique environmental (E) components), which is the full model and thus fits the data best and also provides estimates of the A, C and E parameters.
Results
Of the 3152 children with genotyping data, 2402 families (76%) had provided anthropometric data and recorded age when the children were measured. Data from four children were excluded because they were reported as being <8 years old at the time they were measured; and 80 data points were excluded for implausible anthropometric results. Data from 22 children whose zygosity was unknown were excluded from the analyses because they could not be included in the twin analyses, along with a small number of children (n=27) with severe medical problems. Following exclusions, 2269 children had genotyping and anthropometric data.
The sample characteristics for the children included in the GCTA analysis are shown in Table 1. The average age was 9.9 years, 53% were girls and 39% were from monozygotic (identical) twin pairs. Their average BMI-SDS placed them close to the 1990 reference value, with comparatively low rates of overweight (8.7%) and obesity (3.6%).
Table 1. Characteristics of the GCTA analysis sample (n=2269 children).
| Mean (s.d.) or n (%) | |
|---|---|
| Age (years) | 9.90 (0.84) |
| Sex | |
| Females | 1208 (53.2) |
| Males | 1061 (46.8) |
| Zygosity | |
| Monozygotic | 890 (39.2) |
| Dizygotic | 1379 (60.8) |
| Weight (kg) | 33.28 (7.33) |
| Height (m) | 1.39 (0.08) |
| BMI (kg m−2) | 17.03 (2.59) |
| BMI-SDS | −0.02 (1.13) |
| Weight status | |
| Healthy weight | 1991 (87.7) |
| Overweight | 197 (8.7) |
| Obese | 81 (3.6) |
Abbreviations: BMI, body mass index; BMI-SDS, BMI s.d. scores.
The twin estimate of heritability of BMI-SDS in the sample was 82% (95% confidence interval: 0.74–0.88; P<0.001). Full results from the twin modelling are available from the first author. The GCTA estimate of genetic influence due to the additive effect of common SNPs was 30% (95% confidence interval 0.02–0.59; P=0.02). SNP heritability was therefore equivalent to 37% of the twin-estimated heritability (30%/82%). Figure 1 plots the variance explained in BMI-SDS from the twin analyses and the GCTA.
Figure 1.
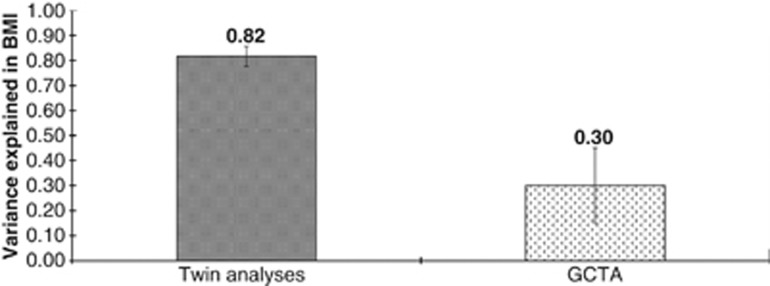
Comparison of variance explained in BMI (and s.e.) by genetic influences from twin analyses and GCTA at age 10.
Discussion
This is the first pediatric study to use GCTA to estimate the genetic influence on adiposity attributable to additive genetic effects from common SNPs across the entire genome. Consistent with findings in adults11, 13 the GCTA method gave an order-of-magnitude increase compared with the GWAS estimates (1.5%), explaining 30% of the variance in BMI-SDS. This equated to 37% of the estimate of heritability derived from the twin design (82%) in the same families, and is comparable to many estimates derived from family studies.3 Given that the 32 SNPs of the largest effect account for only 5% (1.5/30%) of the total common additive genetic SNP variance, these results suggest that 95% of the variation due to common SNPs have been undetected through GWAS. There are therefore likely to be hundreds of additional causal variants influencing childhood adiposity.
This GCTA estimate is likely to be at the lower end of the true additive genomic influence because it is limited to SNPs with a minor allele frequency of ⩾5% rarer variants are therefore excluded. In addition, causal variants that were not genotyped or not highly correlated with the SNPs on the genotyping array will also have been missed.
The GCTA value (30%) was larger than has been reported in studies in adults (10–16.5%),11, 13 suggesting that the additive genetic effect from common SNPs on BMI may be slightly higher for children. This is consistent with the higher estimates of heritability from pediatric than adult twin studies.3, 13 This may reflect the fact that adults are more likely than children to be making deliberate attempts at weight control, thus, limiting the observed genetic effect. The larger value may also be explained by the narrow age range of the sample, which reduces the effect of gene by age interaction.
These results have clinical and public health implications. Although the method used in the GCTA analysis cannot be used to predict obesity risk for any one individual because the genetic variants involved are not identified, the results underline the importance of additive genetic effects in the development of adiposity in childhood. This supports the current convention of using parental weight status as a proxy for childhood obesity risk.25 Targeting children of obese parents for early-life obesity-prevention interventions, given that these children are most at risk, might be a useful direction to take.
This study has limitations. BMI tends to be lower in twins than singletons26 and consistent with this, the average BMI of the sample placed them close to the 1990 reference value, and therefore below contemporary levels of adiposity. The sample size meant that it was not possible to carry out subgroup analyses. Height and weight data were parent-reported, therefore may be less reliable than researcher-measured anthropometrics, although they were found to be reliable in a subsample of families.20 Lastly, the effects of pubertal status were not examined in this study; differences in pubertal status may have resulted in a slightly lower GCTA estimate of additive genetic effects on BMI-SDS. The study's strengths included the opportunity to estimate heritability using the twin design in the same sample for which we carried out the GCTA analysis.
These results find that GCTA analysis explains a substantial proportion of the genetic effect identified as ‘missing heritability'. They provide compelling evidence that additive genetic influence from multiple common SNPs is a powerful determinant of adiposity in childhood.
Acknowledgments
The TEDS is supported by a programme grant to Robert Plomin from the UK Medical Research Council (G0901245, and previously G0500079), with additional support from the US National Institutes of Health (HD044454 and HD046167). The anthropometric data were collected as part of a grant to Jane Wardle from the UK Biological and Biotechnology Research Council (D19086), with additional support from programme grant funding from Cancer Research United Kingdom. Genome-wide genotyping was made possible by grants from the Wellcome Trust Case Control Consortium 2 project (085475/B/08/Z and 085475/Z/08/Z).
The authors declare no conflict of interest.
References
- Keiller SM, Colley JR, Carpenter RG. Obesity in schoolchildren and their parents. Ann Hum Biol. 1979;6:443–455. doi: 10.1080/03014467900003831. [DOI] [PubMed] [Google Scholar]
- Garn SM, Cole PE, Bailey SM. Living together as a factor in family-line resemblances. Hum Biol. 1979;51:565–587. [PubMed] [Google Scholar]
- Elks CE, den HM, Zhao JH, Sharp SJ, Wareham NJ, Loos RJ, et al. Variability in the heritability of body mass index: a systematic review and meta-regression. Front Endocrinol (Lausanne) 2012;3:29. doi: 10.3389/fendo.2012.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27:325–351. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249 796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Bradfield JP, Zhang H, Sleiman PM, Kim CE, Glessner JT, et al. Role of BMI-associated loci identified in GWAS meta-analyses in the context of common childhood obesity in European Americans. Obesity (Silver Spring) 2011;19:2436–2439. doi: 10.1038/oby.2011.237. [DOI] [PubMed] [Google Scholar]
- Bradfield JP, Taal HR, Timpson NJ, Scherag A, Lecoeur C, Warrington NM, et al. A genome-wide association meta-analysis identifies new childhood obesity loci. Nat Genet. 2012;44:526–531. doi: 10.1038/ng.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elks CE, Loos RJ, Hardy R, Wills AK, Wong A, Wareham NJ, et al. Adult obesity susceptibility variants are associated with greater childhood weight gain and a faster tempo of growth: the 1946 British Birth Cohort Study. Am J Clin Nutr. 2012;95:1150–1156. doi: 10.3945/ajcn.111.027870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebebrand J, Volckmar AL, Knoll N, Hinney A. Chipping away the 'missing heritability': GIANT steps forward in the molecular elucidation of obesity - but still lots to go. Obes Facts. 2010;3:294–303. doi: 10.1159/000321537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet. 2011;43:519–525. doi: 10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt SC, Hasstedt SJ, Kuida H, Stults BM, Hopkins PN, Williams RR. Genetic heritability and common environmental components of resting and stressed blood pressures, lipids, and body mass index in Utah pedigrees and twins. Am J Epidemiol. 1989;129:625–638. doi: 10.1093/oxfordjournals.aje.a115175. [DOI] [PubMed] [Google Scholar]
- Vattikuti S, Guo J, Chow CC. Heritability and genetic correlations explained by common SNPs for metabolic syndrome traits. PLoS Genet. 2012;8:e1002637. doi: 10.1371/journal.pgen.1002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth CM, Davis OS, Plomin R. Twins Early Development Study (TEDS): a genetically sensitive investigation of cognitive and behavioral development from childhood to young adulthood. Twin Res Hum Genet. 2012;16:117–125. doi: 10.1017/thg.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzaskowski M, Eley TC, Davis OS, Doherty SJ, Hanscombe KB, Meaburn EL, et al. First genome-wide association study on anxiety-related behaviours in childhood PloS one 2013. In Press. [DOI] [PMC free article] [PubMed]
- Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle J, Carnell S, Haworth CM, Plomin R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87:398–404. doi: 10.1093/ajcn/87.2.398. [DOI] [PubMed] [Google Scholar]
- Cole TJ, Freeman JV, Preece MA. Body-mass index reference curves for the UK, 1990. Arch Dis Child. 1995;73:25–29. doi: 10.1136/adc.73.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TJ.Software for LMS method: LMSGrowth PC 2009. Available at http://www.ucl.ac.uk/ich/research-ich/mrc-cech/research/studies/LMS-method 2009; (accessed on 21 November 2012).
- Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boker S, Neale M, Maes H, Wilde M, Spiegel M, Brick T, et al. OpenMx: an open source extended structural equation modeling framework. Psychometrika. 2011;76:306–317. doi: 10.1007/s11336-010-9200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker KL, Jarvis MJ, Beeken RJ, Boniface D, Wardle J. Comparing maternal and paternal intergenerational transmission of obesity risk in a large population-based sample. Am J Clin Nutr. 2010;91:1560–1567. doi: 10.3945/ajcn.2009.28838. [DOI] [PubMed] [Google Scholar]
- Pietilainen KH, Kaprio J, Rissanen A, Winter T, Rimpela A, Viken RJ, et al. Distribution and heritability of BMI in Finnish adolescents aged 16y and 17y: a study of 4884 twins and 2509 singletons. Int J Obes Relat Metab Disord. 1999;23:107–115. doi: 10.1038/sj.ijo.0800767. [DOI] [PubMed] [Google Scholar]