

Abstract
Campylobacter jejuni is a gram-negative, curved and rod-shaped bacterium that causes human gastroenteritis. Acute disease is associated with C. jejuni invasion of the intestinal epithelium. Epithelial cells infected with C. jejuni strains containing mutations in the FlpA and CadF fibronectin (Fn)-binding proteins exhibit reduced invasion of host cells and a C. jejuni CadF FlpA double mutant is impaired in the activation of epidermal growth factor receptor (EGFR) and Rho GTPase Rac1. Although these observations establish a role for Fn-binding proteins during C. jejuni invasion, their mechanistic contributions to invasion-associated signaling are unclear. We examined FlpA, a C. jejuni Fn-binding protein composed of three FNIII-like repeats D1, D2 and D3, to identify the interactions required for cellular adherence on pathogen-induced host cell signaling. We report that FlpA binds the Fn gelatin-binding domain via a motif within the D2 repeat. Epithelial cells infected with a flpA mutant exhibited decreased Rac1 activation and reduced membrane ruffling that coincided with impaired delivery of the secreted Cia proteins and reduced cell association. Phosphorylation of the Erk1/2 kinase, a downstream effector of EGFR signaling, was specifically associated with FlpA-mediated activation of β1-integrin and EGFR signaling. In vivo experiments revealed that FlpA is necessary for C. jejuni disease based on bacterial dissemination to the spleen of IL-10−/− germ-free mice. Thus, a novel Fn-binding motif within FlpA potentiates activation of Erk1/2 signaling via β1-integrin during C. jejuni infection.
Keywords: adherence, adhesion, extracellular matrix, lipoprotein, membrane, MSCRAMM
INTRODUCTION
Pathogen adherence to the surface of epithelial cells is often critical for host colonization and infection. There are currently ∼100 known bacterial outer surface adherence proteins (adhesins) that bind fibronectin (Fn),1 suggesting that adherence to Fn confers specific benefits to pathogens. Fn is a dimeric multidomain glycoprotein composed of 12 Fn type I repeats (FNI), two Fn type II repeats (FNII) and 15–17 Fn type III repeats (FNIII).2 Pathogen binding to Fn is thought to induce integrin clustering and recruitment of numerous regulatory molecules to cytoplasmic integrin domains.3,4,5 The resulting multiprotein complexes formed, termed focal complexes (FCs), act as signaling platforms that physically link Fn in the extracellular matrix to intracellular signaling machinery and the actin cytoskeleton. By initiating signaling through FCs, Fn on the cell surface can stimulate cytoskeletal and membrane rearrangements involved in cell motility, division, and phagocytosis.6,7,8,9 In addition, Fn provides a surface exposed ligand for bacterial attachment enabling pathogens to engage cellular regulatory pathways, in particular signaling events that promote bacterial internalization.1,3,4,10
Campylobacter jejuni is a gram-negative bacterium and the causative agent of campylobacteriosis, a debilitating gastrointestinal illness associated with abdominal cramps, fever and diarrhea.11 C. jejuni encodes two known Fn-binding proteins, an outer membrane protein termed CadF,12 and a recently identified lipoprotein termed FlpA that harbors three FNIII-like domains.13 Needed are studies to dissect the contribution of Fn binding in C. jejuni–host cell interactions.
Our understanding of the mechanism utilized by C. jejuni to invade cells is incomplete. Specifically, complex interactions at the bacterial host cell interface resulting in C. jejuni internalization are ill defined. We conducted this study to better define the nature of C. jejuni adherence to Fn and the contribution of this interaction to activating host signaling responses to promote invasion. Here, we identify the FlpA Fn-binding residues, the sites on Fn bound by FlpA, and the specific invasion-associated host cell signaling responses to the FlpA–Fn interaction, which contribute to acute disease.
MATERIALS AND METHODS
Bacterial strains/plasmids/primers
C. jejuni were cultured on Mueller-Hinton agar plates supplemented with 5% bovine blood (Mueller-Hinton blood agar) under microaerobic conditions. Escherichia coli XL-1 Blue (Stratagene, Garden Grove, CA, USA) and BL21(DE3) (Novagen, Madison, WI, USA) were maintained on Luria-Bertani agar plates or in Luria-Bertani broth aerobically at 37 °C. Strains harboring pGEX-5X-1 (GE Healthcare, Pittsburgh, PA, USA) were grown on medium supplemented with 100 µg/mL ampicillin. Construction and expression of the recombinant N-terminal glutathione S-transferase (GST)-tagged protein was performed using standard molecular biology techniques described previously.14 The primers used in this study are provided in Supplementary Table S1.
Protein purification
Protein purification was performed as previously described.15 Briefly, E. coli pGEX-5X-1 strains were grown in Luria-Bertani broth supplemented with antibiotics at 37 °C to an OD540=0.6 and induced with 1 mM Isopropyl-β-𝒹-thio-galactoside (Sigma, St Louis, MO, USA) overnight at 22 °C. Cells were harvested by centrifugation (6000 g for 15 min at 4 °C), resuspended in ice-cold 20 mM NaPi, 150 mM NaCl, pH 7.4 buffer (phosphate-buffered saline (PBS)), lysed by sonication and centrifuged at 15 000 g for 30 min at 4 °C to remove insoluble debris. GST-tagged proteins were purified from the clarified lysates using Sepharose 4B GST affinity resin (GE Healthcare) according to the manufacturer's instructions. Fractions containing recombinant GST fusion proteins were pooled, dialyzed in 25 mM Tris pH 7.5 or PBS and concentrated using Amicon Ultra centrifugal filter units (Millipore, Bedford, MA, USA).
Peptide synthesis
FlpA peptides were synthesized using conventional fluorenylmethyloxycarbonyl chloride solid-phase peptide chemistry on an Applied Biosystems 431A Peptide Synthesizer using protocols supplied by the manufacturer (Applied Biosystems, Foster City, CA, USA) by the School of Molecular Biosciences Laboratory for Bioanalysis and Biotechnology at Washington State University (Pullman, WA, USA). Synthesis of CadF FRLS+ and FRLS− peptides was described previously.15
Enzyme-linked immunosorbent assays (ELISAs) with GST-FlpA proteins
Human plasma Fn, the 30-kDa N-terminal domain, and the 40-kDa gelatin binding domain (GBD) were purchased from Sigma. To determine if FlpA or the FlpA domains bound to Fn, 96-well polystyrene plates (Costar, Corning, NY, USA) were coated with 40 nM of Fn or Fn fragments (Sigma) in 20 mM NaPi, 150 mM NaCl, pH 7.4 (PBS) overnight at 4 °C. Plates were washed once with PBS, 0.01% Tween 20 pH 7.4 (PBST) and then blocked with PBS containing 1% bovine serum albumin (BSA) (fraction V; Sigma). While the plates incubated with block solution, serial dilutions of the GST-FlpA, GST-FlpA-D1, GST-FlpA-D2 and GST-FlpA-D3 were made in PBS to produce concentrations that ranged from 1000 nM to 7.815 nM. After washing the wells with PBST, the GST fusion protein samples were added in triplicate and incubated for 2 h with shaking. Wells were washed three times with PBST and GST antibody (Sigma) diluted 1∶1000 in PBS was added for 2 h. Following incubation the wells were washed again, and a horseradish peroxidase antibody specific to rabbit IgG (α-R-HRP; Sigma) was added at a 1∶5000 dilution in PBS for 1.5 h. The wells were washed extensively and developed using the TMB substrate kit (Thermo Scientific, Marietta, OH, USA) according to the manufacturer's instructions. Binding was quantitated spectrophotometrically by measuring the absorbance at 450 nm (A450 nm). All samples were assayed in triplicate and the experiments were conducted at room temperature. Absorbance measurements from wells not treated with GST fusion proteins were subtracted from the sample absorbances to control for nonspecific binding by the primary and secondary antibodies.
Fn-binding ELISAs
To measure the amount of Fn bound to immobilized GST fusion proteins, 96-well plates were coated with 250 nM solutions of GST-FlpA, GST-FlpA-D1, GST-FlpA-D2 and GST-FlpA-D3 in PBS overnight at 4 °C. A concentration of 2.5 µM of the FlpA or CadF synthetic peptides was used to coat plates. Plates were washed with PBST containing 0.1% BSA (PBST-BSA) and blocked with PBS containing 1% BSA for 1 h. Serial dilutions of Fn were made in PBS containing 0.02% BSA to produce concentrations ranging from 80 nM to 0.04 nM. Plates were washed with PBS-BSA and the Fn solutions were added and incubated for 2 h with shaking. Plates were washed extensively and Fn antibody (Sigma) was added at a 1∶1000 dilution in PBS 0.02% BSA for 1 h. After another wash step, α-R-HRP was added and the ELISA was performed as described previously.
Peptide competitive inhibition assay
Fn and BSA fraction V (Sigma) were diluted in 0.1 M sodium carbonate buffer pH 9.5 to a concentration of 250 µg/mL. A flat-bottomed maxisorp 96-well plate (NUNC, Rochester, NY, USA) was coated with 100 µL per well of the diluted protein and incubated overnight at 4 °C. Following incubation, the plate was rinsed three times with PBS. The synthetic peptides were diluted in PBS, added to the wells and incubated for 1 h at 20 °C. The plate was rinsed three times with PBS and 1×107 of the C. jejuni wild-type strain and the flpA mutant were added to each well and incubated for 1 h at 37 °C. The plate was then rinsed three times with PBS and the bacteria were harvested by treatment with 1% trypsin for 15 min. The bacteria were resuspended, serial diluted, and plated on Mueller-Hinton agar for enumeration.
C. jejuni Fn-binding and cell-binding assays
Measurements of C. jejuni adherence to Fn and epithelial cells were performed as previously described.14
Generation of FlpA constructs with altered Fn-binding linear motif (FBLM) residues
The FlpA mutant and FlpA complemented strain of C. jejuni were previously generated using standard techniques.14 The pRY111 FlpA complementation plasmid was used as a template for PCR reactions.14 Primers MEK3316 and MEK3317 were used to generate full FlpA Fn binding domain mutants. MEK3318 and MEK3319 were used to generate proline and aspartic acid mutations. These primers contain a 5′ SphI restriction site. PCR products were digested with SphI and re-ligated to generate the pRY111 flpA Δ158–164 and pRY11 flpA P160A D161C mutants. Plasmid constructs were confirmed by DNA sequencing.
FlpA gels and immunoblots
Bacterial whole-cell lysates and outer membrane protein extracts were separated by SDS–PAGE and transferred to PVDF membranes (Millipore, Temecula, CA, USA) as described previously.16 The following antibodies were used: β1 integrin (4706), pErk (4370) (Cell Signaling Technology, Danvers, MA, USA), actin (sc-1616) (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), rabbit anti-FlpA polyclonal serum (Konkel lab) and rabbit anti-CysM polyclonal serum (Konkel lab).
Cia protein delivery assay
Cia protein delivery assays were performed 30 min post-infection as previously described.17
Scanning electron microscopy
Scanning electron microscopy was performed as previously described.16 Quantification of membrane ruffling was done by two independent observers as described previously.16
Rho GTPase Rac1 activation
Rac1 activation was quantified as previously described.16
Erk1/2 phosphorylation and β1 integrin knockdown
INT 407 cells were seeded in 24-well plates at a density of 1.5×105 cells per well. The following day, the cells were rinsed twice with PBS and serum starved for 3 h in minimal essential medium without fetal bovine serum. For protein depletion assays, INT 407 cells were transfected with 1 pmol siRNA of non-targeting (46–2000; Invitrogen, Grand Island, NY. USA) and β1 integrin-targeting siRNAs (ITGB1HSS105559; Invitrogen) using Lipofectamine RNAi MAX (Invitrogen) according to manufacturer's instructions and incubated for 24 h prior to an assay. β1-integrin depletion was confirmed by immunoblot. The C. jejuni infection assay on INT 407 cells was performed in the following manner. INT 407 cells were infected with C. jejuni suspended in minimal essential medium (OD540=0.03), centrifuged at 800 g for 5 min to promote bacteria-host cell contact and incubated for 45 min. Where indicated, C. jejuni were resuspended in minimal essential medium containing 1024 µg/mL chloramphenicol for 30 min prior to the assay. This same concentration of chloramphenicol was maintained throughout the course of the assay. Treatment of C. jejuni with 1024 µg/mL of chloramphenicol for 30 min completely blocks C. jejuni protein synthesis, as judged by the absence of [35S]-methionine incorporation (not shown). At the end of the assay, the INT 407 cells were lysed and analyzed by immunoblot with an antibody against phosphorylated Erk1/2.
C. jejuni inoculation of IL-10−/− mice and bacterial quantification
All animal experiments were conducted according to National Institutes of Health guidelines under Washington State University Animal Use Form (ASAF 04313). 129S6/SvEv IL-10−/− germ-free mice were obtained from the National Gnotobiotic Rodent Resource Center and maintained and monitored in a specific pathogen-free colony in microisolator cages at Washington State University as previously described.18 Mice were infected with ∼1×1010 colony forming units of C. jejuni by oral gavage and observed daily for clinical signs of disease as previously described.18 Mice were euthanized and necropsied promptly when clinical signs of disease developed or at 14 days post-infection. The colon and spleen of euthanized IL-10−/− mice were removed aseptically and re-suspended in 1∶1 (weight/volume) in Mueller-Hinton broth. The number of C. jejuni in each organ was determined by plating serial dilutions on CampyCefx plates.
Quantitative data and statistical analysis
For ELISAs and bacterial binding experiments, three technical replicates were performed with new reagents on separate days to demonstrate reproducibility. Within each experiment, the samples were measured in triplicate. The plotted values represent mean counts, and error bars show standard deviations. Statistical analyses were performed using GraphPad Prism (La Jolla, CA. USA) and statistical significance was determined by one-way analysis of variance (ANOVA) using Tukey's post-test. Sample data sets were considered statistically different when P≤0.05. The non-parametric Kruskal–Wallis one-way ANOVA, followed by post hoc Dunn's multiple comparisons test of the means, was used for statistical analysis of bacterial colonization of the colon and the spleen.
RESULTS
Domain 2 of FlpA binds Fn
Fn is composed of type I (FNI), type II (FNII) and type III repeats (FNIII) (Supplementary Figure S1) and the FlpA amino acid sequence contains three domains that resemble FNIII repeats FlpA-D1, FlpA-D2 and FlpA-D3 (Figure 1A).14 To determine whether the three FlpA FNIII-like domains harbor Fn-binding activity, each FlpA domain was expressed as an N-terminal fusion with GST: GST-FlpA-D1 (aa 35–132), GST-FlpA-D2 (aa 135–226) and GST-FlpA-D3 (aa 232–410) (Figure 1B). The GST tag was used to purify the GST fusion proteins and an antibody against GST was used for detection. Serial dilutions of GST-FlpA-D1, GST-FlpA-D2 and GST-FlpA-D3 fusion proteins were incubated in wells coated with Fn and the amount of GST protein bound was determined by ELISA. Full-length GST-FlpA (aa 35–410) protein was used as a positive control and wells coated with BSA were used to determine background adherence. Of the three FlpA domain fusion proteins, only GST-FlpA-D2 (aa 135–226) bound to the Fn-coated wells in amounts similar to the full-length GST-FlpA protein (aa 35–410) (Figure 2A). As with full-length GST-FlpA, the amount of GST-FlpA-D2 bound to the Fn-coated wells was dose-dependent and saturable, demonstrating specificity. Compared to GST-FlpA, significantly less GST-FlpA-D1 and GST-FlpA-D3 bound Fn-coated wells and did not exhibit attributes of specific binding. A second ELISA was performed in the opposite orientation, where wells were coated with GST-FlpA, GST-FlpA-D1, GST-FlpA-D2 and GST-FlpA-D3 and serial dilutions of Fn were added (Supplementary Figure S2). As expected, the amount of Fn bound to wells coated with GST-FlpA-D2 was similar to that of GST-FlpA and minimal binding to Fn was measured for D1 and D3.
Figure 1.

FlpA domain organization, GST fusion proteins and synthetic peptides used in this study. (A) Schematic of FlpA structural features. FlpA harbors a signal peptide ‘S' at the amino terminus that is lipidated at A20 and three domains designated FlpA-D1, FlpA-D2 and FlpA-D3, homologous to Fn type three (FNIII) repeats. (B) N-terminally tagged GST fusion proteins used in this study. Each of the three FlpA FNIII-like repeats (FlpA-D1, FlpA-D2 and FlpA-D3) were expressed with GST tags. (C) Sequences of the synthetic peptides used in this study. Peptides are aligned relative to the FlpA-D2 sequence. The 30mer peptides P1–P5 overlap by 15 residues and span the entire FlpA-D2 sequence. Peptides P6 and P7 span predicted β-strands and flanking disordered regions of interest. White double arrowheads indicate the predicted β-strands and vertical gray boxes highlight their location (DOMpro 1.0). Peptides NP1–NP9 are 12mers that overlap by 9 residues. The sequence of the FlpA FBLM 156WRPHPDFRV164 is highlighted in bold.
Figure 2.

FlpA-D2 binds to the GBD of Fn. (A) Binding of GST-tagged FlpA proteins to wells coated with full-length Fn. The GST-FlpA (full-length), GST-FlpA-D1, GST-FlpA-D2 and GST-FlpA-D3 fusion proteins were added in serial dilutions to plates with Fn and Fn fragments. The amount of bound GST protein was determined by ELISA. (B) Binding of GST-tagged FlpA proteins to wells coated with NTD of Fn. (C) Binding of GST-tagged FlpA proteins to wells coated with the GBD of Fn. Mean values and standard deviations from triplicate samples are plotted. Statistically significant differences in the amount of GST fusion protein bound to Fn was determined at 250 nM by one-way ANOVA using Tukey's post-test (*P<0.05).
Domain 2 of FlpA binds the GBD of Fn
The site(s) on Fn bound by FlpA was also examined. Amino acid sequence alignments of FlpA-D2 with the 1–15FNIII repeats using ClustalW19 revealed that FlpA-D2 most closely aligned with 1FNIII (22.9% sequence identity) (Supplementary Figure S3). 1FNIII is involved in intramolecular interactions with the FNI and FNII repeats within the N-terminus of Fn, suggesting FlpA may also bind the N-terminus.7 Digestion of Fn with thermolysin produces fragments that retain their biological activity.8 One of these Fn fragments, termed the N-terminal domain (NTD), is approximately 30 kDa in size and composed of 1–5FNI. Another fragment of approximately 40 kDa contains the GBD and is composed of 6–9FNI and 1,2FNII.8 To evaluate the binding of FlpA to the N-terminal region of Fn (i.e., either the NTD or GBD fragment), 96-well plates were coated with full-length Fn, the 30 kDa NTD fragment or the 40 kDa GBD Fn fragment. Serial dilutions of the GST-FlpA, GST-FlpA-D1, GST-FlpA-D2 and GST-FlpA-D3 proteins were incubated in NTD- and GBD-coated wells and the amount of GST-FlpA fusion proteins bound to the Fn fragments was determined by ELISA (Figures 2B and 2C). FlpA and FlpA-D2 exhibited dose-dependent and saturable binding to wells coated with the 40 kDa GBD, indicative of a specific interaction, and low levels of nonspecific binding to wells coated with the 30 kDa NTD. The equilibrium binding constants (KD) calculated from the saturation curves for FlpA and FlpA-D2 were 28.7 nM and 2.3 nM to immobilized full-length Fn and 11.5 nM and 5.5 nM to the immobilized GBD of Fn, respectively. These results indicate that FlpA and FlpA-D2 bind to Fn with high affinity, similar to that of other Fn-binding proteins such as BBK32 from B. burgdorferi.20 FlpA-D1 and FlpA-D3 did not exhibit specific or high affinity binding to the GBD, and the amount of Fn bound by FlpA-D1 and FlpA-D3 was significantly reduced compared to FlpA and FlpA-D2 (P<0.05). While we cannot exclude the possibility that FlpA-D1 and FlpA-D3 contribute to Fn-binding, these data indicate that FlpA-D2 mediates the interaction of FlpA with the GBD of Fn.
FlpA amino acids N150–F164 have maximal Fn-binding activity
Experiments were then performed to localize the Fn-binding residues within FlpA-D2. Five overlapping peptides were designed spanning the FlpA-D2 sequence: P1 (R135–V164), P2 (N150–F179), P3 (D165–I194), P4 (K180–I209) and P5 (D195–V224) (Figure 1C). As above, microtiter plates were coated with each of the five 30mer peptides, Fn was added to the wells, and the amount of Fn bound to peptide-coated wells determined (Supplementary Figure S4). Previously characterized CadF peptides, FRLS+ (aa 128–143) positive for Fn-binding, and FRLS− (aa 118–143) negative for Fn-binding, were included as controls.15 The amount of Fn bound to wells coated with peptides P1 and P2 was comparable to FRLS+ control peptide, whereas the amount of Fn bound to peptides P3, P4 and P5 was equivalent or less than that bound by the FRLS− peptide. No difference was observed in the amount of Fn bound by P1 (R135–V164) and P2 (N150–F179). Together, these data suggest that: (i) the Fn-binding residues are shared by P1 and P2, which corresponds to residues N150–V164; or (ii) P1 and P2 each have unique residues responsible for Fn-binding, indicating that the Fn-binding site is non-contiguous and extends beyond the overlapping region of P1 and P2.
Fn binding is localized to a predicted disordered domain within FlpA-D2
Fn FNIII repeats are composed of seven β-strands arranged in two anti-parallel β-sheets connected by flexible loops.21 The integrin binding motif, Arg-Gly-Asp (RGD) within 10FNIII is located on a loop/turn connecting two β-strands.22 Similar to the FNIII repeats of Fn, the secondary structure of FlpA-D2 contains β-strands that alternate with disordered loop domains (DOMpro 1.0 and DisEMBL).23 To assess whether FlpA Fn-binding is localized to the β-strands (K153–R157) or disordered loops within the N150–V164 span, two more peptides were synthesized (P6 and P7) (Figure 1C). The amount of Fn bound by P6 was equivalent to P1 and P2 (Figure 3), but little Fn was bound to P7 (not shown). These data suggested that the Fn-binding activity is localized to a predicted β-strand or the flanking disordered regions (L148–I152 and P158–V164) (Figure 1C). Given that neither P7 nor P3 bound significant amounts of Fn, we concluded that the residues unique to P6 158PHPDFRV164 were likely involved in Fn-binding activity.
Figure 3.

Fn-binding is localized to residues 158Pro-His-Pro-Asp-Phe-Arg-Val164 encoded within the P6 peptide. Binding of Fn to 96-well plates coated with peptides P1, P2, P5 and P6. Peptide-coated plates were incubated with serial dilutions of Fn and the amount of Fn bound was determined by ELISA. P1, P2 and P5 are 30mer peptides used above and P6 was designed to span predicted β strands and flanking disordered regions within the region shared by P1 and P2 (N150–V164). P5 was included as a negative control for Fn-binding. Data are displayed as mean values and standard deviations from triplicate samples. Statistical significance was determined at the 80 nM concentration by one-way ANOVA using Tukey's post-test (*P<0.05).
Identification of a FlpA peptide encoding residues required for C. jejuni adherence to Fn
To confirm the residues localized within N150–V164 of FlpA-D2 are required for Fn-binding, binding assays were conducted with a set of new peptides (NP1–9) of 12 residues. Each of the peptides overlapped by nine amino acids to localize the Fn-binding activity to three amino acids. The P6 30mer used above was included as a positive control, and the amount of Fn bound to NP1–9 was measured by ELISA (Figure 4A). Of the new peptides, NP5 bound the largest amount of Fn (P<0.05). NP3, NP4 and NP6 also bound Fn, but significantly less than NP5. The amount of Fn bound was statistically indistinguishable for the NP3, NP4 and NP6 peptides, but all three peptides bound more Fn (P<0.05) than for the NP1, NP2, NP7, NP8 and NP9 peptides. Collectively, these data indicate that the FlpA residues 156WRPHPDFRV164 bind to Fn. We termed these nine residues the FlpA FBLM.
Figure 4.

The 12mer peptide NP5 competitively inhibits FlpA-mediated adherence of C. jejuni. (A) Binding of Fn to 96-well plates coated with peptides NP1–9. Ninety-six-well plates were coated with 12mer peptides (NP1–9) spanning the N150–V164 region of FlpA-D2. Fn was added to the peptide coated wells and the amount of Fn bound was measured by ELISA. The P6 30mer peptide used above was included as a positive control. Data are displayed as mean values and stan dard deviations from triplicate samples and statistical significance was determined by one-way ANOVA using Tukey's post-test (*P<0.05). (B) C. jejuni binding to wells coated with Fn pretreated with FlpA peptides. Ninety-six-well plates were coated with Fn and then blocked with peptides NP5 or NP2 at the indicated concentrations. A suspension of C. jejuni was added to the wells. After incubation and washing, the bacteria were collected by trypsinization and plated to determine CFU. The C. jejuni flpA mutant was also included as a negative control. Triplicate samples were used to calculate means and standard deviations plotted. The number of C. jejuni bound to the no peptide control wells was compared to the peptide treated samples and statistical difference was determined by one-way ANOVA using Tukey's post-test (*P<0.05).
The experiments to this point had been performed to localize the Fn-binding activity with the FlpA protein. We then performed experiments to determine the importance of the FlpA FBLM in C. jejuni adherence to Fn and host cells. We first examined whether NP5 could inhibit binding of C. jejuni to immobilized Fn. NP5 harbors all nine residues of the FlpA FBLM and had bound more Fn than the other 12mer peptides. Plates coated with Fn were incubated with C. jejuni and serial dilutions of NP5, NP2 (non-binding control) or no peptide. The number of C. jejuni bound to the peptide treated wells was determined (Figure 4B). NP5 blocked the adherence of C. jejuni to Fn-coated wells in a dose-dependent manner, whereas the highest concentration of the NP2 peptide tested (50 µM) did not inhibit C. jejuni adherence. Furthermore, the amount of C. jejuni bound to wells treated with 50 µM NP5 was statistically indistinguishable (P>0.05) from the amount bound by the C. jejuni flpA mutant. These findings indicate that the FlpA FBLM is required for C. jejuni binding to Fn.
Deletion of conserved residues abrogates FlpA-mediated adherence of C. jejuni to Fn and INT 407 epithelial cells
We previously showed complementation of the C. jejuni flpA mutant with flpA in trans restores bacterial adherence to INT 407 epithelial cells.14 Thus, we generated two constructs for the synthesis of FlpA proteins containing mutations in the FBLM residues and examined whether they could rescue the binding defect of the flpA mutant. In the first construct, FlpA Δ158–164, residues 158PHPDFRV164 were deleted and replaced with alanine and cysteine (Figure 5A). In the second construct, FlpA P160A D161C, a smaller mutation was made in central FBLM residues by substituting P160 and D161 with alanine and cysteine. This smaller alteration was made to examine the sensitivity of the FBLM to small changes in residue composition. For comparison, we also analyzed a C. jejuni flpA mutant harboring a wild-type copy of flpA. Equivalent protein synthesis of FlpA, FlpA Δ158–164 and FlpA P160A D161C and insertion within the outer membrane of the C. jejuni flpA mutant was confirmed by immunoblot analysis of bacterial lysates and outer membrane extracts, respectively (Figure 5B). Surface exposure of FlpA, FlpA Δ158–164 and FlpA P160A D161C was confirmed by indirect immunofluorescence microscopy (Supplementary Figure S5A). Introduction of cysteine residues within the altered FlpA proteins created the possibility of introducing disulfide bonds, but both wild-type and the altered FlpA proteins exhibited similar mobility under reducing and non-reducing conditions (Supplementary Figure S5B). Assays were then conducted to measure binding by flpA mutants synthesizing the FlpA, FlpA Δ158–164 and FlpA P160A D161C to immobilized Fn and epithelial cells (Figures 5C and 5D). The C. jejuni flpA mutant synthesizing FlpA wild-type protein bound to Fn-coated wells and epithelial cells at or above wild-type levels, indicating functional complementation (Figures 5C and 5D). Synthesis of the FlpA Δ158–164 and FlpA P160A D161C proteins did not restore the binding of the flpA mutant to Fn-coated wells or INT 407 cells. Failure of FlpA Δ158–164 and FlpA P160A D161C to complement the C. jejuni flpA mutant is consistent with the peptide inhibition results (Figure 4B), and support the conclusion that the FlpA FBLM is required for C. jejuni to bind to Fn.
Figure 5.

Mutation of the FlpA FBLM abrogates FlpA-mediated adherence. (A) Sequence modifications for generation of FlpA Δ158–164 and FlpA P160A D161. Non-native residues (gray) result from enzyme restriction sites introduced for removal of the FBLM residues. (B) FlpA protein synthesis and outer membrane insertion. WCLs and OMP extracts from C. jejuni flpA mutant strains synthesizing FlpA, FlpA Δ158–164 and FlpA P160A D161C proteins were immunoblotted with FlpA-specific rabbit serum. OMP extracts were stained with Coomassie Brilliant Blue (CBB-R250) to show equal loading. (C) Binding of C. jejuni flpA mutant strains synthesizing the FlpA, FlpA Δ158–164, and FlpA P160A D161C proteins to immobilized Fn. C. jejuni were added to 96-well plates coated with Fn and the CFU bound to each well were determined. C. jejuni wild-type and the flpA mutant were included as positive and negative controls, respectively. (D) Binding of C. jejuni flpA mutant strains synthesizing the FlpA, FlpA Δ158–164 and FlpA P160A D161C proteins to INT 407 epithelial cells. All C. jejuni binding experiments were performed three times with triplicate samples per experiment. The means and standard deviations are plotted and statistical difference was determined by ANOVA using Tukey's post-test (*P<0.05). CFU, colony-forming unit; OMP, outer membrane protein; WCL, whole-cell lysate.
FlpA-mediated adherence facilitates C. jejuni–host cell interactions
After identifying the Fn-binding residues in FlpA, we shifted our focus to examination of how FlpA-mediated adherence contributes to C. jejuni–host cell interactions. Others have found that C. jejuni invasion of cells is associated with ruffling of the cytoplasmic membrane.16,24 To determine if the FlpA protein is involved in membrane ruffling, INT 407 cells were inoculated with the C. jejuni wild-type strain, flpA mutant, flpA mutant synthesizing the FlpA wild-type protein, FlpA Δ158–164 or FlpA P160A D161C proteins, and the C. jejuni ciaC mutant. The cells were then examined by scanning electron microscopy (Figure 6A). Uninfected cells were used to calculate the basal level of membrane ruffling and the C. jejuni ciaC mutant was used as a negative control, as this mutant is deficient for activation of levels of membrane ruffling relative to wild-type C. jejuni.16 Membrane ruffling was significantly reduced in cells inoculated with the C. jejuni flpA mutant compared to cells inoculated with the C. jejuni wild-type strain and was restored by synthesis of FlpA wild-type protein in the C. jejuni flpA mutant. Synthesis of the FlpA Δ158–164 or FlpA P160A D161C proteins in the C. jejuni flpA mutant did not rescue the membrane ruffling phenotype observed with the C. jejuni wild-type strain. Given that C. jejuni internalization is dependent upon the activation of Rac1 GTPase, we measured the amount of Rac1-GTP (activated Rac1) produced by cells in response to infection with the C. jejuni strains (Figure 6B). Compared to cells inoculated with the C. jejuni wild-type strain, cells produced significantly less activated Rac1 when inoculated with the flpA mutant or the flpA mutant synthesizing the FlpA Δ158–164 or FlpA P160A D161C proteins. In contrast, cells inoculated with the flpA mutant synthesizing the FlpA wild-type protein (complemented strain) produced similar levels of activated Rac1 to cells inoculated with the wild-type strain. These findings suggest that the FlpA FBLM is involved, either directly or indirectly, in C. jejuni engagement of host signaling pathways.
Figure 6.

Decreased epithelial cell membrane ruffling and Rac1 GTPase activation in response to C. jejuni flpA mutant strains synthesizing FlpA with altered FBLM residues. (A) Representative scanning electron micrographs of INT 407 cell membrane ruffles stimulated in response to infection with C. jejuni strains: (a) cells only; (b) C. jejuni F38011 wild-type strain; (c) C. jejuni flpA mutant; (d) C. jejuni flpA complement; (e) C. jejuni flpA Δ158–164; (f) C. jejuni flpAP160A D161C; (g) C. jejuni ciaC mutant. The number of cells exhibiting membrane ruffles is indicated as a percentage of total cells (×7000 magnification, scale bar=10 µm). Boxes within images ‘a' through ‘g' indicate the magnified area shown adjacent to the right ‘a-1' through ‘g-1' (×50 000 magnification, scale bar=2 µm). Arrowheads indicate C. jejuni. (B) Rac1-GTP production in INT 407 cells infected with C. jejuni. INT 407 cells were infected with C. jejuni F38011 wild-type strain, C. jejuni flpA mutant, C. jejuni flpA complement, C. jejuni flpA Δ158–164 and C. jejuni flpA P160A D161C. Activated Rac1 (GTP bound) in lysates from infected cells was measured by G-LISA and expressed as relative optical density. Means and standard deviation of total Rac1-GTP are plotted. Statistical significance was determined by one-way ANOVA using Tukey's post-test (*P<0.05).
Adherence facilitates Cia protein delivery
To determine if FlpA is required for the delivery of Cia proteins to host cells, we performed protein delivery assays. To measure protein delivery, the C. jejuni isolates were transformed with a plasmid encoding CiaC fused to the adenylate cyclase domain (ACD) of the Bordetella pertussis protein CyaA.17 Translocation of CiaC-ACD into the host cell cytosol catalyzes the production of cAMP only when bound by calmodulin in the eukaryotic cell cytosol. For these experiments, translocation of CiaC into the host cell cytosol by the C. jejuni flpA mutant was compared to the C. jejuni wild-type strain. As a control, we also included a C. jejuni capA mutant.13,25 The C. jejuni capA mutant displays a similar reduction in epithelial cell adherence as the flpA mutant (Supplementary Figure S6A). The cells inoculated with the C. jejuni flpA and capA mutants synthesizing the CiaC-ACD resulted in significantly less cAMP than cells inoculated with the C. jejuni wild-type strain (P<0.05) (Supplementary Figure S6B). Moreover, equivalent reductions in cAMP concentrations were observed in response to the C. jejuni flpA and capA mutants. These data indicate that bacterial attachment to the host cell, and not specifically FlpA–Fn ligation, is required for CiaC delivery.
FlpA-mediated adherence triggers Erk1/2 phosphorylation in the absence of Cia synthesis
Previous work has demonstrated that binding of C. jejuni to Fn on host cells induces activation of the FC. One possible consequence of FC activation is phosphorylation of mitogen-activated protein kinases, including Erk1/2.26 Therefore, we compared Erk1/2 phosphorylation in epithelial cells inoculated with the C. jejuni wild-type strain and flpA mutant. The cells were also inoculated with a C. jejuni capA mutant, as the reduction in binding of this mutant to cells is comparable to that of the flpA mutant. To avoid the signaling responses initiated by the Cia proteins, host cell infections were performed in the presence of chloramphenicol, as the synthesis of the Cia proteins is completely inhibited by chloramphenicol.27 Cells infected with the C. jejuni wild-type strain and the capA mutant exhibited increased Erk1/2 phosphorylation compared to uninfected cells, while cells infected with the flpA mutant did not exhibit increased Erk1/2 phosphorylation (Figure 7A). These findings suggest that FlpA-mediated adherence to Fn contributes to the activation of Erk1/2.
Figure 7.

FlpA and β1-integrin are required for phosphorylation of Erk1/2 during C. jejuni infection. (A) Levels of Erk1/2 phosphorylation in INT 407 epithelial cells infected with C. jejuni treated with chloramphenicol. Cell lysates were immunoblotted with antibodies against pErk and actin (loading control). Uninfected cells treated with chloramphenicol served as a negative control. Band intensities were quantitated by densitometry (n=4, error bars indicate standard error of the mean). (B) Erk1/2 phosphorylation in epithelial cells depleted of β1 integrin in response to C. jejuni infection. Uninfected cells treated with NT show basal levels of Erk1/2 phosphorylation, and C. jejuni-infected cells treated with NT were used as a positive control. Erk1/2 phosphorylation was measured by densitometric analysis (n=4, error bars indicate standard error of the mean) and statistical significance was determined by ANOVA using Tukey's post-test (*P<0.05). NT, non-targeting siRNA; pErk, phosphorylated Erk1/2.
While other integrin receptors also bind Fn, outside-in signal transduction is predominantly associated with the α5β1 integrin receptor.28 Thus, engagement of Fn during C. jejuni infection could activate Erk1/2 by a β1-integrin-dependent pathway. To examine the requirement of β1-integrin for Erk1/2 phosphorylation during C. jejuni infection, we knocked down cellular β1-integrin with specific siRNA and examined Erk1/2 activation upon C. jejuni infection. Cells treated with non-targeting siRNA, either infected with C. jejuni or uninfected, were used as positive or negative controls, respectively. Efficient siRNA knockdown of β1-integrin protein levels were confirmed by immunoblot (Figure 7B). Increased Erk1/2 phosphorylation was observed in cells treated with non-targeting siRNA and infected with the C. jejuni wild-type strain (Figure 7B). However, in C. jejuni infected cells depleted of β1-integrin, we did not observe an increase in Erk1/2 phosphorylation. Collectively, these results indicate that Erk1/2 is activated by FlpA-mediated adherence, and suggest that FlpA binding to Fn stimulates outside-in signaling.
FlpA is required for maximal disease progression in IL-10−/− mice
Since the C. jejuni flpA mutant is reduced in its ability to adhere to fibronectin, adhere to epithelial cells and to stimulate host cell signaling, we wanted to determine if FlpA contributes to disease in vivo. IL-10−/− germ-free mice were inoculated with a C. jejuni wild-type strain, flpA mutant and flpA complemented strain and housed for 14 days prior to necropsy. Uninfected mice (PBS sham-inoculated) were included as a negative control. We determined the number of bacteria in the colon in order to determine if there is a relationship between intestinal colonization and invasion of the spleen. The colons of the mice inoculated with the C. jejuni wild-type strain and C. jejuni flpA complemented strain showed signs of edema and apparent stool softening compared to mice inoculated with the C. jejuni flpA mutant and uninoculated mice (Supplementary Figure S7). Noteworthy is that all of the C. jejuni inoculated mice were colonized at statistically indistinguishable levels (Figure 8). In contrast to the levels of colonization, fewer bacteria were recovered from the spleen of mice inoculated with the C. jejuni flpA mutant versus the C. jejuni wild-type and flpA complemented strains (Figure 8). Together, these data indicate that FlpA is necessary for the development of severe disease (bacterial dissemination) in IL-10−/− germ-free mice.
Figure 8.
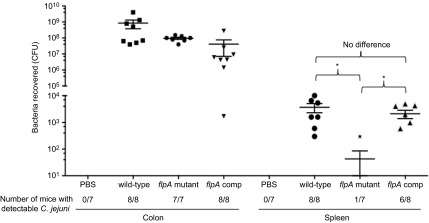
FlpA is required for C. jejuni dissemination to the spleen. IL-10−/− germ-free mice were inoculated with the C. jejuni wild-type strain, flpA mutant and the flpA complemented strain. No difference was observed in bacterial colonization of the colon. However, significant differences were observed in bacterial dissemination to the spleen between the C. jejuni wild-type strain and the flpA mutant. Similarly, the flpA complemented strain of C. jejuni had levels of dissemination to the spleen indistinguishable from the levels obtained for the C. jejuni wild-type strain. Results are expressed as mean±s.e.m. for at least seven animals per group (n≥7). Significance (*P<0.05) between groups was determined by non-parametric Kruskal–Wallis one-way ANOVA, followed by Dunn's multiple comparison test of the means. A second experiment, which was performed to ensure reproducibility, yielded identical results.
DISCUSSION
This study increases our understanding of the mechanism used by C. jejuni to bind to and invade epithelial cells. We demonstrated that FlpA binds the GBD of Fn via the FBLM within the second FNIII-like repeat (FlpA-D2) and that the phosphorylation of Erk1/2 requires FlpA-mediated adherence and β1-integrin engagement. These findings are consistent with the hypothesis that the FlpA–Fn interaction triggers outside-in signaling during C. jejuni infection of epithelial cells.
FlpA is comprised of three FNIII-like repeats and is ≥99% conserved among C. jejuni sequenced strains (Supplementary Figure S3), implying that FlpA plays a critical role in pathogenesis. Other bacterial adhesins have been identified with multiple Fn-like domains. Brown et al.29 reported that the Fn-binding protein Streptococcal C5a peptidase of S. agalactiae (SCPB) contains three Fn-like domains and two RGD motifs predicted to stabilize integrin interactions with Fn.29,30 Unlike the Fn-binding protein Streptococcal C5a peptidase of S. agalactiae, the three FNIII-like repeats within FlpA do not share significant homology with known Fn-binding proteins, as judged by BLAST (reviewed in Henderson et al., 2011).1 In the current study, high affinity FlpA–Fn interactions were found between FlpA-D2 and the GBD of Fn. Including FlpA, there are currently 12 known Fn-binding proteins reported to bind to the GBD of Fn.1 Although we did not observe Fn binding to FlpA-D1 or FlpA-D3, the possibility remains that these domains contribute to FlpA Fn-binding. Given the scaffolding role of FNIII repeats for the display of binding motifs in other proteins,31 it is possible that FlpA D1 and/or D3 provide structural support for interactions between FlpA-D2 and the GBD of Fn.
We found that peptides harboring residues 156WRPHPDFRV164 (termed the FlpA FBLM) exhibited the greatest Fn-binding affinity and inhibited C. jejuni adherence to Fn and cells. FBLM residues 157RPHPD161 are flanked on either end by hydrophobic residues and comprise the core of the Fn-binding motif in which charged residues alternate with proline residues. Similar to the FBLM of FlpA, proline-rich motifs have been identified within eukaryotic proteins recognized by SH3 domains during the assembly of signal transduction complexes.32 The conformational constraints of the proline cyclic side chain confers secondary structure beneficial for proper alignment of residues within binding motifs.33 The substitution of proline and aspartate within the FBLM (P160A D161C) drastically impaired FlpA Fn-binding affinity. Disruption of secondary structure and charge distribution localized to the FBLM likely accounts for the loss of FlpA P160A D161C binding activity, as assessments of outer-membrane localization, surface exposure, and electrophoretic gel mobility indicated the altered FlpA protein behaves like wild-type protein. Sensitivity of the FBLM to minor changes in amino acid composition is a characteristic shared with the linear binding motifs in other systems.34 Together, these results are consistent with the conclusion that FBLM residues are required for FlpA-mediated adherence.
Bacterial adherence to Fn, in addition to having a critical role for host cell attachment, can initiate integrin-dependent activation of regulatory proteins that coordinate cytoskeletal rearrangements and bacterial uptake by non-phagocytic cells.3,5,10 Here, reduced epithelial cell membrane ruffling and Rac1 activation were observed in response to infection with the flpA mutant. Based on these results, we concluded that the loss of FlpA-mediated adherence is sufficient to disrupt the activation of host signaling pathways.
Epidermal growth factor receptor (EGFR) activation can be initiated by extracellular ligands or via an integrin-dependent mechanism. Interestingly, treatment of epithelial cells with inhibitors of EGFR phosphorylation (PD168393 and erlotinib) blocks C. jejuni invasion and the addition of exogenous EGF to cells rescues the invasiveness of a C. jejuni ciaC mutant.16 We examined Erk1/2, which is a signaling molecule downstream of the EGFR, in cells infected with C. jejuni. We found that Erk1/2 phosphorylation was reduced in cells inoculated with the C. jejuni flpA mutant, but not with the C. jejuni capA mutant. These findings indicate that Erk1/2 is phosphorylated in response to FlpA binding to Fn. As stated above, C. jejuni stimulates host signaling during infection by multiple mechanisms,16,24,35 but these data suggested that FlpA binding to Fn specifically contributes to Erk1/2 phosphorylation during C. jejuni infection. Consistent with this observation, knockdown of β1 integrin with specific siRNA also inhibited Erk1/2 phosphorylation in cells infected with the C. jejuni wild-type strain. While others have shown maximal C. jejuni invasion requires β1 integrin36,37 and Erk1/2 activation,38 we demonstrate that C. jejuni stimulates integrin-dependent signaling by a mechanism requiring FlpA adherence for Erk1/2 phosphorylation. Our findings suggest that outside-in signaling is initiated by FlpA–Fn binding, which through the β1 integrin sets the stage for C. jejuni invasion of host cells (proposed pathway: FlpA→Fn→β1-integrin→EGFR→Erk1/2 signaling pathway). Dissection of the precise mechanism of Erk1/2 activation and the role of mitogen-activated protein kinases in Campylobacter pathogenesis is a topic of ongoing investigation.
The development of acute disease caused by C. jejuni has been studied in multiple animal models, however the IL-10−/− germ-free mouse model has emerged as a reproducible and reliable model system for the study of campylobacteriosis.39,40 We used bacterial invasion of the spleen as an indicator of C. jejuni disease.41,42 IL-10−/− germ-free mice inoculated with C. jejuni wild-type strain, flpA mutant and flpA complemented strain were colonized equally. Colonization of IL-10−/− germ-free mice by the flpA mutant was inconsistent with the previous finding that FlpA is required for colonization of chicks.13 While there are a number of possible explanations for this difference, we speculate that the response is both host-specific (avian versus murine) and that IL-10−/− germ-free mice are permissive of flpA mutant colonization due to reduced numbers of resident microflora in the gut and thus reduced binding requirements for establishing C. jejuni residency. In addition, the compromised innate immunity of the IL-10−/− germ-free mice could contribute to permissiveness of the flpA mutant colonization. FlpA does appear to participate in C. jejuni infection of mice, as greater numbers of the C. jejuni wild-type strain and flpA complemented strain were recovered from the spleen than for the flpA mutant. We speculate that the attenuation of the flpA mutant is due to a reduction in C. jejuni: (i) adherence to fibronectin; (ii) adherence to epithelial cells; (iii) stimulation of host cell signaling pathways; and (iv) invasion of epithelial cells. Other factors that may contribute to the loss in virulence of the C. jejuni flpA mutant include its inability to effectively: (i) translocate across the intestinal epithelium; and (ii) combat the host innate and acquired immune responses. While additional work is needed to define the route of C. jejuni dissemination from the intestine and to dissect the complexity of systemic disease initiated by oral inoculation, we concluded that FlpA contributes to disease in the IL-10−/− germ-free mouse model.
We identified the FBLM in FlpA and discovered that FlpA binds to the gelatin-binding domain of Fn. We discovered that FlpA is required for the maximal binding of host intestinal epithelial cells, and is required for the Cia-independent activation of Erk1/2. We demonstrate that FlpA is necessary for C. jejuni invasion of the spleen using IL-10−/− germ-free mice.
Acknowledgments
We thank Dr Jason M Neal-McKinney (School of Molecular Biosciences, College of Veterinary Medicine, Washington State University, USA) for proofreading this manuscript. We would also like to thank Dr Magnus Hook (Professor of Biochemistry and Biophysics, of the IBT-Houston, Veterinary Anatomy and Public Health, Director, Center for Extracellular Matrix Biology) and Dr Laurent Vuillard (BioXtal, PX Unit, c/o AFMB, UMR 6098, Case 932, 163 Avenue de Luminy, 13288 Marseille CEDEX 09, France) for helpful discussions. Finally, we thank Dr Maureen Bower at the Gnotobiotic Core Facility of the Center for Gastrointestinal and Dr Sue Tonkonogy at the College of Veterinary Medicine, North Carolina State University (Raleigh, NC, USA). This project was supported by Agriculture and Food Research Initiative Competitive (Grant NO. 2011-67015-30772) from the USDA National Institute of Food and Agriculture awarded to Michael E Konkel. Derrick R Samuelson and Tyson P Eucker were supported by Award Numbers T32GM083864 and T32GM008336 from the National Institute of General Medical Sciences, respectively. Jason L O'Loughlin was supported by a National Institutes of Health T32 Training program in Infectious Diseases and Microbial Immunology (Award Number 5 T32 AI 7025-33). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the National Institutes of Health.
Supplementary Information for this article can be found on Emerging Microbes and Infections's website (http://www.nature.com/emi).
Supplementary Information
References
- Henderson B, Nair S, Pallas J, Williams MA. Fibronectin: a multidomain host adhesin targeted by bacterial fibronectin-binding proteins. FEMS Microbiol Rev. 2011;35:147–200. doi: 10.1111/j.1574-6976.2010.00243.x. [DOI] [PubMed] [Google Scholar]
- Xu J, Bae E, Zhang Q, Annis DS, Erickson HP, Mosher DF. Display of cell surface sites for fibronectin assembly is modulated by cell adherence to 1F3 and C-terminal modules of fibronectin. PLoS ONE. 2009;4:e4113. doi: 10.1371/journal.pone.0004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjenberg ZR, Ellis IR, Hagan RM, et al. Cooperative binding and activation of fibronectin by a bacterial surface protein. J Biol Chem. 2011;286:1884–1894. doi: 10.1074/jbc.M110.183053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz-Linek U, Hook M, Potts JR. The molecular basis of fibronectin-mediated bacterial adherence to host cells. Mol Microbiol. 2004;52:631–641. doi: 10.1111/j.1365-2958.2004.04027.x. [DOI] [PubMed] [Google Scholar]
- Talay SR, Zock A, Rohde M, et al. Co-operative binding of human fibronectin to Sfbl protein triggers streptococcal invasion into respiratory epithelial cells. Cell Microbiol. 2000;2:521–535. doi: 10.1046/j.1462-5822.2000.00076.x. [DOI] [PubMed] [Google Scholar]
- Broussard JA, Webb DJ, Kaverina I. Asymmetric focal adhesion disassembly in motile cells. Curr Opin Cell Biol. 2008;20:85–90. doi: 10.1016/j.ceb.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005;24:389–399. doi: 10.1016/j.matbio.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Amelung S, Nerlich A, Rohde M, et al. The FbaB-type fibronectin-binding protein of Streptococcus pyogenes promotes specific invasion into endothelial cells. Cell Microbiol. 2011;13:1200–1211. doi: 10.1111/j.1462-5822.2011.01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allos BM. Campylobacter jejuni Infections: update on emerging issues and trends. Clin Infect Dis. 2001;32:1201–1206. doi: 10.1086/319760. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Garvis SG, Tipton SL, Anderson DE, Jr, Cieplak W., Jr Identification and molecular cloning of a gene encoding a fibronectin-binding protein (CadF) from Campylobacter jejuni. . Mol Microbiol. 1997;24:953–963. doi: 10.1046/j.1365-2958.1997.4031771.x. [DOI] [PubMed] [Google Scholar]
- Flanagan RC, Neal-McKinney JM, Dhillon AS, Miller WG, Konkel ME. Examination of Campylobacter jejuni putative adhesins leads to the identification of a new protein, designated FlpA, required for chicken colonization. Infect Immun. 2009;77:2399–2407. doi: 10.1128/IAI.01266-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Larson CL, Flanagan RC. Campylobacter jejuni FlpA binds fibronectin and is required for maximal host cell adherence. J Bacteriol. 2010;192:68–76. doi: 10.1128/JB.00969-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Christensen JE, Keech AM, Monteville MR, Klena JD, Garvis SG. Identification of a fibronectin-binding domain within the Campylobacter jejuni CadF protein. Mol Microbiol. 2005;57:1022–1035. doi: 10.1111/j.1365-2958.2005.04744.x. [DOI] [PubMed] [Google Scholar]
- Eucker TP, Konkel ME. The cooperative action of bacterial fibronectin-binding proteins and secreted proteins promote maximal Campylobacter jejuni invasion of host cells by stimulating membrane ruffling. Cell Microbiol. 2012;14:226–238. doi: 10.1111/j.1462-5822.2011.01714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal-McKinney JM, Konkel ME. The Campylobacter jejuni CiaC virulence protein is secreted from the flagellum and delivered to the cytosol of host cells. Front Cell Infect Microbiol. 2012;2:31. doi: 10.3389/fcimb.2012.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield LS, Bell JA, Wilson DL, et al. C57BL/6 and congenic interleukin-10-deficient mice can serve as models of Campylobacter jejuni colonization and enteritis. Infect Immun. 2007;75:1099–1115. doi: 10.1128/IAI.00833-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakaran S, Liang X, Skare JT, Potts JR, Hook M. A novel fibronectin binding motif in MSCRAMMs targets F3 modules. PLoS ONE. 2009;4:e5412. doi: 10.1371/journal.pone.0005412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson CD, Gay DA, Parello J, Ruoslahti E, Ely KR. Crystals of the cell-binding module of fibronectin obtained from a series of recombinant fragments differing in length. J Mol Biol. 1994;238:123–127. doi: 10.1006/jmbi.1994.1272. [DOI] [PubMed] [Google Scholar]
- Krammer A, Lu H, Isralewitz B, Schulten K, Vogel V. Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch. Proc Natl Acad Sci USA. 1999;96:1351–1356. doi: 10.1073/pnas.96.4.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Krause-Gruszczynska M, Rohde M, Hartig R, et al. Role of the small Rho GTPases Rac1 and Cdc42 in host cell invasion of Campylobacter jejuni. Cell Microbiol. 2007;9:2431–2444. doi: 10.1111/j.1462-5822.2007.00971.x. [DOI] [PubMed] [Google Scholar]
- Ashgar SS, Oldfield NJ, Wooldridge KG, et al. CapA, an autotransporter protein of Campylobacter jejuni, mediates association with human epithelial cells and colonization of the chicken gut. J Bacteriol. 2007;189:1856–1865. doi: 10.1128/JB.01427-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Cieplak W., Jr Altered synthetic response of Campylobacter jejuni to cocultivation with human epithelial cells is associated with enhanced internalization. Infect Immun. 1992;60:4945–4949. doi: 10.1128/iai.60.11.4945-4949.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Brown CK, Gu ZY, Matsuka YV, et al. Structure of the streptococcal cell wall C5a peptidase. Proc Natl Acad Sci USA. 2005;102:18391–18396. doi: 10.1073/pnas.0504954102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q. The Group B streptococcal C5a peptidase is both a specific protease and an invasin. Infect Immun. 2002;70:2408–2413. doi: 10.1128/IAI.70.5.2408-2413.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom L, Calabro V. FN3: a new protein scaffold reaches the clinic. Drug Discov Today. 2009;14:949–955. doi: 10.1016/j.drudis.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science. 1993;259:1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- Zarrinpar A, Bhattacharyya RP, Lim WA. The structure and function of proline recognition domains. Sci STKE. 2003;2003:RE8. doi: 10.1126/stke.2003.179.re8. [DOI] [PubMed] [Google Scholar]
- Davey NE, Trave G, Gibson TJ. How viruses hijack cell regulation. Trends Biochem Sci. 2011;36:159–169. doi: 10.1016/j.tibs.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Watson RO, Galan JE. Signal transduction in Campylobacter jejuni-induced cytokine production. Cell Microbiol. 2005;7:655–665. doi: 10.1111/j.1462-5822.2004.00498.x. [DOI] [PubMed] [Google Scholar]
- Boehm M, Krause-Gruszczynska M, Rohde M, et al. Major host factors involved in epithelial cell invasion of Campylobacter jejuni: role of fibronectin, integrin beta1, FAK, Tiam-1, and DOCK180 in activating Rho GTPase Rac1. Front Cell Infect Microbiol. 2011;1:17. doi: 10.3389/fcimb.2011.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause-Gruszczynska M, Boehm M, Rohde M, et al. The signaling pathway of Campylobacter jejuni-induced Cdc42 activation: role of fibronectin, integrin beta1, tyrosine kinases and guanine exchange factor Vav2. Cell Commun Signal. 2011;9:32. doi: 10.1186/1478-811X-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, McDaniel JP, Kopecko DJ. Signal transduction events involved in human epithelial cell invasion by Campylobacter jejuni 81–176. Microb Pathog. 2006;40:91–100. doi: 10.1016/j.micpath.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Haag LM, Fischer A, Otto B, et al. Campylobacter jejuni induces acute enterocolitis in gnotobiotic IL-10-/- mice via Toll-like-receptor-2 and -4 signaling. PloS ONE. 2012;7:e40761. doi: 10.1371/journal.pone.0040761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert E, Karrasch T, Sun X, et al. Gnotobiotic IL-10; NF-kappaB mice develop rapid and severe colitis following Campylobacter jejuni infection. PloS ONE. 2009;4:e7413. doi: 10.1371/journal.pone.0007413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen LN, Luijkx TA, Vegge CS, et al. Identification of immunogenic and virulence-associated Campylobacter jejuni proteins. Clin Vaccine Immunol. 2012;19:113–119. doi: 10.1128/CVI.05161-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Liu B, Sartor RB, Jobin C. Phosphatidylinositol 3-kinase-γ signaling promotes Campylobacter jejuni-induced colitis through neutrophil recruitment in mice. J Immunol. 2013;190:357–365. doi: 10.4049/jimmunol.1201825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.