

Abstract
Rationale: Shifts in the gene expression of nuclear protein in chronic obstructive pulmonary disease (COPD), a progressive disease that is characterized by extensive lung inflammation and apoptosis, are common; however, the extent of the elevation of the core histones, which are the major components of nuclear proteins and their consequences in COPD, has not been characterized, which is important because extracellular histones are cytotoxic to endothelial and airway epithelial cells.
Objectives: To investigate the role of extracellular histones in COPD disease progression.
Methods: We analyzed the nuclear lung proteomes of ex-smokers with and without the disease. Further studies on the consequences of H3.3 were also performed.
Measurements and Main Results: A striking finding was a COPD-specific eightfold increase of hyperacetylated histone H3.3. The hyperacetylation renders H3.3 resistant to proteasomal degradation despite ubiquitination; when combined with the reduction in proteasome activity that is known for COPD, this resistance helps account for the increased levels of H3.3. Using anti-H3 antibodies, we found H3.3 in the airway lumen, alveolar fluid, and plasma of COPD samples. H3.3 was cytotoxic to lung structural cells via a mechanism that involves the perturbation of Ca2+ homeostasis and mitochondrial toxicity. We used the primary human airway epithelial cells and found that the antibodies to either the C or N terminus of H3 could partially reverse H3.3 toxicity.
Conclusions: Our data indicate that there is an uncontrolled positive feedback loop in which the damaged cells release acetylated H3.3, which causes more damage, adds H3.3 release, and contributes toward the disease progression.
Keywords: chronic obstructive pulmonary disease, histone H3.3, acetylation, cytotoxicity, proteomics
At a Glance Commentary
Scientific Knowledge on the Subject
Several mechanisms involved in the development of chronic obstructive pulmonary disease (COPD) have been described, including the expression of proinflammatory molecules, increased lung cell death, and tissue remodeling. However, the mechanism behind the progression of the disease has not been established.
What This Study Adds to the Field
We found increased histone 3 expression in COPD lungs, which correlated with the progression of the disease. Analysis of posttranslational modifications identified a specific histone 3.3 (H3.3) isoform with hyperacetylation, methylation, and ubiquitination patterns. The hyperacetylated H3.3 was resistant to proteasomal degradation. Extracellular H3.3 was present in the lumen of the airway and was also prominent in the bronchoalveolar lavage fluid and the plasma of subjects with COPD. Moreover, H3.3 was cytotoxic to lung structural cells by a mechanism that was driven by perturbation of Ca2+ homeostasis and mitochondrial toxicity. These points suggest that free H3.3 participates in a self-sustaining cell death process that contributes to COPD progression; we observed that blocking this process could be therapeutic.
The prevalence of chronic obstructive pulmonary disease (COPD) is increasing in industrialized countries (1, 2). COPD is the third leading cause of death worldwide (3). In 2011, the CDC estimated that 15 million adults in the United States had COPD, which contributed to $49 billion in direct and indirect healthcare costs (4, 5). Disease management can relieve symptoms and prolong life, although there are no treatments to stop the disease progression, which ultimately results in death.
Although the molecular pathophysiology that underlies COPD has not been established, it is known that the expression of proinflammatory molecules, lung cell death, and tissue remodeling play critical roles (2, 6–8). Shifts in transcription factor activation and epigenetic markers, such as altered histone acetylation, are associated with changes in signaling proteins upstream of regulatory genes and in the assembly of nuclear chromatin complexes (9, 10). Core histones H2A, H2B, H3, H4 and their variants are key components of nuclear chromatin, the scaffolding that controls interactions between DNA with transcription factors and RNA polymerase, thereby regulating gene expression (11). Post-translational modification of histones causes chromatin remodeling, which alters these interactions (12–14). COPD is associated with a reduced histone deacetylase (HDAC) activity, which results in increased histone acetylation that might contribute to COPD-associated lung inflammation (15–17). Hyperacetylation of histone lysine is reportedly associated with inflammatory gene expression (15–17), but the nature of other post-translational modifications and amino acids involved is poorly understood. In addition to their role in chromatin regulation, histones released in the circulation can induce lung inflammation, and extracellular histones are cytotoxic to human lung cells (18). Significantly, extracellular histones are mediators of trauma-associated lung injury (19). Although the elevation of extracellular nuclear protein, such as HMGB1 in COPD, has been reported (20), the role and consequences of extracellular histones have not been examined. Our studies are designed to address these gaps in knowledge with an overall goal of understanding the disease progression in COPD. Our results support the hypothesis that extracellular, degradation-resistant, hyperacetylated H3.3 is elevated in COPD. The H3.3 binds to the cell membranes and induces cellular calcium influx and endoplasmic reticulum (ER) calcium depletion which, results in mitochondrial toxicity by repetitive alteration of the mitochondria membrane potential. Several results that support our hypothesis have been previously reported in abstract form (21).
Methods
Human Lung Tissue Samples
We obtained frozen lung tissues (n = 82) from the region’s transplant network, Life Inc., and the Temple University Lung Tissue Bank, both under a protocol approved by the Temple University Institutional Review Board. The subjects’ demographic information, including smoking history and lung function, are shown in Table E1 in the online supplement. Additionally, we obtained bronchoalveolar lavage fluid (BALF) (n = 20) and plasma (n = 15) samples from 15 subjects with COPD (Global Initiative for Chronic Obstructive Lung Disease [GOLD] stage II for BALF [n = 10] and stage IV for plasma [n = 5]); there were 15 subjects without COPD (BALF [n = 10] and plasma never-smokers [n = 5] and current smokers [n = 5]) (see Table E3).
Lung Tissue Proteomic Analysis
Lung tissues were subjected to nuclear protein extraction and were processed for gel electrophoresis–liquid chromatography–mass spectroscopy analysis. The levels of the protein identified were assessed by label-free quantitation. The mass spectroscopy analysis was used for the identification of histone post-translational modifications. Extensive details are provided in the online supplement.
Immunochemistry
We evaluated H3.3 protein levels in the lung tissue, BALF, and plasma using Western blots, slot blots, ELISA, immunohistochemistry, and immunoprecipitation analysis. Extensive details are provided in the online supplement.
Mechanism of Increased H3.3 Levels in COPD
We evaluated the H3 family mRNA expression levels by TaqMan quantitative polymerase chain reaction assays. The impaired proteosomal degradation of hyperacetylated H3.3 in subjects with COPD and control subjects was determined using the Western blot analysis. Extensive details are provided in the online supplement.
Histones Cytotoxicity Assays
We used the primary human bronchial epithelial cells to evaluate in vitro the cytotoxicity effect of H3.3 purified from subjects with COPD and human recombinant histones H2B, H3.3, and H4 (New England Biolabs, Ipswich, MA). We performed the proteomic analysis to elucidate the mechanisms that are implicated in H3 cytotoxicity. Extensive details are provided in the online supplement.
H3.3-mediated Ca2+ Signaling and Mitochondrial Toxicity
We performed simultaneous measurements of cytoplasmic and mitochondrial Ca2+ changes ([Ca2+]c and [Ca2+]m) in human lung endothelial cells by confocal microscopy. Extensive details are provided in the online supplement.
Statistical Analysis
Significant variation in the data within groups was determined using either the Kruskal-Wallis test or analysis of variance. The Mann-Whitney U test with Bonferroni correction was used to compare the difference between both groups, unless otherwise specified. Extensive details are provided in the online supplement.
Results
H3.3 Levels Are Elevated in COPD
Because the COPD-associated changes likely affect the nuclear proteome, we examined the nuclear proteins from the lung samples of subjects that were classified by GOLD to have advanced disease (GOLD IV), and we compared the results with those from a control group composed of ex-smokers who had normal lung function. To eliminate complications caused by the direct effects of smoking, both cohorts comprised ex-smokers with similar smoking histories (see Table E1).
When the GOLD IV subjects were compared with the ex-smoker control subjects, we found proteome elevations in several nuclear proteins, including several proteins that were involved in gene expression control, several proteins that were major components of the nucleosome core, and proteins that mediated the inflammatory processes and ion transport (see Table E2). The eightfold increase in histone H3.3 was the most marked change (see Table E2). Because the proteomic analysis in individual samples was not feasible, we used the methods dependent on anti-H3 antibodies. The H3 family is composed of H3.1, H3.2, and H3.3 variants, which are highly conserved (see Figure E2). The available antibodies are likely to cross-react among the three variants, and we used the monoclonal antibody that recognizes C-term of these variants. We verified our pooled proteomic results by the Western blot analysis in each of the five individual samples. The H3.3 was increased by eightfold (i.e., similar to the proteomic results) (Figures 1A and 1B). To confirm this increase and to determine the H3.3 levels in other COPD stages, we used an ELISA assay to examine the lung tissue from subjects classified as GOLD IV (n = 24); GOLD I–III (n = 12); and ex-smokers (n = 9). We also included never-smokers (n = 14) and current smokers (n = 11). The results showed COPD-significant H3.3 elevations between the GOLD IV and the four control groups (P < 0.02 using the Kruskal-Wallis test) and between GOLD I–III and control subjects (P < 0.02 using the Kruskal-Wallis test) (Figure 1C). Because the severity of pathologic changes in COPD is classified by spirometric measurements, we also correlated the H3.3 levels to FEV1 and FEV1/FVC by performing linear regression analysis. The H3.3 levels correlated poorly with either FEV1 or FEV1/FVC (R2 = 0.06648 and R2 = 0.1169, respectively) (see Figure E3).
Figure 1.

(A) Verification of the proteomic results by Western blotting of the individual samples used to comprise the pool of the discovery groups. (B) Quantitation of the Western blot results from the verification groups (*P < 0.001). (C) H3.3 is elevated in chronic obstructive pulmonary disease (COPD) and is not altered in other lung inflammatory diseases. The H3.3 levels were determined in Global Initiative for Chronic Obstructive Lung Disease (GOLD) IV and GOLD I–III, never-smokers, ex-smokers, and smokers using ELISA. The differences in H3.3 between the cohorts were assessed using the Kruskal-Wallis test; P < 0.02 values were obtained when GOLD I–III were compared with the four control groups and GOLD IV was compared with the four control groups.
H3.3 Is Present Extracellularly in the Airway in COPD
Because extracellular histones exert biologic effects that could be relevant to COPD (22), the presence of extracellular H3.3 in the lung was assessed with immunohistochemistry using the anti-H3 antibody that was used for ELISA studies (Figure 2A).
Figure 2.

(A) Immunohistochemical analysis of the lungs of Global Initiative for Chronic Obstructive Lung Disease (GOLD) IV and ex-smokers, showing the presence of extracellular H3.3 in the lumen of the airway inside the plug of mucus together with cell debris; these free histones are attached to the cilia of the epithelial cells (red arrowheads). (B) The Western blot quantitation of H3.3 in the bronchoalveolar lavage fluid (BALF) of GOLD II and ex-smoker subjects (see Figure E3). (C) Plasma H3.3 levels in subjects with GOLD IV as compared with current and never-smokers as determined by Western blot analysis (see Figure E3). P < 0.05 using the Kruskal-Wallis test.
As expected, all of the cell nuclei demonstrated H3.3 immunoreactivity. The specificity of the immunohistochemistry was assessed by preincubation of the H3 antibody with 2 μg of human recombinant H3.3 (New England Biolabs) protein, and no signal was observed. Notably, the H3.3 staining was also present in the mucus plugs in the airway lumen and in the cell debris attached to the cilia of airway epithelial cells in severe COPD (n = 4). Additionally, monocytes also demonstrated cytoplasmic and nuclear staining for H3.3 in severe COPD. In contrast, H3.3 immunoreactivity was present in the control subjects in only the lung cell nuclei (n = 4). These data indicate the extracellular and intracellular presence of H3.3 in the airways and lung parenchyma of subjects with COPD.
H3.3 Is Present in the Cell-Free BALF and Plasma in COPD
Given the presence of increased amounts of H3.3 in the COPD lung and the presence of extracellular H3.3 in advanced COPD, we sought to determine whether this histone was also present in BALF in subjects with milder COPD. Accordingly, we obtained BALF from the stable subjects with COPD with moderate disease (GOLD II; n = 10), and from normal, never-smokers (n = 10) who served as control subjects (see Table E3). There was a significant increase in H3.3 in the BALF supernatant among the subjects with COPD relative to the never-smokers (Figure 2B; see Figure E4A).
Because H3.3 was present in BALF, we also sought to determine whether H3.3 was secreted into the systemic circulation. In fact, H3.3 was present threefold higher in the plasma of the subjects with COPD (n = 5) compared with the current (n = 5) and never-smokers (Figure 2C; see Figure E4B).
H3.3 Gene Expression in COPD
To explore the mechanisms that underlie an increase in H3.3, we compared the mRNA levels of histone H3.3 (H3F3A and H3F3B), H3.1 (1H3B), and H2B (H2B2) in the lung tissue samples from patients who were classified as ex-smokers (n = 8), GOLD II (n = 8), and GOLD IV (n = 8). Using the validated commercial TaqMan quantitative polymerase chain reaction assays we observed twofold increases in H3F3A mRNA and H3F3B mRNA (P < 0.05 using the Kruskal-Wallis test) in GOLD IV relative to ex-smokers (Figure 3), but no significant differences between GOLD II and ex-smokers for these mRNAs or between other mRNAs and any groups, which is further supported by previous microarray studies (23, 24). Collectively, these results indicate that gene expression alone does not fully explain the increase in the levels of H3.3 proteins seen in the entire spectrum of COPD severity.
Figure 3.

Comparison of gene expression levels of H3.3, H3.1, and H2B in the individual lung tissue from subjects with chronic obstructive pulmonary disease (Global Initiative for Chronic Obstructive Lung Disease [GOLD] II and IV) and control subjects (ex-smokers), as determined by the TaqMan quantitative polymerase chain reaction assay system, to measure mRNA. Gene expression levels were not different when GOLD II was compared with control subjects. Only twofold increase in H3.3 genes expression (H3F3A and H3F3B) was observed when GOLD IV was compared with the control subjects. P < 0.05 by Kruskal-Wallis.
Mechanism of H3.3 Increase in COPD Involves Impaired Degradation
To further understand the mechanism by which H3.3 is increased, we focused on the impaired degradation of H3.3. Because it is known that the proteasomal activity is reduced in COPD (25) and our proteomic-based histone modification data revealed H3.3 ubiquitination at lysine 57 and 123 (Figures 4 and 5; see Table E4 and Figure E7), modifications are expected to reduce sensitivity to proteasome degradation. The combination of reduced proteasome activity with resistance to proteasome degradation supports the hypothesis that impaired degradation leads to increased H3.3. To explore the ubiquitinated H3.3 accumulation, we purified nuclear histones from the pooled samples of five GOLD IV and five ex-smoker subjects and subsequently prepared the Western blots using antiubiquitin and anti-H3 antibodies. The accumulation of ubiquitinated H3.3 (H3.3-Ubi2) in GOLD IV samples shown by the Western blot was confirmed by reciprocal immunoprecipitation (Figure 4A). Of note, the 37-kD band showing ubiquitination and H3.3 identity observed in the Western blots is consistent with the mass spectroscopy data for H3.3 (Figure 4B; see Table E4 and Figure E7).
Figure 4.

(A) Western blots showing a comparison of the ubiquitination pattern of H3.3 in the chronic obstructive pulmonary disease and ex-smokers groups. (B) Identification of H3.3 ubiquitination by mass spectroscopy. The tandem mass spectrometry profile of each of the two identified peptides that were differentially ubiquitinated in chronic obstructive pulmonary disease and their representative sequence are shown at the top left of each panel.
Figure 5.

Ribbon diagram of histone H3.3 showing a comparison of acetylated (blue spheres) or ubiquitylated (red spheres) residues in Global Initiative for Chronic Obstructive Lung Disease IV (chronic obstructive pulmonary disease [COPD]) and ex-smokers (Control), as identified by mass spectroscopy.
Previous COPD studies using the Western blot analysis and chromatin immunoprecipitation (ChIP) assays demonstrated increased acetylation of histones 3 and 4, which was attributed to the decreased HDAC expression and activity (10, 16, 22). Because our mass spectroscopy analyses revealed missed trypsin cleavages that correlated with acetylation sites (see Table E5 and Figure E8), we investigated whether H3.3 might be differentially acetylated in COPD.
We confirmed a previously reported COPD-associated histone N-terminal hyperacetylation, identifying the specific residues that are acetylated as those that are at Lys 10, 15, 19, and 24 in H3.3 (Figures 5 and 6), and the extent of this acetylation in COPD (Figure 7). Our results indicated approximately 2.5-fold increase in H3.3 acetylation in COPD. We also found a previously unreported C-terminal acetylation at Lys 116 (Figure 5; see Tables E4 and Table E5 and Figure E8). We confirmed the effect of acetylation on proteasome degradation, using histones that were purified from A549 cells treated with a combination of HDAC inhibitors (trichostatin, MS-275, and suberoylanilide hydroxamic acid) to produce hyperacetylated histones (26). In Figure 8, the Western blots show that hyperacetylated H3.3, which is purified from the treated cells, is less sensitive to degradation by 20S proteasomes than H3.3 from the control cells. The confirmation of proteasomal dependency was demonstrated using the specific proteasome inhibitor MG132. Figure 8B shows that the effect of acetylation remains prominent even after 16 hours of proteasome exposure. Using matrix-assisted laser desorption ionization time-of-flight mass spectroscopy, we also determined that all four N-terminal H3.3 acetylation played a role in obtaining maximal resistance to proteosomal degradation (see Figures E5A and E5B).
Figure 6.

(A) Identification of H3.3 acetylation by mass spectroscopy. The tandem mass spectrometry profile of two representatives of the acetylated peptides identified in chronic obstructive pulmonary disease (COPD); their sequences are shown at the top left of each panel. (B) The Western blots showing the expression levels of H3 AcK24 and H3.3 in individual lung lysates (n = 5) from Global Initiative for Chronic Obstructive Lung Disease IV and ex-smokers. These results confirm the mass spectrometry data in A.
Figure 7.

Histone 3.3 acetylation detection by slot blotting. (A) Detection of the expression levels of H3.3, H3 AcK10, H3 AcK19, and H3 AcK24 in individual lung lysates (n = 6, each) from Global Initiative for Chronic Obstructive Lung Disease (GOLD) IV, current smokers, never-smokers, and ex-smokers as determined by slot blot. (B) The total acetylation levels were determined by quantitative densitometry; the values were normalized against the total H3.3 and are shown as the average of each group. The blue diamond represent H3 AcK24, red circles H3 AcK19, and black triangles H3 AcK10; P < 0.005 by Kruskal-Wallis test.
Figure 8.

Impaired proteasomal degradation of hyperacetylated H3.3 in chronic obstructive pulmonary disease. (A) Western blots showing in vitro evaluation of H3.3 degradation with and without hyperacetylation. Both acetylated and nonacetylated histones were purified from A549 cells treated with and without histone deacetylase inhibitors. Hyperacetylation of H3.3 was confirmed by Western blotting (Lanes 1 and 2). A 20S proteasome degradation assay was performed on these two samples, which showed that hyperacetylated H3.3 was resistant to degradation (Lanes 3 and 4). Proteasomal-specific degradation of H3.3 was determined by the addition of proteasome inhibitor MG132 (Lane 5). (B) Comparison of the effect of H3.3 proteasomal degradation over 16 hours with and without hyperacetylation, as quantitated by Western blotting and densitometry analysis. All of the data are expressed as the mean ± SEM for at least three independent experiments. For all of the comparisons P < 0.001. (C) Western blots showing the effect of cigarette smoke condensate 200 μg/ml (Lane 2), dexamethasone 1 μM (Lane 3), and theophylline 5 μg/ml (Lane 4) on H3.3 and its acetylation in the primary lung cells. Untreated cells and the cells treated with a combination of histone deacetylase inhibitors (Lanes 1 and 5) were used as negative and positive controls, respectively.
H3.3 Levels Are Not Affected by Glucocorticoids and Cigarette Smoke Condensate
The H3.3 expression was similar in ex-smokers and current smokers without COPD, which suggests that the change may not be a direct consequence of smoking per se. To further investigate the effect of smoking, we treated the primary lung cells with cigarette smoke condensate and found that the H3.3 levels were unchanged in the treated group compared with the untreated control subjects (Figure 8C).
Because several subjects in the COPD groups were treated with glucocorticoids, bronchodilators that can alter histone acetylation (27, 28), we compared the effect of theophylline and dexamethasone on H3.3 and its acetylation. The untreated primary lung cells and the cells treated with a combination of HDAC inhibitors were used as negative and positive controls, respectively. These drugs had no effect on either the H3.3 levels or Ac K10H3 (Figure 8C).
Extracellular H3.3 Induces Apoptosis by Perturbing Calcium Homeostasis and Inducing Unfolded Protein Response and Mitochondrial Toxicity
Extracellular histones are products of cell death and are known to have diverse effects. Although not usually considered to be noxious, they can induce lung inflammation, and cultured human pulmonary artery endothelial cells can undergo apoptosis when exposed to free core histones (18, 19, 29), effects that could relate to the pathogenesis of COPD. Using primary human airway epithelial cells exposed to H3.3 that is purified from GOLD IV lung tissue and using commercially sourced human H3.3, we found that both histones were equally cytotoxic to these primary human lung cells (Figure 10A). These data indicated that acetylation alone might not play a role in the toxicity of histones. Therefore, for the remainder of the studies, we used commercially available H3.3. We found that H3.3 was localized in the cell membrane (Figure 9) and could induce concentration-dependent apoptosis (Figure 10B). We used a H3.3 concentration (10 μg/ml) that was similar to the concentrations found in the epithelial lining fluid of COPD lungs. On comparing histones elevated in COPD, we found the following relations: the toxicity of H3.3 greater than H4 greater than H2B (Figure 10C). To address whether the intracellular H3.3 or extracellular H3.3 was responsible for cytotoxicity, we compared the extent of cell death caused by the primary lung cells endogenously overexpressing recombinant H3.3 to the cells transfected to secrete recombinant H3.3. Figure 10D shows the dramatic increase in cell death associated with the secreted recombinant H3.3 compared with the intracellular H3.3 and normal cells (P < 0.001 using the analysis of variance test).
Figure 10.

(A) Comparison of the cytotoxicity of enriched H3.3 from chronic obstructive pulmonary disease (COPD) nuclear lung lysates and recombinant H3.3. The primary lung cell damage was measured by a fluorescent detection of annexin V binding, 24 hours after incubation, with H3.3 extracted from the COPD lungs and recombinant H3.3 (10 μg/ml). (B) The primary cells were treated with increasing concentrations of recombinant H3.3 or (C) human recombinant histones H2B, H3.3, and H4 (10 μg/ml) for 24 hours. Cytotoxicity was evaluated by measuring bound fluorescent annexin V using a fluorescent reader and was normalized against the control cells without histone H3.3. (D) Comparison of the cell death in primary lung cells endogenously overexpressing recombinant H3.3 and the cells transfected to secrete recombinant H3.3. Cells transfected with empty plasmid were used as a control. P < 0.001 using the analysis of variance test. Bottom panel shows the secreted H3.3 in primary cells culture media as detected by Western blotting. (E) Partial reduction of H3.3 cytotoxicity in the presence of antibodies against the N- or C-terminal sequence of the H3 proteins. Isotype IgG mouse antibody was used as a control. Primary human epithelial cells were treated with purified H3.3 (10 μg/ml) for 24 hours. All of the data are expressed as the mean ± SEM of at least three independent experiments.
Figure 9.

(A) H3.3 cytotoxicity in the primary lung cells. Indirect immunofluorescence detecting H3.3 attached to the membrane of the primary lung cells. The cells were incubated with purified recombinant H3.3 (10 μg/ml) for 1 hour and were extensively washed with phosphate-buffered saline. (B) The primary lung cell damage was measured 24 hours after incubation with recombinant H3.3 (10 μg/ml) using fluorescent detection of annexin V binding and propidium iodide incorporation.
If the extracellular histones exert toxicity by binding to the cell membrane and thereby inducing apoptosis, we hypothesized that this mechanism could be blocked by anti-H3 antibodies. For a preliminary test, we used primary human airway epithelial cells and found that antibodies to either the C or N terminus of H3 could partially block H3.3 toxicity (Figure 10E).
Previous studies indicated that histones in the extracellular milieu induced apoptosis of human pulmonary endothelial cells by the activation of TLR2 and TLR4 (29), affected Ca2+ homeostasis (19, 30), and effect the mitochondrial function (30); the full pathway has not been described. To study this pathway, we analyzed proteome changes in the human primary airway epithelial cells that were exposed to H3.3. The data indicated that apoptosis is driven by the unfolded protein response (UPR), eukaryotic translation initiation factor-2α (eIF2α), and Ca2+ signaling (see Table E6). The UPR involvement is indicated by the elevation of chaperones GRP78, GRP94, and foldase protein disulfide isomerase (PDI) A4 (Figure 11; see Table E6). PERK-dependent initiation is revealed by increased phosphorylated eIF2α, C/EBP-homologous protein (CHOP), and cleaved caspase 7 with cytoplasmic translocation in a time-dependent manner (Figure 11B). Increases in cleaved caspases 3 and 9 (Figure 11A) provide additional evidence of UPR-driven apoptosis. UPR can be triggered by perturbation of Ca2+ flux. Our results showed elevations of primary endothelial cell cytoplasmic (Figure 12A) and mitochondrial Ca2+ levels (Figure 12B) in response to treatment with H3.3. The [Ca2+]c and [Ca2+]m elevations are persistent even after the depletion of the extracellular Ca2+ (see Figure E6), which indicates that UPR is involved in H3.3-mediated Ca2+ signaling. Significantly, the increase in mitochondrial Ca2+ resulted in a loss of mitochondrial membrane potential in a dose-dependent manner (Figures 12C and 12D). These results supported and extended earlier work on lung tissue lysates, which linked UPR to COPD (25); they also connected Ca2+ signaling, impaired mitochondrial function, and H3.3 toxicity (18, 19, 30–32).
Figure 11.

(A) Western blots showing the cleavage of the apoptotic markers caspase 3 and 9, and the unfolded protein response apoptotic markers CHOP, eIF2α, and GRP78, from primary cells that were treated with H3.3. The concentration of H3.3 (10 μg/ml) that was used was similar to the concentration that was present in the epithelial lining fluid of chronic obstructive pulmonary disease lungs, based on the calculations from the amounts in the bronchoalveolar lavage fluid. (B) Measurement of time-dependent caspase 7 activation by Western blotting of endoplasmic reticulum (ER)-enriched and cytoplasmic fractions to determine the mechanism that is implicated for H3.3 cytotoxicity. The primary cells were incubated with H3.3 (10 μg/ml) for the time period indicated and were subsequently subjected to subcellular fractionation, to separate ER-enriched and cytoplasmic fractions.
Figure 12.
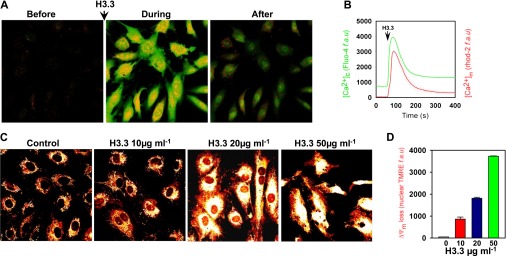
Changes in human endothelial cell cytoplasmic and mitochondrial matrix [Ca2+] in response to H3.3 were simultaneously measured by fluo-4 and rhod-2 imaging, respectively. (A) Representative images of human endothelial cells showing cytosolic (green) and mitochondrial (red) [Ca2+] before, during, and after histone 3.3 exposure. (B) Changes in cytoplasmic (green) and mitochondrial matrix (red) [Ca2+] responses in response to H3.3 (10 μg). n = 3. (C) Representative images showing TMRE (mitochondrial membrane potential indicator) fluorescence in human endothelial cells treated with 10, 20, or 50 μg/ml of H3.3 for 24 hours. Untreated cells served as controls. (D) Bar chart represents the quantitation of nuclear TMRE fluorescence (indicator of membrane potential loss). n = 3.
Endoplasmic reticulum stress that is associated with UPR is also a major producer of reactive oxygen species (33–35), which can cause tissue damage similar to that seen in COPD. The cells defend against reactive oxygen species with pathways that are initiated by the Nrf-2 network, and the proteome data (see Table E6) showed that this network is activated in human epithelial lung cells that are exposed to H3.3, thereby providing a further tie between extracellular H3.3 and COPD.
Discussion
COPD is characterized by extensive lung inflammation, cell apoptosis, and tissue remodeling, changes that are commonly associated with shifts in gene expression that are induced by increases in proinflammatory transcription factors (e.g., NfκB), impaired expression of antioxidant transcription factors (e.g., Nrf2), and epigenetic alterations in the chromatin structure that affects histone acetylation (36, 37). Because such changes suggest histone proteome shifts that alter cell function, we predicted that a better understanding of the histone expression would contribute to a better understanding of COPD disease progression and perhaps new approaches to treatment. We tested this prediction by comparing the protein composition of a nuclear-enriched fraction of peripheral lung lysates from subjects with advanced COPD (i.e., GOLD IV) to control preparations from ex-smokers with normal lung function. To eliminate the complications that can be caused by current smoking behavior, both groups comprised ex-smokers with similar smoking histories.
Our results showed that several nuclear proteins that are known to affect gene expression are elevated in the lungs of subjects with COPD, the most notable being those that belong to the core histones, especially H3.3. Relative to control subjects, the lung samples from subjects with COPD showed increased H3.3 in extracellular spaces, cellular debris, airway lumen mucous, BALF, and plasma. We were unable to demonstrate the origin of extracellular H3.3 in COPD lung samples; however, the following points suggested that they are products of apoptosis. Core histones H2A, H2B, H3, and H4 are known to increase early in apoptosis and can be found in the cytoplasm and exposed on the cell surface (38). The lungs affected by COPD showed increased apoptosis among the structural and inflammatory cells (39). Histones are reported to be elevated in neutrophils that are found in expectorated sputum and in alveolar pneumocytes and pulmonary artery endothelial cells, which provides additional evidence of increased apoptosis (18, 40). Moreover, the circulating core histones have been demonstrated to be mediators of trauma-associated lung injury (19). Core histone acetylation affects gene expression by enhancing the transcriptional complex access to chromatin DNA (11). Previous ChIP assays of COPD lung tissue demonstrated increased acetylation of H3 and H4, which are changes that are attributed to decreased HDAC expression and activity (10, 15, 16). Using mass spectroscopy, a completely different methodology, we were able to map the entire sequence and characterize with fine detail the post-translational modifications of H3.3 in ex-smokers with and without COPD; we found hyperacetylation of H3.3 with a pattern of N-terminal lysine hyperacetylation (Lys 10, 15, 19, and 24) in four of the six acetylation targets (41). We also reported a novel observation of C-terminal lysine acetylation (Lys 116). Although we have not studied the effect of Lys 116 acetylation directly, a reduction in the histone-positive charge from acetylation is known to enhance chromatin unwinding and lead to an increased transcription of the inflammatory proteins that are associated with COPD (41). Acetylation reduces the sensitivity to proteasome degradation (42, 43); thus, Lys 116 acetylation could contribute to the observed H3.3 increase. Resistance to proteasome degradation caused by H3.3 acetylation is supported by our in vitro assays (Figure 8; see Figures E5A and E5B), which demonstrated that histone modifications, especially acetylations, promote resistance to 20S proteasome degradation; these results were in agreement with a previous report (25).
Because previous studies have shown that core histones are cytotoxic to endothelial cells and our results show 8.2-fold increase for H3.3 (see Table E2), compared with 1.8- and 1.9-fold for H4 and H2b (Figure 10C), and H4 and H2b were 26% and 89% less toxic than H3.3, we chose to pursue the mechanism of cytotoxicity of H3.3. It is possible that H4 and H2b could play a minor role in inducing cellular toxicity and their effect has to be pursued in future studies. Post-translational modification analysis indicates a greater modification of H3.3 (see Table E4; Figure 5), which suggests greater resistance to proteasome degradation and helps to account for the higher levels of H3.3. Histones H3.1, H3.2, and H3.3 have cell cycle functions with expression patterns (44), but H3.3 has been reported to be expressed independent of the cell cycle stage, with protein translocation to active gene transcription regions (45).
Our data suggested that molecular modifications of histones and their release from apoptotic cells could trigger further apoptosis, thus creating a pathogenic cascade. The addition of H3.3 to cultured human airway epithelial cells produces dose- and time-dependent caspase activation (Figure 11), a finding that is consistent with earlier studies that reported that free core histones cause cultured human pulmonary artery endothelial cells to undergo apoptosis (18, 19, 29, 40).
Core histones bind to and disrupt cell membranes, causing an increase in Ca2+ influx (19) and mitochondrial toxicity (30). Our results support these studies and show specifically that H3.3-induced apoptosis involves binding to cell membrane, induction of Ca2+ influx, emptying of ER Ca2+ pools, induction of UPR, elevation of [Ca2+]m, and mitochondrial toxicity.
UPR has prosurvival and proapoptotic signaling pathways. If prosurvival signaling allows resolution of the stress condition, the cell survives; if prosurvival signaling fails, the proapoptotic pathways dominate and the cell undergoes apoptosis. One prosurvival response is the increase in ER-resident chaperones, such as GRP78/calreticulin/calnexin/GRP94 and GRP170, and foldases, such as PDI (46). We find that H3.3 exposure causes an elevation in ER chaperones GRP78 and GRP94, and the foldase PDI A4 (see Table E6). One proapoptotic pathway is triggered by the activation of PERK, which leads to enhanced translation of transcription factor CHOP (47). CHOP inhibits the expression of the antiapoptotic factor BCL-2 and activates the ER-resident caspase 7 (48, 49), an apoptosis enzyme. We show that H3.3 exposure results in the activation of CHOP and also phosphorylation of eIF2α (Figure 11), which is evidence of apoptosis induction by PERK signaling. Phosphorylated eIF2α is crucial for the proapoptosis path because it facilitates the translation of selected mRNAs simultaneously globally repressing the translation of others. Inhibition of eIF2α allows up-regulation of the prosurvival factor ATF4, which enhances ER chaperone expression, thus helping to resolve the stress of misfolded proteins in the ER. Our proteome data also indicate the activation of Nrf2, which up-regulates the expression of antioxidant and xenobiotic metabolizing enzymes (50). PERK also directly phosphorylates Nrf2, which facilitates its dissociation from the cytoplasmic inhibitor, KEAP1, and its translocation to the nucleus (51).
We reported several COPD-specific changes in lung histones that include an overall increase in H3.3, the presence of extracellular H3.3, H3.3 hyperacetylation, and a novel H3.3 acetylation site. We confirmed that hyperacetylation reduces H3.3 sensitivity to proteasome degradation which, in combination with the reported reduction of proteasome activity in COPD lungs and increased H3.3 transcription, helps to account for the increase in lung H3.3. We also confirmed the presence of H3.3 on lung cell membranes and the ability of this histone to induce apoptosis in the primary cultures of human airway epithelial cells and lung endothelial cells. Our data for the first time indicated that H3.3-induced apoptosis is driven, at least in part, by a UPR response and, in part, by Ca2+-induced mitochondrial toxicity. We conclude that COPD disease progression includes a positive feedback, self-sustaining cascade of apoptosis that results in the release of degradation-resistant histones, especially H3.3, which induce further lung cell apoptosis.
Acknowledgments
Acknowledgment
The authors thank the NIH for supporting this work through grants RC2-HL101713, R03-HL095437, and R03-HL095450. The authors also thank the Temple Lung Center Biobank for facilitating the lung tissue specimens, bronchoalveolar lavage fluids, and plasma samples. They also thank E. Jay Bienen for helping in editing the manuscript.
Footnotes
Supported by NIH grants RC2-HL101713, R03-HL095437, and R03-HL095450.
Author Contributions: C.A.B. designed and performed experiments, performed protein identification and post-translational modification analysis using mass spectrometry, and wrote the manuscript. O.P.-L., M.A., R.J., and Y.L. performed reverse transcriptase polymerase chain reaction, immunohistochemistry, Western blots, ELISA, and immunofluorescence. C.M. performed statistical analysis of identified proteins. K.M. and M.M. performed confocal microscopy for intracellular [Ca2+] and mitochondrial membrane potential measurements. S.G.K. and G.J.C. provided clinical parameters and tissue sample selection for the study. S.M. designed and supervised this study, analyzed the data, and wrote the manuscript.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201302-0342OC on August 7, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Maclay JD, Rabinovich RA, MacNee W. Update in chronic obstructive pulmonary disease 2008. Am J Respir Crit Care Med. 2009;179:533–541. doi: 10.1164/rccm.200901-0134UP. [DOI] [PubMed] [Google Scholar]
- 2.Stockley RA, Mannino D, Barnes PJ. Burden and pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:524–526. doi: 10.1513/pats.200904-016DS. [DOI] [PubMed] [Google Scholar]
- 3.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2013;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. Chronic obstructive pulmonary disease among adults—United States, 2011. MMWR Morb Mortal Wkly Rep. 2012;61:938–943. [PubMed] [Google Scholar]
- 5.NHLBI. Bethesda, MD: NIH Heart, Lung and Blood Institute–National Institutes of Health; 2009. Morbidity and mortality: 2009 chart book on cardiovascular, lung and blood diseases. [Google Scholar]
- 6.Lee G, Walser TC, Dubinett SM. Chronic inflammation, chronic obstructive pulmonary disease, and lung cancer. Curr Opin Pulm Med. 2009;15:303–307. doi: 10.1097/MCP.0b013e32832c975a. [DOI] [PubMed] [Google Scholar]
- 7.Postma DS, Timens W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:434–439. doi: 10.1513/pats.200601-006AW. [DOI] [PubMed] [Google Scholar]
- 8.Salazar LM, Herrera AM. Fibrotic response of tissue remodeling in COPD. Lung. 2011;189:101–109. doi: 10.1007/s00408-011-9279-2. [DOI] [PubMed] [Google Scholar]
- 9.MacNee W.Pathogenesis of chronic obstructive pulmonary disease Proc Am Thorac Soc 20052258–266.discussion 290–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajendrasozhan S, Yao H, Rahman I. Current perspectives on role of chromatin modifications and deacetylases in lung inflammation in COPD. COPD. 2009;6:291–297. doi: 10.1080/15412550903049132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamond AI, Earnshaw WC. Structure and function in the nucleus. Science. 1998;280:547–553. doi: 10.1126/science.280.5363.547. [DOI] [PubMed] [Google Scholar]
- 12.Voigt P, Reinberg D. Histone tails: ideal motifs for probing epigenetics through chemical biology approaches. ChemBioChem. 2011;12:236–252. doi: 10.1002/cbic.201000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood C, Snijders A, Williamson J, Reynolds C, Baldwin J, Dickman M. Post-translational modifications of the linker histone variants and their association with cell mechanisms. FEBS J. 2009;276:3685–3697. doi: 10.1111/j.1742-4658.2009.07079.x. [DOI] [PubMed] [Google Scholar]
- 14.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes PJ. Histone deacetylase-2 and airway disease. Ther Adv Respir Dis. 2009;3:235–243. doi: 10.1177/1753465809348648. [DOI] [PubMed] [Google Scholar]
- 16.Barnes PJ. Role of hdac2 in the pathophysiology of COPD. Annu Rev Physiol. 2009;71:451–464. doi: 10.1146/annurev.physiol.010908.163257. [DOI] [PubMed] [Google Scholar]
- 17.Barnes PJ. Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:693–696. doi: 10.1513/pats.200907-071DP. [DOI] [PubMed] [Google Scholar]
- 18.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, Wang SS, Brohi K, Kipar A, Yu W, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med. 2013;187:160–169. doi: 10.1164/rccm.201206-1037OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou C, Zhao H, Liu L, Li W, Zhou X, Lv Y, Shen X, Liang Z, Cai S, Zou F. High mobility group protein b1 (HMGB1) in asthma: comparison of patients with chronic obstructive pulmonary disease and healthy controls. Mol Med. 2011;17:807–815. doi: 10.2119/molmed.2010.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrero CA, Kelsen SG, Aksoy M, Ji R, Merali S. Extracellular histones in the COPD lung are cytotoxic [abstract] Am J Respir Crit Care Med. 2011;183:A3018. [Google Scholar]
- 22.Szulakowski P, Crowther AJ, Jimenez LA, Donaldson K, Mayer R, Leonard TB, MacNee W, Drost EM. The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174:41–50. doi: 10.1164/rccm.200505-725OC. [DOI] [PubMed] [Google Scholar]
- 23.Spira A, Beane J, Pinto-Plata V, Kadar A, Liu G, Shah V, Celli B, Brody JS. Gene expression profiling of human lung tissue from smokers with severe emphysema. Am J Respir Cell Mol Biol. 2004;31:601–610. doi: 10.1165/rcmb.2004-0273OC. [DOI] [PubMed] [Google Scholar]
- 24.Tilley AE, Harvey BG, Heguy A, Hackett NR, Wang R, O'Connor TP, Crystal RG. Down-regulation of the notch pathway in human airway epithelium in association with smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179:457–466. doi: 10.1164/rccm.200705-795OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malhotra D, Thimmulappa R, Vij N, Navas-Acien A, Sussan T, Merali S, Zhang L, Kelsen SG, Myers A, Wise R, et al. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: the role of nrf2-regulated proteasomal activity. Am J Respir Crit Care Med. 2009;180:1196–1207. doi: 10.1164/rccm.200903-0324OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Varghese S, Gupta D, Baran T, Jiemjit A, Gore SD, Casero RA, Jr, Woster PM. Alkyl-substituted polyaminohydroxamic acids: a novel class of targeted histone deacetylase inhibitors. J Med Chem. 2005;48:6350–6365. doi: 10.1021/jm0505009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ford PA, Durham AL, Russell RE, Gordon F, Adcock IM, Barnes PJ. Treatment effects of low-dose theophylline combined with an inhaled corticosteroid in COPD. Chest. 2010;137:1338–1344. doi: 10.1378/chest.09-2363. [DOI] [PubMed] [Google Scholar]
- 28.Inamochi Y, Mochizuki K, Osaki A, Ishii T, Nakayama T, Goda T. Histone h3 methylation at lysine 4 on the slc2a5 gene in intestinal caco-2 cells is involved in slc2a5 expression. Biochem Biophys Res Commun. 2010;392:16–21. doi: 10.1016/j.bbrc.2009.12.136. [DOI] [PubMed] [Google Scholar]
- 29.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through tlr2 and tlr4 in mouse fatal liver injury. J Immunol. 2011;187:2626–2631. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cascone A, Bruelle C, Lindholm D, Bernardi P, Eriksson O. Destabilization of the outer and inner mitochondrial membranes by core and linker histones. PLoS ONE. 2012;7:e35357. doi: 10.1371/journal.pone.0035357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gamberucci A, Fulceri R, Marcolongo P, Pralong WF, Benedetti A. Histones and basic polypeptides activate Ca2+/cation influx in various cell types. Biochem J. 1998;331:623–630. doi: 10.1042/bj3310623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleine TJ, Lewis PN, Lewis SA. Histone-induced damage of a mammalian epithelium: the role of protein and membrane structure. Am J Physiol. 1997;273:C1925–C1936. doi: 10.1152/ajpcell.1997.273.6.C1925. [DOI] [PubMed] [Google Scholar]
- 33.Hung CC, Ichimura T, Stevens JL, Bonventre JV. Protection of renal epithelial cells against oxidative injury by endoplasmic reticulum stress preconditioning is mediated by erk1/2 activation. J Biol Chem. 2003;278:29317–29326. doi: 10.1074/jbc.M302368200. [DOI] [PubMed] [Google Scholar]
- 34.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 35.Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–S109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 36.Adcock IM, Tsaprouni L, Bhavsar P, Ito K. Epigenetic regulation of airway inflammation. Curr Opin Immunol. 2007;19:694–700. doi: 10.1016/j.coi.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 37.Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J. 2009;33:1165–1185. doi: 10.1183/09031936.00128008. [DOI] [PubMed] [Google Scholar]
- 38.Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol. 2004;172:6692–6700. doi: 10.4049/jimmunol.172.11.6692. [DOI] [PubMed] [Google Scholar]
- 39.Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31:1334–1356. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- 40.Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. 2012;7:e32366. doi: 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito K, Charron CE, Adcock IM. Impact of protein acetylation in inflammatory lung diseases. Pharmacol Ther. 2007;116:249–265. doi: 10.1016/j.pharmthera.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 42.Buchwald M, Kramer OH, Heinzel T. HDACI–targets beyond chromatin. Cancer Lett. 2009;280:160–167. doi: 10.1016/j.canlet.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 43.Arendt CS, Hochstrasser M. Eukaryotic 20s proteasome catalytic subunit propeptides prevent active site inactivation by N-terminal acetylation and promote particle assembly. EMBO J. 1999;18:3575–3585. doi: 10.1093/emboj/18.13.3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant h3.3. Cell Res. 2011;21:421–434. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ray-Gallet D, Almouzni G. Nucleosome dynamics and histone variants. Essays Biochem. 2010;48:75–87. doi: 10.1042/bse0480075. [DOI] [PubMed] [Google Scholar]
- 46.Jager R, Bertrand MJ, Gorman AM, Vandenabeele P, Samali A. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol Cell. 2012;104:259–270. doi: 10.1111/boc.201100055. [DOI] [PubMed] [Google Scholar]
- 47.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Natl Rev. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 48.Faitova J, Krekac D, Hrstka R, Vojtesek B. Endoplasmic reticulum stress and apoptosis. Cell Mol Biol Lett. 2006;11:488–505. doi: 10.2478/s11658-006-0040-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho HY, Reddy SP, Debiase A, Yamamoto M, Kleeberger SR. Gene expression profiling of nrf2-mediated protection against oxidative injury. Free Radic Biol Med. 2005;38:325–343. doi: 10.1016/j.freeradbiomed.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 51.Cullinan SB, Diehl JA. Perk-dependent activation of nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279:20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]