

Abstract
Antenatal maternal hypoxia (AMH) can lead to intrauterine growth restriction (IUGR), as well as idiopathic pulmonary hypertension of newborn and adult, the latter of which may be a consequence of alterations in the local pulmonary renin-angiotensin system (RAS). Little is known of these adaptations, however. Thus, we tested the hypothesis that antenatal maternal hypoxia is associated with alterations in gene and protein expression of the pulmonary renin-angiotensin system, which may play an important role in pulmonary disorders in the offspring. In FVB/NJ mice, we studied messenger RNA (mRNA) and protein expression, as well as promoter DNA methylation and microRNA (miRNA) levels in response to 48 hours hypoxia (10.5% O2) at 15.5 day post coitum (DPC). In response to AMH, the pulmonary mRNA levels of angiotensin-converting enzyme (ACE) 1.2, ACE-2, and angiotensin II type 1b (AT-1b) receptors were increased significantly, as compared to controls (N = 4). In response to antenatal hypoxia, pulmonary protein levels of renin and ACE-2 also were increased significantly, whereas ACE-1 protein expression was reduced. In fetal lungs, we also observed reduced expression of the miRNAs: mmu-mir −199b, −27b, −200b, and −468 that putatively increase the translation of renin, ACE-1, ACE-2, and AT-1 receptors, respectively. In response to AMH, promoter methylation of ACE was unchanged. We conclude that AMH leads to changes in expression of pulmonary RAS of fetal mice. The possible implications of these changes for the regulation of pulmonary vascular contractility in later life remain to be explored.
Keywords: DNA methylation, microRNA, developmental origins
Introduction
In response to reduced oxygenation, the fetus redistributes cardiac output to the brain and heart, at the expense of the other tissues, and this relative hypoxia/ischemia can lead to various disorders during postnatal life. Of importance, antenatal maternal hypoxia (AMH) has been shown to cause altered heart growth and neonatal vascular function,1 permanent neurological deficits,2 pulmonary arterial dysfunction,3 atherosclerosis,4 and intrauterine growth restriction (IUGR).5
Some of these changes may be a consequence of relative tissue hypoxia/ischemia. In association with maternal antenatal stress, other studies suggest dysregulation of various genes during fetal life.6,7 In particular, the renin-angiotensin system (RAS) has received increased attention, and our understanding of this system has changed radically over the past several decades. Originally, RAS was regarded as a systemic cascade, in which the α2-globulin angiotensinogen (AGT) is produced constitutively and released into the circulation, chiefly by the liver. A substrate for renin (secreted by kidneys), AGT, is converted into the decapeptide angiotensin (Ang) I and subsequently by angiotensin-converting enzyme (ACE) 1 to the octapeptide Ang II. By the action of ACE-2, Ang II activity is terminated by its conversion to Ang 1-7. Of note, both ACE-1 and ACE-2 are secreted by the lung.8 Ang II is an important regulator of peripheral vascular resistance, as well as sodium and water metabolism in the body.
As noted, the RAS exists not only as a systemic pathway, but as a whole in various tissues and organ systems that function independently.9–12 Nonetheless, ACE is present in a high concentration in the lungs, and its activity is increased further by hypoxia.13 Ang II also may contribute to the development of pulmonary pathologies via its vasoconstrictor effects or by its action on vascular smooth muscle cell migration and growth with remodeling.14 Of importance, in rats, ACE inhibition has been shown to reduce hypoxia-induced pulmonary hypertension and pulmonary vascular remodeling.14
Hypoxia with resulting hypercapnia also can modulate the plasma renin activity during late gestation.15 However, the extent to which maternal hypoxia can lead to changes in the expression of the RAS components in fetal lungs are not known. Thus, in the current study, we tested the hypothesis that antenatal maternal hypoxia is associated with alterations in gene and protein expression of the pulmonary renin-angiotensin system, which may play an important role in pulmonary disorders in the offspring.
Materials and Methods
Experimental Animal and Tissues
All experimental procedures were approved by the Animal Care and Use Committee (IACUC) of Loma Linda University. We conducted studies on FVB/NJ mice; a strain chosen because of the relatively large size of their fetuses. At 16 weeks of age, we bred the mice by keeping the males and females together for 12 hours (overnight). In the morning, we confirmed the mating by examination of vaginal plugs and considered that 0.5 days postcoitum (DPC). Pregnancy was confirmed by increase in the weight at 7 DPC. As previously described,16 following breeding, at 17.5 DPC the dams were killed by cervical dislocation. The uterus was removed rapidly and placed in a Petri dish containing physiological buffered saline (PBS) solution at 4°C. Fetal lungs were isolated under a dissection microscope, and other tissues were removed. The isolated and cleaned fetal lungs were snap frozen in liquid nitrogen and stored at −80°C for later analysis. For each study, tissues obtained from the fetuses from 1 mother were used for either real-time polymerized chain reaction (PCR) or western immunoblots assay and considered n = 1. We determined the developmental stages of the embryos by visual inspection according to a modified Theiler-staging system.17 Details of the staging system are available online at http://genex.hgu.mrc.ac.uk/Databases/Anatomy/MAstaging.shtml.
Hypoxic Exposure
As described previously,16 at 15.5 DPC, the pregnant mice were placed in a custom-sealed Plexiglas cage. A mixture of compressed air and nitrogen was infused, and O2 concentration was measured using the Oxychecq Expedition, an in-line oxygen meter (Oxychecq, Fort Pierce, Florida). Mice were maintained in an environment of 10.5% O2 (one-half atmosphere equivalent) for 48 hours. To ensure a consistent hypoxic exposure, the O2 concentration in the cages was checked at hourly intervals during the day and every 3 hours at night.
Messenger RNA and Protein Quantification
Western immunoblot assays and real-time PCR were conducted, as described and validated previously by our laboratory.6,10,16 We isolated and quantified RNA and protein by Allprep DNA/RNA Mini Kit according to the manufacturer’s instructions (Qiagen Inc, Valencia, CA Cat # 80204). Isolated messenger RNA (mRNA) was analyzed using a Beckman Spectrophotometer at 260/280 wavelength UV rays to check for quality and quantity. The 260/280 ratio of 1.8 to 2 was accepted for quantification with real-time PCR. The mRNA was reverse transcribed using First Strand Reverse Transcriptor kit (Roche, Inc, Indianapolis, Indiana). Real-time PCR was performed on Light Cycler 1.5 (Roche Inc) using hydrolysis Taqman probes and primers (Table 1 ), designed using the Universal Probe Library, a Web-based software (Roche Inc), and the Fast-Start real time PCR master-mix (Roche Inc). Cycles required (based on initial mRNA of a particular gene) to reach a threshold detection limit by the real-time PCR were recorded and normalized to a housekeeping gene (18 S ribosomal RNA). We have observed that these normalized cycle threshold values (ΔCT-values) are reliable and repeatable. Moreover, these values are more accurate representative of the initial mRNA amount of a particular gene and are provided in results. We determined the fold changes with ΔΔCT method18 after performing validation curves.
Table 1.
Primers Used in the Study
| No | Name | Gene Acc. No | Primer/Probe | Sequence |
|---|---|---|---|---|
| 1 | Angiotensinogen | NM_007428.3 | Left primer | caacacctacgttcacttccaa |
| Right primer | cagacaccgagatgctgttg | |||
| Probe | ctggagtc | |||
| 2 | Renin | NM_031192.2 | Left primer | acggatcagggagagtcaaa |
| Right primer | cacagtgattccacccacag | |||
| Probe | ctcagcca | |||
| 3 | ACE-1 | NM_207624.4 | Left primer | ccctaggacctgccaatct |
| Right primer | tgctcatgttgcttagcagag | |||
| Probe | ggcagcag | |||
| 4 | ACE-2 | NM_027286.3 | Left primer | tgaaaaagtggtgggagatga |
| Right primer | aacagagatgcagggtcaca | |||
| Probe | tggtggag | |||
| 5 | AT-1a | NM_177322.3 | Left primer | actcacagcaaccctccaag |
| Right primer | ctcagacactgttcaaaatgcac | |||
| Probe | catcacca | |||
| 6 | AT-1b | NM_175086.3 | Left primer | cgccagcagcactgtaga |
| Right primer | ggagggggtgaattcaaaa | |||
| Probe | gggagcag | |||
| 7 | AT-2 | NM_007429.4 | Left primer | ggagctcggaactgaaagc |
| Right primer | ctgcagcaactccaaattctt | |||
| Probe | cttcagcc |
Abbreviations: ACE, angiotensin-converting enzyme; AT, angiotensin.
For western immunoblot experiments, frozen samples were homogenized in the 1x cell lysing buffer (Cell Signaling Technology, Beverly, Massachusetts) containing 1× phosphatase and protease inhibitors cocktail (Sigma, St. Louis, MO). Nuclei and debris were pelleted by centrifugation at 1000g for 10 minutes. The supernatant was collected and stored at –80°C. Sodium dodecyl sulfate (SDS)-gel and western blot were performed by using appropriate antibodies (Table 2 ).19 All secondary antibodies were obtained from Abcam (Cambridge, MA). Twenty µg protein from each sample was loaded on an SDS-gel and electrophoresed at 100 V for 3 hours. Proteins were transferred to a nitrocellulose membrane and subjected to immunoblotting with antibodies (Table 2). Bands were detected with enhanced chemiluminescence using a ChemiImager (Alpha-Innotech, San Leandro, California). The results are expressed as fraction of control. We performed control experiments with glyceraldehyde-3-phosphate dehydrogenase (GAPDH), beta-tubulin, alpha-actin, and extracellular signal-regulated kinase-1/2 (ERK1/2); our results demonstrated ERK1/2 protein expression was unaltered with hypoxia in fetal murine lungs. Moreover, ERK1/2 integrated density (arbitrary unit) on densitometry analysis correlated well with different amount of protein loaded and was used as an internal control to account for uniform protein loading.
Table 2.
Antibodies Used in the Study
| No. | Name | Company | Cat. No. | Dilution |
|---|---|---|---|---|
| 1 | Angiotensinogen | Swant Inc Switzerland | 138 | 1:1000 |
| 2 | Renin | Santa Cruz Biotechnologies | sc-27320 | 1:300 |
| 3 | ACE-1 | Abcam Inc | ab39172 | 1:1000 |
| 4 | ACE-2 | Abcam Inc | ab59351 | 1:1000 |
| 5 | AT-1 | Abcam Inc | ab59018 | 1:250 |
| 6 | AT-2 | Abcam Inc | ab19134 | 1:250 |
| 7 | GAPDH | Abcam Inc | ab9485 | 1:2000 |
| 8 | Alpha-Actin | Abcam Inc | Ab1801 | 1:1000 |
| 9 | ERK1/2 | Cell Signaling Technology | #9102 | 1:1000 |
| 10 | Beta-tubulin | Santa Cruz Biotechnology | sc-5274 | 1:1000 |
Abbreviations: ACE, angiotensin converting enzyme; AT, angiotensin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ERK, extracellular signal-regulated kinase.
Epigenetic Studies
For those RAS genes showing significant alterations in expression, we examined the role of transcriptional gene regulation (DNA methylation status of the cytosine guanine [CpG] islands of the promoter region) as well as posttranscriptional gene regulation (microRNA analysis).
Promoter Methylation Analysis
We examined methylation status of the CpG islands of ACE-1 genes, as described previously.20 Briefly, DNA was isolated, analyzed, and cytosine bases were converted to uracil (except the methyl-cytosine bases) using bisulfite modification kit (Qiagen Inc, Valencia, California). After conversion, genomic DNA was cleaned and amplified by heminested-PCR, by the use of specially designed bisulfite specific primers of the promoter regions. Primers were designed using Methyl Primer Express Software v1.0 (Applied Biosystems, Foster City, California). The isolated promoter DNA was inserted into a vector and cloned using TOPO – TA cloning kit (Invitrogen, Inc, Carlsbad, California). The cloned product was cleaned of plasmid DNA using Miniprep Kits (Qiagen Inc.), and sequenced using the commercial services available from Retrogen Inc (San Diego, California). The sequences obtained were analyzed using Web-based software QUMA.21
MicroRNA Studies
MicroRNAs (miRNA) were identified for the 3′ untranslated region (UTR) of the RAS mRNAs, using the Web-based bioinformatics software—TargetScan 4.2 (http://www.targetscan.org). Of 170 miRNA suggested by the bioinformatics software, we choose 17 miRNA for the current study, by the use of the Context Score of more than 90th percentile, as described by Grimson et al.22 The identified miRNA levels were measured by the use of Real-Time Taqman microRNA PCR assays, according to manufacturer’s instructions (Applied Biosystems).
Statistical Analysis
To determine significant differences between groups, we analyzed the data using unpaired, 2-tailed Student t test and chi-square test, by the use of GraphPad Prism software (GraphPad Software Inc, San Diego, California). The hypothesis was accepted at P < .05. For each study, the fetuses from 1 mother were considered n = 1. For each experiment, n was equal to 4 or more, as noted.
Results
Antenatal hypoxic stress has been shown to cause altered heart growth and neonatal vascular function,1 permanent neurological deficits,2 pulmonary arterial dysfunction,3 atherosclerosis,4 and IUGR.5 Studies also demonstrate changes in the renin-angiotensin system as a consequence of antenatal hypoxia.13–15 In addition, it is also established that ACE inhibition can reduce hypoxia-induced pulmonary hypertension and pulmonary vascular remodeling.14 To further extend our knowledge, in the current study, we demonstrate that the renin-angiotensin system is expressed as a local pathway in lungs, and several of its components are altered significantly as a consequence of antenatal hypoxic stress.
Antenatal Maternal Hypoxia and Angiotensinogen Expression in the Fetal Lung
Our results indicate existence of local AGT expression in fetal lungs. However, we observed no difference in mRNA expression in the lung of fetuses from hypoxic and control mice dams (Table 3 ). Similarly, we observed no change in protein expression by western immunoblot (data not shown), the relative integrated density values (IDV; arbitrary units) being 0.91 ± 0.04 (N = 6) and 1.03 ± 0.14 (N = 4) for fetuses from control and hypoxic dams, respectively. Furthermore, we examined miRNAs that can regulate AGT translation. Bioinformatics analysis suggested several miRNAs that can bind at 3′ UTR of AGT mRNA and decrease its translation. Based on context score (see methods) of >90 percentile, we selected mmu-mir-201, mmu-mir-678, and mmu-mir-698. We observed no change in any of these miRNA examined in fetal lung from hypoxic dams, as compared to the control (Table 4 ). Thus, the experiments suggest that AGT is expressed in lungs locally; however, the levels are unaltered as a consequence of AMH.
Table 3.
Changes in mRNA Expression of the Renin-Angiotensin System in Fetal Lung as a Consequence of Antenatal Hypoxic Stress
| Gene | ΔCt | Fold Change | P Value | |
|---|---|---|---|---|
| Control | Hypoxic | |||
| AGT | 10.66 ± 0.49, N = 8 | 9.75 ± 0.80, N = 6 | – | – |
| Renin | 20.47 ± 0.57, N = 8 | 19.70 ± 0.62, N = 5 | – | – |
| ACE-1.1 | 8.47 ± 0.11, N = 8 | 7.45 ± 0.76, N = 5 | – | – |
| ACE-1.2 | 11.63 ± 0.30, N = 8 | 9.46 ± 0.83, N = 6 | 4.490 | <.05 |
| ACE-2 | 13.68 ± 0.31, N = 7 | 11.45 ± 0.89, N = 5 | 3.175 | <.05 |
| AT-1a | 14.38 ± 0.18, N = 8 | 13.21 ± 0.75, N = 6 | – | – |
| AT-1b | 23.13 ± 0.46, N = 8 | 21.13 ± 0.55, N = 6 | 4.000 | <.05 |
| AT2 | 22.38 ± 0.46, N = 4 | 23.28 ± 0.89, N = 4 | – | – |
Abbreviations: AGT, α2-globulin angiotensinogen; ACE, angiotensin converting enzyme; AT, angiotensin.
Table 4.
Changes in miRNA Expression Complementary to the Renin-Angiotensin System Genes in Fetal Lung as a Consequence of Antenatal Hypoxic Stress
| Regulating Gene | miRNA | ΔCt | Fold Change | P Value | |
|---|---|---|---|---|---|
| Control | Hypoxic | ||||
| AGT | 201 | 15.65 ± 0.13 | 15.12 ± 0.26 | – | – |
| 678 | 18.99 ± 0.19 | 19.68 ± 0.90 | – | – | |
| 698 | 11.74 ± 1.21 | 11.54 ± 0.36 | – | – | |
| Renin | 742 | 14.43 ± 0.98 | 16.00 ± 0.23 | – | – |
| 199a | 13.33 ± 0.28 | 14.03 ± 0.33 | – | – | |
| 199b | 14.23 ± 0.40 | 16.40 ± 0.16 | 0.22 | <.05 | |
| ACE-1 | 27a | 9.89 ± 0.37 | 10.89 ± 0.41 | – | – |
| 27b | 9.46 ± 0.45 | 11.54 ± 0.43 | 0.24 | <.05 | |
| ACE-2 | 200b | 5.02 ± 0.71 | 7.79 ± 0.55 | 0.15 | <.05 |
| 200c | 6.53 ± 0.34 | 7.60 ± 0.48 | – | – | |
| 429 | 12.18 ± 0.66 | 13.55 ± 0.36 | – | – | |
| AT-1 | 216b | 6.16 ± 1.38 | 5.65 ± 0.35 | – | – |
| 468 | 13.61 ± 0.34 | 15.09 ± 0.23 | 0.35 | <.05 | |
| 369-3p | 6.28 ± 0.33 | 6.74 ± 0.09 | – | – | |
| AT-2 | 330 | 10.33 ± 0.31 | 10.28 ± 0.37 | – | – |
| 770-3p | 7.02 ± 0.15 | 6.82 ± 0.29 | – | – | |
| 291b-5p | 12.55 ± 0.61 | 11.46 ± 0.72 | – | – | |
Abbreviations: AGT, α2-globulin angiotensinogen; ACE, angiotensin converting enzyme; AT, angiotensin; miRNA; microRNA.
Antenatal Maternal Hypoxia and Renin Expression in the Fetal Lung
Our results also provide evidence for the expression of renin locally in fetal lungs; however, we observed no difference in the renin mRNA expression in the fetus as a consequence of AMH (Table 3). In contrast, protein expression of renin was significantly greater (P < .05) in lung from hypoxic dams, as compared to the control (Figure 1 ), relative IDVs being 0.49 ± 0.02 (N=4) and 1.31 ± 0.14 (N=6) for fetuses from control and hypoxic dams, respectively (Figure 1). Increase in the protein from similar mRNA levels can be a result of reduced miRNA expression. Thus, we examined miRNA complementary to the 3′UTR of renin mRNA. We observed ~4-fold decrease of miRNA mmu-mir-199b in fetal lung from hypoxic dams, as compared to the control (Table 4). We observed no change in expression of miRNA mmu-mir-742 and mmu-mir-199a. Thus, these experiments suggest that renin is expressed locally in fetal lungs, and as a consequence of AMH, protein expression is increased despite no change in mRNA levels. Also, miRNA mmu-mir-199b may be involved in posttranscriptional (translational) regulation of renin in response to AMH.
Figure 1.
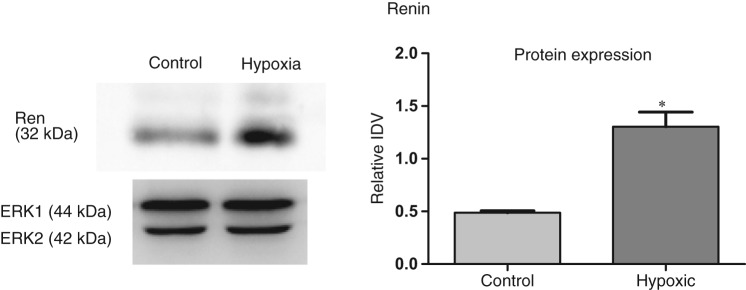
Acute antenatal hypoxia and renin expression in fetal lungs. To left, representative Western immunoblot showing protein expression of renin with extracellular signal-regulated kinase-1/2 (ERK1/2) as control. To right, bar graph demonstrating normalized relative integrated density value (IDV) of renin protein expression in control and hypoxic fetal lung. *Denotes significant difference by an unpaired Student t test (P < .05).
Antenatal Maternal Hypoxia and ACE-1 Expression in the Fetal Lung
Two transcripts of ACE-1 are known to exist, ACE-1.1 and ACE-1.2, and both ACE transcripts are present in fetal lungs. Of interest, ACE-1.2 but not ACE-1.2 expression, was significantly higher (~ 4-fold) in fetal lung from hypoxic dams, as compared to the control (Table 3). However, only 1 antibody exists for ACE-1 and is unable to differentiate between the ACE-1.1 and ACE-1.2 proteins. We observed an overall reduction in ACE-1 protein expression (P < .05) in lung from hypoxic dams, as compared to control (Figure 2A) the relative IDVs being 1.22 ± 0.17 (N = 4) and 0.25 ± 0.06 (N = 9) for fetuses from control and hypoxic dams, respectively. Moreover, with hypoxic stress, we also observed ~4-fold decrease in miRNA mmu-mir-27b in fetal lung from hypoxic dams, as compared to control. Such a decrease in miRNA should increase the protein translation; clearly this is not the case with ACE, suggesting involvement of other posttranslational mechanisms. We observed no change in expression of miRNA 27a (Table 4). We also performed promoter methylation assay of ACE-1 promoter region. Again, we observed no change in the 23 CpG sites of the ACE-1 promoter in lungs as a consequence of maternal hypoxic stress (data now shown). The percentage of CpG islands unmethylated were 70.92 ± 5.14 (N = 5) and 67.63 ± 8.27 (N = 5) from fetal lung from control and hypoxic dams, respectively. Thus, the result of these experiments suggests regulation of ACE as a consequence of AMH.
Figure 2.

Acute antenatal hypoxia and expression of angiotensin-converting enzyme 1 and 2 (ACE-1 and ACE-2) in fetal lungs. A, to left, representative Western immunoblot showing protein expression of ACE-1. To right, bar graph demonstrating normalized relative integrated density value (IDV) for ACE-1 protein in control and hypoxic fetal lung. B, acute antenatal hypoxia and ACE-2 expression in the fetal lungs. To left, representative Western immunoblot showing protein expression of ACE-2. To right, bar graph demonstrating normalized relative integrated density value (IDV) in control and hypoxic fetal lung. *Denotes significant difference by an unpaired Student t test (P < .05).
Antenatal Maternal Hypoxia and ACE-2 Expression in the Fetal Lung
As shown in Table 3, ACE-2 mRNA was significantly greater in fetal lung from hypoxic dams. Similarly, we observed a marked increase in ACE-2 protein expression (P < .05) in lung from hypoxic dams, as compared to control, the relative IDVs being 1.0 ± 0.4 (N = 4) and 4.5 ± 0.43 (N = 4) for fetuses from control and hypoxic dams, respectively (Figure 2B). We also observed a 6.6-fold decrease in miRNA mmu-mir-200b with hypoxic stress (Table 4). This indicates that hypoxia upregulates mRNA, miRNA, and protein expression of ACE-2, a protective element of the RAS. We observed no changes in expression of miRNA mmu-mir-200c or mmu-mir-429 (Table 4). Thus, these experiments suggest upregulation of ACE-2 production as a consequence of AMH.
Antenatal Maternal Hypoxia and AT-1 Expression in the Fetal Lung
Two angiotensin II type 1 (AT-1) receptors transcripts are known to exist in mice, AT-1a and AT-1b; and our results indicate the presence of both AT-1 transcripts in fetal lungs. Of interest, AT-1b but not AT1a mRNA expression, was significantly higher in fetal lung from hypoxic dams (Table 3 ). However, only 1 antibody exists for AT-1 and is unable to differentiate between the isoforms AT-1a and AT-1b (Figure 3). We observed no change in the protein expression in lung from control and hypoxic dams; the relative IDVs being 0.99 ± 0.15 (N = 4) and 0.73 ± 0.08 (N = 6), P = 0.1, respectively. Furthermore, we observed a ~3-fold decrease in miRNA mmu-mir-468 with hypoxic stress, but no change in expression of miRNA mmu-mir-216b or mmu-mir-369-3p (Table 4).
Figure 3.
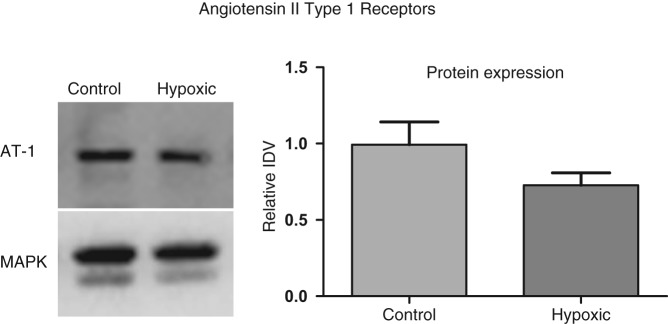
Acute antenatal hypoxia and angiotensin II type 1 receptor expression in the fetal lungs. To left, representative western immunoblot showing protein expression of AT-1. To right, bar graph demonstrating normalized relative integrated density value (IDV) for AT-1 in control and hypoxic fetal lung.
Antenatal Maternal Hypoxia and AT-2 Expression in the Fetal Lung
Angiotensin II type 2 also is expressed locally in fetal lungs; however, no difference in mRNA expression was observed in the lung of fetuses from hypoxic or control mice dams (Table 3). Similarly, protein expression of AT-2 receptors also was unchanged in lung from control and hypoxic dams, the relative IDVs being 1.50 ± 0.56 (N = 4), and 1.99 ± 0.33, (N = 4), respectively (Data not shown). We also analyzed several miRNA such as mmu-mir-330, mmu-mir-770-3p, and mmu-mir-291b-5p. However, no change in any of these miRNA levels was observed as a consequence of AMH (Table 4).
Discussion
Growing evidence suggests the presence of a local renin-angiotensin system in lungs, supported by its expression in rats,23–26 humans,27 and mice (current study). Surprisingly, because of its importance, studies aimed to demonstrate alterations of the pulmonary RAS with hypoxic stress have been lacking, these were the scope of the current study. Antenatal maternal hypoxia is linked to several disorders such as altered heart growth and neonatal vascular function,1 permanent neurological deficits,2 pulmonary arterial dysfunction,3 atherosclerosis,4 and IUGR.5 In the current study, we examined several aspects of the pulmonary RAS in response to AMH to establish the changes that may be responsible for such disorders or as protective mechanisms during fetal life. For instance, AT-1 receptor activation, which produces vasoconstriction, was downregulated as a consequence of AMH. This may be an important protective effect compensating for hypoxic pulmonary vasoconstriction.
In addition, we observed several important changes in fetal lungs as a consequence of AMH. Of importance, ACE is abundantly secreted by lung vasculature and is elevated in a number of interstitial lung diseases.28 In the current study, however, we observed reduced ACE-1 expression as a consequence of AMH. In humans and many other species, ACE-1 is known to exist in 2 splice variants ACE-1.1 and ACE-1.2,29,30 however, the roles of these transcript variants are unknown. In the current study with AMH, we observed increased mRNA levels of ACE-1.2 but not of ACE-1.1. Thus, further studies are required to elucidate the mechanisms of such a shift in splicing as a result of hypoxic stress. Additionally, in lungs from the hypoxic fetuses, ACE-1 protein levels were reduced, despite normal ACE-1.1 and increased ACE-1.2 mRNA levels, as compared to control. Several reasons may account for reduced protein expression, despite increased mRNA levels. One of these could be the translational regulation by miRNA. Another could be reduced mRNA half-life. Additionally, the overexpression of translational repressors or underexpression of translational activators also could account for the observed discrepancy. To study the epigenetic regulation of translational control, we examined several miRNA that are partially complementary to the 3′UTR of the several RAS genes. MicroRNAs are single-stranded RNA molecules of about 22 nucleotides in length, which regulate gene expression. MicroRNAs are encoded (transcribed) but are not translated into protein, ie, they are noncoding RNAs. MicroRNA molecules are partially complementary to one or more mRNA molecules, their main function being to downregulate gene expression without degrading the mRNA.31 We performed a bioinformatics analysis, which suggested several miRNA that may suppress the protein translation of ACE-1. In the current study, 1 of those miRNAs, mmu-mir-27b, was significantly reduced in the fetal lungs from hypoxic dams. Moreover, our finding of reduced miRNA mmu-mir-27b in fetal lung (complementary to ACE-1) indicates posttranscriptional epigenetic programming to increase protein translation. However, during fetal life, the programmed upregulated protein translation remains suppressed, but in postnatal life, due to some unknown factors, the suppression is released with increased ACE-1 protein expression. Furthermore, other studies have shown alterations in ACE expression with developmental maturation in lungs.32–34 To address the issue of increased mRNA transcription, we examined CpG islands of the ACE-1 promoter. At about 20 sites examined, we observed no change in the methylation status of the CpG islands. However, other mechanisms such as histone modification maybe involved in epigenetic regulation of the ACE-1 transcription, and this will require further investigation.
Importantly, RAS activity can be divided into either vasoconstrictive/proliferative/fibrogenesis actions, which are mediated by ACE-1 activation leading to Ang II production and activation of AT1 receptors, or a vasoprotective action triggered by ACE-2 expression (leading to degradation of Ang II) or upregulation of AT-2 receptors. Angiotensin-converting enzyme 2 acts to convert Ang II to Ang 1-7 by the action of ACE-2, which is known to play a protective role in ischemia-induced cardiac dysfunction.35 Angiotensin-converting enzyme 2 overexpression also has been shown to prevent and reverse right ventricular systolic pressure and associated pathophysiology in monocrotaline-induced pulmonary hypertension. This also occurs by a mechanism involving a shift from the vasoconstrictive, proliferative, and fibrotic axes to the vasoprotective axis of the RAS, with inhibition of proinflammatory cytokines.36 Consistent with these studies, we observed significant overexpression of both ACE-2 mRNA and protein in fetal lungs, as a consequence of maternal hypoxic stress. Moreover, we also observed reduced expression of miRNA mmu-mir-200b, which may be responsible for increased translation of ACE-2 protein from mRNA. Overall, these findings suggest that during fetal life, AMH leads to upregulation of ACE-2 at both transcriptional and translational levels. Of note, ACE-2 leads to conversion of Ang II to Ang 1-7 and terminates the activity of Ang II. The current study indicates that during fetal life ACE-2 overexpression may shift the balance from the RAS vasoconstrictive, proliferative axis (ACE-Ang II-AT-1) to the vasoprotective axis (ACE-2-Ang 1-7). As noted above, in postnatal life, due to some unknown factors, this protection may disappear and the programmed disease becomes manifest. This important question requires further investigation.
Following ACE-1-mediated conversion of Ang I to Ang II, Ang II produces its effects by interacting with AT-1 (subdivided into AT-1a and AT-1b) and AT-2 receptors. Of note, previous reports suggest a regulatory role for the AT-2 receptors,37 whereas most of the effects of the RAS are mediated by AT-1 receptors.38 However, 2 subtypes of AT-1 receptors (AT-1a and AT-1b) exist in mouse,39 rat,40 and humans.41 A study of AT-1a and AT-1b–deficient mice suggests that brain AT-1a are involved primarily in blood pressure regulation, whereas AT-1b are involved in the dipsogenic response.42 In the current study, we observed increased mRNA expression of AT-1b with no change in protein expression. We also observed reduced expression of RNA mmu-mir-468, which can regulate AT-1b mRNA translation. At present, however, the role of AT-1b is unknown in the lungs, and it is difficult to interpret a direct rationale for such a programming during fetal life. We speculate that the AT-1b receptor plays an important role in AMH-mediated fetal programming of postnatal disease.
Another important finding of the current study was that of increased renin protein expression with AMH, as compared to control. This finding of increased renin is consistent with a study in which hypoxia and resulting hypercapnia increased the plasma-renin activity during late gestation.15 This increased renin can lead to increased conversion of AGT to Ang I. Furthermore, Ang I can be converted to Ang II by the action of ACE-1, and Ang II is a potent mitogen and a vasoconstrictor.
Perspective and Speculations
Figure 4 summarizes the significant findings of the current study. The figure shows that maternal hypoxic stress during late gestation leads to alterations in genetic (mRNA and protein) and epigenetic (miRNA) expression of several genes of the pulmonary RAS. As the current study demonstrates alterations in several miRNA, it is of importance to appreciate that posttranscriptional mechanisms such as miRNA-mediated regulation finally determine the expression of proteins from mRNA. Discrepancies in mRNA and protein levels, as a result of programming at the transcriptional and posttranscriptional level, may be responsible for the manifestation of disorders at a later age, even though these genes become programmed in fetal life. The current study raises a number of important questions. These include: What is the role of increased renin protein during fetal life? What is the role of the ACE-1.2 transcript? What is the role of AT-1b receptors in lungs? In the current study, ACE-1 promoter DNA methylation was not changed, this necessitates investigation of histone modifications in future studies. What are the posttranscriptional regulatory mechanisms other than miRNA? Also of importance, in future studies in the offspring of dams that experienced antenatal hypoxia, we must determine the extent to which genetic and epigenetic changes in the RAS system occur and their possible relationship to persistent pulmonary hypertension.
Figure 4.

Summarized findings of the present study for mRNA, miRNA, and proteins in fetal lungs in response to antenatal hypoxic stress.
Despite intensive research, morbidity and mortality due to idiopathic pulmonary hypertension in newborns and adults remain high. In contrast to systemic hypertension, we lack substantial therapeutic targets for these disorders. The current study provides important miRNA targets for possible future therapeutic interventions. The observation that maternal hypoxia may result in differential programming of gene and protein expression in the offspring provides increased mechanistic understanding of pulmonary pathology, which requires further investigation.
Acknowledgment
We thank Brenda Kreutzer for assistance in the preparation of this manuscript.
Footnotes
R01 HD003807 HDNICHD NIH HHSUnited States, R01 HD003807-39 HDNICHD NIH HHSUnited States
The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
The authors received no financial support for the research and/or authorship of this article.
References
- 1. Williams SJ, Campbell ME, McMillen IC, Davidge ST. Differential effects of maternal hypoxia or nutrient restriction on carotid and femoral vascular function in neonatal rats. Am J Physiol Regul Integr Comp Physiol. 2005;288(2):R360–R367 [DOI] [PubMed] [Google Scholar]
- 2. Golan H, Kashtutsky I, Hallak M, Sorokin Y, Huleihel M. Maternal hypoxia during pregnancy delays the development of motor reflexes in newborn mice. Dev Neurosci. 2004;26(1):24–29 [DOI] [PubMed] [Google Scholar]
- 3. Hernandez-Diaz S, Van Marter LJ, Werler MM, Louik C, Mitchell AA. Risk factors for persistent pulmonary hypertension of the newborn. Pediatrics. 2007;120(2):e272–e282 [DOI] [PubMed] [Google Scholar]
- 4. Wang Z, Huang Z, Lu G, Lin L, Ferrari M. Hypoxia during pregnancy in rats leads to early morphological changes of atherosclerosis in adult offspring. Am J Physiol Heart Circ Physiol. 2009;296(5):H1321–H1328 [DOI] [PubMed] [Google Scholar]
- 5. Regnault TR, de Vrijer B, Galan HL, Wilkening RB, Battaglia FC, Meschia G. Development and mechanisms of fetal hypoxia in severe fetal growth restriction. Placenta. 2007;28(7):714–723 [DOI] [PubMed] [Google Scholar]
- 6. Gheorghe CP, Goyal R, Holweger JD, Longo LD. Placental gene expression responses to maternal protein restriction in the mouse. Placenta. 2009;30(5):411–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao Y, Portugal AD, Negash S, Zhou W, Longo LD, Usha Raj J. Role of Rho kinases in PKG-mediated relaxation of pulmonary arteries of fetal lambs exposed to chronic high altitude hypoxia. Am J Physiol Lung Cell Mol Physiol. 2007;292(3):L678–L684 [DOI] [PubMed] [Google Scholar]
- 8. Fleming I, Kohlstedt K, Busse R. The tissue renin-angiotensin system and intracellular signalling. Curr Opin Nephrol Hypertens. 2006;15(1):8–13 [DOI] [PubMed] [Google Scholar]
- 9. Valencia JC, Pacheco-Rodriguez G, Carmona AK, et al. Tissue-specific renin-angiotensin system in pulmonary lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. 2006;35(1):40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goyal R, Galffy A, Field SA, Gheorghe CP, Mittal A, Longo LD. Maternal protein deprivation: changes in systemic renin-angiotensin system of the mouse fetus. Reprod Sci. 2009;16(9):894–904 [DOI] [PubMed] [Google Scholar]
- 11. Goyal R, Goyal D, Leitzke A, Gheorghe CP, Longo LD. Brain renin-angiotensin system: fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod Sci. 2010;17(3):227–238 [DOI] [PubMed] [Google Scholar]
- 12. Goyal R, Yellon SM, Longo LD, Mata-Greenwood E. Placental gene expression in a rat “model” of placental insufficiency. Placenta. 2010;31(7):568–575 [DOI] [PubMed] [Google Scholar]
- 13. Mascitelli L, Pezzetta F. Inhibition of the renin-angiotensin system in patients with COPD and pulmonary hypertension. Chest. 2007;131(3):938; author reply 938-939 [DOI] [PubMed] [Google Scholar]
- 14. Nong Z, Stassen JM, Moons L, Collen D, Janssens S. Inhibition of tissue angiotensin-converting enzyme with quinapril reduces hypoxic pulmonary hypertension and pulmonary vascular remodeling. Circulation. 1996;94(8):1941–1947 [DOI] [PubMed] [Google Scholar]
- 15. Wood CE, Kane C, Raff H. Peripheral chemoreceptor control of fetal renin responses to hypoxia and hypercapnia. Circ Res. 1990;67(3):722–732 [DOI] [PubMed] [Google Scholar]
- 16. Gheorghe CP, Mohan S, Oberg KC, Longo LD. Gene expression patterns in the hypoxic murine placenta: a role in epigenesis?. Reprod Sci. 2007;14(3):223–233 [DOI] [PubMed] [Google Scholar]
- 17. Downs KM, Davies T. Staging of gastrulating mouse embryos by morphological landmarks in the dissecting microscope. Development. 1993;118(4):1255–1266 [DOI] [PubMed] [Google Scholar]
- 18. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao Y, Long W, Zhang L, Longo LD. Extracellular signal-regulated kinases and contractile responses in ovine adult and fetal cerebral arteries. J Physiol. 2003;551(pt 2):691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89(5):1827–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008;36:W170–W175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grady EF, Sechi LA, Griffin CA, Schambelan M, Kalinyak JE. Expression of AT2 receptors in the developing rat fetus. J Clin Invest. 1991;88(3):921–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Campbell DJ, Habener JF. Angiotensinogen gene is expressed and differentially regulated in multiple tissues of the rat. J Clin Invest. 1986;78(1):31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Entzeroth M, Hadamovsky S. Angiotensin II receptors in the rat lung are of the AII-1 subtype. Eur J Pharmacol. 1991;206(3):237–241 [DOI] [PubMed] [Google Scholar]
- 26. Cassis L, Shenoy U, Lipke D, Baughn J, Fettinger M, Gillespie M. Lung angiotensin receptor binding characteristics during the development of monocrotaline-induced pulmonary hypertension. Biochem Pharmacol. 1997;54(1):27–31 [DOI] [PubMed] [Google Scholar]
- 27. Phillips MI, Speakman EA, Kimura B. Levels of angiotensin and molecular biology of the tissue renin angiotensin systems. Regul Pept. 1993;43(1-2):1–20 [DOI] [PubMed] [Google Scholar]
- 28. Marshall RP. The pulmonary renin-angiotensin system. Curr Pharm Des. 2003;9(9):715–722 [DOI] [PubMed] [Google Scholar]
- 29. Roy SN, Kusari J, Soffer RL, Lai CY, Sen GC. Isolation of cDNA clones of rabbit angiotensin converting enzyme: identification of two distinct mRNAs for the pulmonary and the testicular isozymes. Biochem Biophys Res Commun. 1988;155(2):678–684 [DOI] [PubMed] [Google Scholar]
- 30. Bernstein KE, Martin BM, Bernstein EA, Linton J, Striker L, Striker G. The isolation of angiotensin-converting enzyme cDNA. J Biol Chem. 1988;263(23):11021–11024 [PubMed] [Google Scholar]
- 31. Pasquinelli AE, Ruvkun G. Control of developmental timing by micrornas and their targets. Annu Rev Cell Dev Biol. 2002;18:495–513 [DOI] [PubMed] [Google Scholar]
- 32. Wallace KB, Bailie MD, Hook JB. Angiotensin-converting enzyme in developing lung and kidney. Am J Physiol. 1978;234(3):R141–R145 [DOI] [PubMed] [Google Scholar]
- 33. Wallace KB, Bailie MD, Hook JB. Development of angiotensin-converting enzyme in fetal rat lungs. Am J Physiol. 1979;236(1):R57–R60 [DOI] [PubMed] [Google Scholar]
- 34. O'Connor SJ, Fowden AL, Holdstock N, Giussani DA, Forhead AJ. Developmental changes in pulmonary and renal angiotensin-converting enzyme concentration in fetal and neonatal horses. Reprod Fertil Dev. 2002;14(7-8):413–417 [DOI] [PubMed] [Google Scholar]
- 35. Ferreira AJ, Santos RA, Almeida AP. Angiotensin-(1-7): cardioprotective effect in myocardial ischemia/reperfusion. Hypertension. 2001;38(3 pt 2):665–668 [DOI] [PubMed] [Google Scholar]
- 36. Yamazato Y, Ferreira AJ, Hong KH, et al. Prevention of pulmonary hypertension by angiotensin-converting enzyme 2 gene transfer. Hypertension. 2009;54(2):365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin KS, Chan JY, Chan SH. Involvement of AT2 receptors at NRVL in tonic baroreflex suppression by endogenous angiotensins. Am J Physiol. 1997;272(5 pt 2):H2204–H2210 [DOI] [PubMed] [Google Scholar]
- 38. Timmermans PB, Wong PC, Chiu AT, et al. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev. 1993;45:205–251 [PubMed] [Google Scholar]
- 39. Sasamura H, Hein L, Krieger JE, Pratt RE, Kobilka BK, Dzau VJ. Cloning, characterization, and expression of two angiotensin receptor (AT-1) isoforms from the mouse genome. Biochem Biophys Res Commun. 1992;185(1):253–259 [DOI] [PubMed] [Google Scholar]
- 40. Iwai N, Inagami T. Identification of two subtypes in the rat type I angiotensin II receptor. FEBS Lett. 1992;298(2-3):257–260 [DOI] [PubMed] [Google Scholar]
- 41. Konishi H, Kuroda S, Inada Y, Fujisawa Y. Novel subtype of human angiotensin II type 1 receptor: cDNA cloning and expression. Biochem Biophys Res Commun. 1994;199(2):467–474 [DOI] [PubMed] [Google Scholar]
- 42. Davisson RL, Oliverio MI, Coffman TM, Sigmund CD. Divergent functions of angiotensin II receptor isoforms in the brain. J Clin Invest. 2000;106(1):103–106 [DOI] [PMC free article] [PubMed] [Google Scholar]