

Abstract
Diabetic retinopathy remains one of the most debilitating chronic complications, but despite extensive research in the field, the exact mechanism(s) responsible for how retina is damaged in diabetes remains ambiguous. Many metabolic pathways have been implicated in its development, and genes associated with these pathways are altered. Diabetic environment also facilitates epigenetics modifications, which can alter the gene expression without permanent changes in DNA sequence. The role of epigenetics in diabetic retinopathy is now an emerging area, and recent work has shown that genes encoding mitochondrial superoxide dismutase (Sod2) and matrix metalloproteinase-9 (MMP-9) are epigenetically modified, activates of epigenetic modification enzymes, histone lysine demethylase 1 (LSD1), and DNA methyltransferase are increased, and the micro RNAs responsible for regulating nuclear transcriptional factor and VEGF are upregulated. With the growing evidence of epigenetic modifications in diabetic retinopathy, better understanding of these modifications has potential to identify novel targets to inhibit this devastating disease. Fortunately, the inhibitors and mimics targeted towards histone modification, DNA methylation, and miRNAs are now being tried for cancer and other chronic diseases, and better understanding of the role of epigenetics in diabetic retinopathy will open the door for their possible use in combating this blinding disease.
1. Introduction
Diabetic retinopathy remains the leading cause of blindness in young adults affecting over 90% patients with 20 years of diabetes. The disease carries a heavy burden on our society as it is responsible for 4.8% of the 37 million cases of eye disease-related blindness worldwide. With the incidence of diabetes increasing at an alarming rate, the number of people with diabetic retinopathy is expected to grow from 126.6 million in 2010 to 191.0 million by 2030 [1]. This slow progressing disease is mainly characterized by damage of the microvasculature of the retina. Some of the early histopathological changes of this slow progressing disease include microaneurysms, hemorrhages, cotton wool spots, intraretinal microvascular abnormalities, and venous bleeding. But, in more advanced stages, new fragile vessels are formed along the retina and on the posterior surface of the vitreous, and if not treated, they result in the detachment of the retina, leading to blindness [2]. Although hyperglycemia is the major initiator of diabetic retinopathy, hypertension and dyslipidemia are also considered significant risk factors in its development and progression [3–5].
Despite extensive research in the field to understand the pathogenesis of diabetic retinopathy, the exact mechanism(s) remains elusive making inhibition of the progression of this disease a daunting task. Some of the possible mechanisms implicated in its development include oxidative stress, increased formation of advanced glycation end products, and activation of protein kinase-C, polyol production, and hexosamine pathways [2, 6, 7]. These pathways appear to be interlinked [8], which further complicates the strategies to prevent the development/progression of this devastating complication of diabetes.
2. Genetics and Diabetic Retinopathy
Pathogenesis of a disease is also influenced by genetic factors, and due to variability in the severity of retinopathy among diabetic patients with similar risk factors, the role of genetic factors in diabetic retinopathy should be somewhat predictable. However, such genetic associations are not yet clearly established. Landmark “Genome-Wide Association Studies” have identified number of genetic variants that could explain some of the interindividual variations in the susceptibility of diabetes [9]. A meta-analysis study to investigate possible genetic associations with diabetic retinopathy, which incorporated reports published between January 1990 and August 2008, has revealed a significant variation in the AKR1B1 gene. This gene encodes aldo-keto reductase family 1 member B1, which is the rate limiting enzyme of the polyol pathway [10], one of the pathways implicated in the development of diabetic retinopathy [2, 6, 7]. Another meta-analysis study has shown that Ala allele of the Pro12Ala polymorphism in the peroxisome proliferator-activated receptor-γ2 gene acts as a protective gene in the incidence of retinopathy in type 2 diabetic patients [11]. In contrast, recent studies have failed to find any associations between VEGF-related SNPs (rs6921438 and rs10738760) and the risk of diabetic retinopathy and nephropathy in diabetic patients [12]. Thus, the association between genetic factors and diabetic retinopathy remains to be clarified.
3. Epigenetic Modifications and Gene Regulation
Diabetic environment disturbs metabolic homeostasis and also alters various genes, including genes associated with oxidative stress, apoptosis and inflammation [13–16]. Control of gene expression (i.e., the ability of a gene to produce a biologically active protein) in mammals, in addition to being modulated by transcriptional and translational initiation, can also be controlled by changes “on the top” of the genes without altering the nucleotide composition of the genome [17]. These “epigenetic” modifications are stable, but potentially reversible, and can be passed from generation to generation. Recent studies have shown that epigenetic changes play a major role in many chronic diseases such as cancer and diabetes where small changes in the epigenome over time are considered to lead to disease manifestation. Three major epigenetic mechanisms considered to regulate gene expression are DNA methylation, histone modifications, and noncoding RNA activity [18–21].
DNA methylation, addition of a methyl group on position 5 of cytosine residues of the cluster of CpG dinucleotides (CpG Island), which is the regulatory region of most genes, is typically associated with transcriptional repression [22, 23]. The methylation process brings in the unintended changes brought up by the environmental exposures or other life style, and these changes can be passed on for multiple generations. Cytosine 5 methylation generates 5-methylcytosine (5mC), and the reaction is catalyzed by a family of enzymes—DNA (cytosine-5) methyltransferases (Dnmts). The conversion of 5mC to 5-hydroxymethylcytosine (5hmC) is facilitated by Ten-eleven-translocation enzymes (TETs) [24, 25], making these enzymes future targets of pharmacological regulation. However, how the process of DNA methylation-demethylation is balanced remains somewhat unclear. A subfamily of DNA glycosylases are considered to promote active DNA demethylation by removing the 5-methylcytosine base, followed by cleavage of the DNA backbone at the abasic site, and the methylated cytosine is replaced by an unmethylated cytosine. In contrast, the passive process involves absence/inactivation of Dnmt1 resulting in hypomethylated DNA [26].
Gene expression pattern is also dictated by chromatin, which is a composite structure of histones and nucleic acid. Histones act as spools around which DNA winds, and among the 4-histone proteins, histone 2A and B (H2A and H2B), H3 and H4 form a tetrameric structure, the nucleosome [27]. Despite such sophisticated DNA packaging, N-terminal of histones remains vulnerable for posttranslational modifications, and can be acetylated, methylated, and phosphorylated. Such epigenetic modifications alter the chromatin structure which subsequently affects the binding of transcription factors, and can regulate the selective expression of genes in a particular tissue by acting like switches to control gene activity [15, 28–31].
Acetylation is one of the most common modifications which is generally associated with gene activation, and the process relaxes the chromatin structure allowing recruitment and binding of transcription factor and RNA polymerase II [32]. Acetylation is regulated by fine balance between histone acetylating and deacetylating enzymes; histone acetyltransferases (HATs) add acetyl group while histone deacetylase (HDAC) removes the acetyl group. Methylation of histones, however, shows greater variability as methylation can occur at both lysine and arginine residues, and can be associated with either gene activation or repression. Methylation of lysine 4 of histone 3 (H3K4) is typically associated with gene activation, while methylation of lysine 9 (H3K9) is associated with gene repression [33–35]. Furthermore, the outcome of methylation is also dictated by the specific histone residue being modified; for example, mono- or tri-methylation of H3K4 induces activation of gene expression [36, 37]. While monomethylation of H3K9 induces gene activation, its tri-methylation suppresses the gene activity [38]. As with acetylation, methylation status of histones depends on the final balance between histone methylating and demethylating enzymes. Histone methylating enzymes, SET1/7/9 help in H3K4 methylation, and suppressors of variegation 3-9 homolog (SUV39H) methylates lysine 9 of histone 3 [39, 40]. Subsequently, in order to protect from such kind of relatively stable methyl modifications and constitutive expression of genes, lysine specific demethylase (LSD1) specifically removes methyl groups from methylated H3K4 and H3K9 [41, 42].
In addition to DNA methylation and histone modifications, gene expression is also regulated by microRNAs (miRNA); the small noncoding RNAs that regulate gene expression posttranscriptionally by binding to complementary sequences in the 3′ untranslated regions of messenger RNAs [43, 44]. This cleaves mRNA resulting in decreased protein synthesis and expression of the targeted gene. Genes with epigenetic functions in chromatin can be regulated by miRNAs affecting chromatin remodeling and gene expression. Regulation of genes by DNA methylation and miRNA appears to be interrelated as the function of Dnmts depends on histone modification patterns, such as H3K9 methylation and histone deacetylation. In addition, inhibition of Dnmts, which causes DNA demethylation, can reactivate some of the miRNAs [45]. Overall, these epigenetic modifications are not permanent, but their continuous response to the changing environment, such as diabetes, and the chances of them being inherited to successive generation, make them attractive targets for chronic diseases.
4. Role of Epigenetic Modifications in Diabetes
It has become clear that both genetic predisposition and environmental factors play critical roles in the development of metabolic diseases including obesity and type 2 diabetes, and epigenetic modifications have important roles in altering gene expressions in various chronic diseases [46–48]. Recent studies have demonstrated the role of epigenetic mechanisms in islet function, and have provided an avenue whereby dietary components could accelerate or prevent age-related diseases through their effects on epigenetic modifications [49, 50]. Increased DNA methylation at the promoter of the peroxisome proliferator-activated receptor coactivator 1 gene in the pancreatic islets is shown to play a key role in regulating mitochondrial genes [51]. Our studies have shown that in diabetes hyper methylation of the CpG sites at the regulatory region of DNA polymerase gamma affects its binding to the mtDNA, and this compromises the transcriptional activity resulting in decreased copy number [52]. Hyper acetylation of histone H4 at the insulin gene promoter influences glucose-induced insulin secretion [53]. Diabetic patients with nephropathy have altered DNA methylation at the key gene promoters compared to those without nephropathy [54]. Tissue specific DNA methylation is observed in streptozotocin-induced diabetic rats with hypomethylation in the liver but not in kidney, and in the leukocytes of diabetic patients, Dnmt is altered [55, 56]. Furthermore, hyperglycemia induces specific and long-lasting epigenetic modifications [14, 15, 28, 29, 52, 57, 58]; glucose-induced persistent transcriptional activation of p65 in vascular cells is associated with methylation of H3K4 and hypomethylation of H3K9, and the concomitant activation of NF-kB is subsequently associated with increased inflammatory response [14].
Studies using zebrafish as a model of diabetes have shown that hyperglycemia induces global DNA hypo methylation and aberrant gene expression in the tissue after limb amputation, and the process is associated with poor wound healing process [59]. Furthermore, β-cells from young IUGR animals, a model of type 2 diabetes, present changes in DNA methylation resulting in dysregulation of genes associated with cellular memory of intrauterine that is associated with adult susceptibility to diabetes [60]. Thus, epigenetic changes can account for chronic and persistent complications of diabetes.
Diabetes also affects miRNAs; it is shown to downregulate miR133a, the miRNA which is implicated in the regulation of the key genes associated with cardiomyocyte hypertrophy [61]. In contrast, upregulation of miR-320 is observed in cardiac microvascular endothelial cells in type 2 diabetic rats [62]. miR-25, the miRNA which targets NADPH oxidase 4, is implicated in the pathogenesis of diabetic nephropathy via promoting oxidative stress [63]. In addition, decreased levels of miR-192 are also linked with the severity of nephropathy and fibrosis in diabetic patients [64]. Better understanding of regulatory roles of miRNAs in diabetic cardiomyopathy and other disease processes is expected to elucidate new avenues for RNA-based therapeutics.
5. Diabetic Retinopathy and Epigenetic Modifications
Diabetic retinopathy is a multifactorial disease and a number of metabolic abnormalities have been associated with its development [7, 65]. However, the role of epigenetic modifications in diabetic retinopathy is still not clear. SUV39H2 is a gene that encodes histone methyltransferase which catalyzes the methylation of H3K9, and recent results from the Finnish Diabetic Nephropathy Study with ~3000 diabetic patients have found an association between the polymorphism in SUV39H2 and diabetic microvascular complications, including retinopathy. These studies have suggested the role of histone modifications in the development of diabetic retinopathy [66]. Experimental evidence using in vitro and in vivo models of diabetic retinopathy have shown that the activities of HDACs are increased and that of HATs are decreased in the retina and its capillary cells in diabetes, and global acetylation of histones is decreased [58]. However, contrary to this, Kadiyala et al. have shown significant increase in retinal histone acetylation in diabetes [67]; strengthening further investigation into the role of histone modifying enzymes in the development of diabetic retinopathy.
Mitochondria are dysfunctional in the retina and its capillary cells in diabetes, and superoxide radicals are elevated and the enzyme responsible for scavenging superoxide radicals in the mitochondria, MnSOD, is impaired [68–70]. Regulation of mitochondrial superoxide levels by maintaining MnSOD protects capillary cell apoptosis and the development of diabetic retinopathy in mice [69, 71]. In the pathogenesis of diabetic retinopathy, Sod2, the MnSOD encoding gene, is epigenetically modified with increased H4K20me3, acetyl H3K9, and p65 subunit of NF-kB (p65) at its promoter/enhancer. We have shown that increased acetyl H3K9 and p65 at Sod2 interact with H4K20me3, and this results in its inactivation [15]. However, diabetes demethylates H3K4, and the recruitment of LSD1 at Sod2 promoter is increased [28]. These results have strongly suggested that the demethylation of H3K4 by LSD1 is also an important factor in the downregulation of Sod2. Furthermore, overexpression of MnSOD prevents increase in H4K20me3 at Sod2 suggesting that superoxide radicals have regulatory role in the epigenetic modification of Sod2 and SUV4-20h2 [15]. This raises the possibility to utilize therapeutic modalities targeted towards regulation of methylation status of histones to prevent inhibition of MnSOD and protect mitochondrial damage.
Matrix metalloproteinase-9 (MMP-9), an enzyme which is proapoptotic in the development of diabetic retinopathy, is also epigenetically modified in the retina in diabetes [29, 72, 73]. We have shown that at its promoter, H3K9me2 is decreased, and acetyl H3K9 and the recruitment of LSD1 and p65 of NF-kB are increased. In addition, regulation of LSD1 ameliorates decreases in H3K9me2 and increases in p65 at MMP-9 promoter, prevents the activation of MMP-9, and also inhibits the cell apoptosis. The results have suggested that the activated LSD1 hypomethylated H3K9 at MMP-9 promoter and this frees up the lysine 9 for acetylation. Acetylated H3K9 opens up the chromatin, and the accessibility to recruit p65 is increased [29]. This ultimately culminates in the activation of MMP-9 and mitochondria damage. Thus, the regulation of LSD1 by molecular or pharmacological means could possibly have potential to prevent/retard the development and progression of retinopathy by regulating Sod2 and MMP-9 and preventing mitochondrial damage and the accelerated capillary cell apoptosis in diabetic patients. Epigenetic modifications of thioredoxin interacting protein, an endogenous inhibitor of antioxidant thioredoxin, are considered responsible for sustained Cox2 expression seen in the retina in diabetes [74]. Furthermore, in diabetes, the CpG sites at the regulatory region of DNA polymerase gamma are hypermethylated affecting their binding to the mtDNA, and the activity of Dnmt is increased [52]. Because of this modification, the transcriptional activity is compromised and copy number is decreased. Thus, there is growing evidence that histone modifications and DNA methylation play an important role in the development of diabetic retinopathy.
Alterations in miRNA expression have also been observed in diabetic eyes, and miRNAs in the eye, including miR200b (one of the VEGF regulating miRNAs) is downregulated [75]. Diabetes also upregulates some of the key NF-kB responsive miRNAs including miR-146, miR-155, miR-132, and miR-21 [76]; upregulation of miR-29b in the early stages of diabetes is considered to be protective against apoptosis of the retinal ganglion cells [77]. These diabetes-induced alterations in miRNAs suggest that they can be used as biomarkers to detect early stages of the disease progression.
Thus, understanding and characterizing the epigenetic regulators and their role in the pathogenesis of diabetic retinopathy could help identify novel targets to combat this disease which is the major cause of blindness in young adults.
6. Oxidative Stress and Epigenetic Modifications
The disruption of the normal redox potential in a cellular environment has detrimental effects on cellular components, including proteins, lipids, and DNA [78–80]. Oxidative stress has been shown to alter histone acetylation/deacetylation and methylation/demethylation. Histone demethylase LSD1 is FAD-dependent amine oxidases, and requires oxygen to function [81]. Oxidative damage to DNA can also result in epigenetic changes in chromatin organization by inhibiting the binding of the methyl-CpG binding domain of methyl-CpG binding protein 2 [82]. Furthermore, disruption of epigenetic mechanisms can result in oxidative stress [83]. In the pathogenesis of diabetic retinopathy, increased oxidative stress is considered to play a major role [7, 65], and our studies have shown that the control of oxidative stress prevents epigenetic changes in retinal Sod2 [15]. Thus, it is plausible that increased oxidative stress could be one of the mechanisms responsible for modulating epigenetic modifications in diabetic retinopathy (Figure 1).
Figure 1.
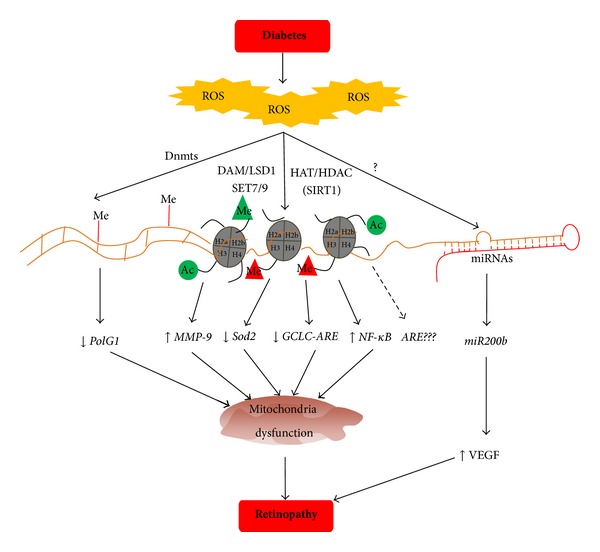
Epigenetic modifications and diabetic retinopathy: diabetes increases oxidative stress in the retina, reactive oxygen species (ROS) modify enzymes responsible for DNA methylation (Dnmts) and histone modifications (SAT, LSD1, HAT and HDAC, etc.), and also miRNAs. Due to epigenetic modifications of various genes and transcription factors, the expression of the targeted genes is altered resulting in mitochondrial dysfunction, and ultimately, in the development of diabetic retinopathy.
7. Epigenetic Modifications of Mitochondrial DNA
Although epigenetic changes in nuclear DNA (nDNA) are now emerging as important areas of investigation in many acquired chronic diseases, epigenetic modification of mtDNA is still in its incipient stages. Recent work has shown that the common epigenetic modifications in nDNA, such as the presence of 5mC and 5hmC, are also observed in the mtDNA, and mitochondria are also equipped with Dnmts and TETs [84]. Furthermore, mtDNA methylation decreases with age, and this age-related hypo methylation is linked with the decreased transcription capacity of proteins encoded by mtDNA [85]. Increase in Dnmt1 in the mitochondria is shown to upregulate the first mtDNA-encoded gene after its initiation, ND1, and downregulate Cytochrome b, the last gene encoded by mtDNA [84]. How diabetes affects mtDNA methylation and the transcription remains to be investigated.
In addition to dysfunction of the mitochondria and their structural abnormalities, mtDNA is also damaged in diabetes, and the DNA polymerase gamma (POLG1), a key member of the DNA replication machinery, is downregulated [13, 52, 71, 86, 87]. The CpG sites at the regulatory region of POLG in the retina are hypermethylated, and this possibly compromises its transcriptional activity [52].
Mitochondrial DNA is very small in size and does not contain histones; instead this DNA is covered with proteins, mainly the mitochondrial transcriptional factor A (TFAM), to form nucleoid [88]. In diabetic retinopathy, the binding of TFAM to a nonspecific region of mtDNA is decreased resulting in decreased levelsn of mtDNA-encoded proteins, and damaging the mtDNA [89, 90]. Since posttranslational modification of histones affects nDNA transcription, it is plausible that such changes in TFAM structure might affect mtDNA transcription. This suggests that by maintaining mitochondria homeostasis by modulating epigenetic changes using pharmaceutical or molecular means could help retard further progression of diabetic retinopathy.
8. Therapies Targeting Epigenetic Modifications
It is now becoming clear that epigenetic factors have a role in altering gene expression in chronic diseases, suggesting that epigenetic mechanisms play a pivotal role in an acquired phenotype. As mentioned above, DNA methylation is one of the most common epigenetic modifications, and methylation at specific CpG islands can provide insights into disease diagnosis. Thus, methylation of genes associated with that disease can be used as a predictor of efficacy for particular drug treatments. The most commonly used DNA methylation inhibitors are nucleoside analogs; these analogs incorporate into the DNA and trap all DNA methyltransferases. Fortunately, FDA has approved Dnmt inhibitors 5-azacytidine (5-Aza-CR; azacitidine; Vidaza) and 5-aza-20-deoxycytidine (5-Aza-CdR; decitabine; Dacogen) for myeloid cancers and cutaneous T cell lymphoma. Furthermore, DNA demethylation agent can also enhance the sensitivity of highly chemoresistant cells to chemotherapeutic drugs, suggesting their use as a promising approach in treating certain cancers [91]. However, these agents have significant toxicity at high concentrations due to DNA damage, and a group of more potent and selective Dnmt inhibitors, potentially with less toxicity, is now being under development. Zebularine (1-(β-D-ribofuranosyl)-2(1H)-pyrimidinone), a cytidine lacking 4-amino group of the pyrimidine ring, is less toxic and acts by forming a covalent complex with Dnmt and cytidine deaminase when incorporated into DNA [92]. In addition, recent work has shown that the status of methylation of VEGFR promoter dictates the efficacy of the VEGF-targeted drugs on the proliferation of cancer tissue, suggesting that the epigenetic alteration of VEGFRs could influence the efficacy of VEGF-specific tyrosine kinase inhibitors [93]. This is very significant for diabetic patients experiencing possibility of losing vision due to retinopathy as VEGF is considered as one of the major growth factor in the neovascularization associated with proliferative diabetic retinopathy.
As mentioned above, histone modifications can alter gene expression modifying the risk for a disease, and the process is homeostatically balanced by groups of cellular enzymes that add an acetyl or methyl group or remove them. Epigallocatechin-3-gallate is a strong HAT inhibitor, and it inhibits p65 acetylation-dependent NF-kB activation [94] and in the pathogenesis of diabetic retinopathy, activation of NF-kB is considered to accelerate apoptosis of capillary cells, suggesting that inhibitor has potential to inhibit the development of diabetic retinopathy. Histone deacetylases inhibitor can suppress the growth and/or survival of tumor cells [95, 96], and Vorinostat and Romidepsin are the first and only histone deacetylases inhibitors approved by FDA for clinical use [97]. However, other inhibitors of histone deacetylases are in clinical trials for other types of cancer treatment.
Since miRNAs can regulate gene expression via different ways, including translational repression, mRNA cleavage, and deadenylation, miRNAs have rapidly emerged as promising targets for the development of novel therapeutics. Double-stranded miRNA mimics and antimRNA antisense oligodeoxyribonucleotide are the two commonly investigated techniques to target specific miRNA. miRNAs have a specific and defined target in the pathogenic mechanism of the disease, miRNA-based therapy comes with the advantage that they target multiple genes involved in the same pathway process [98]. One of the major caveats with the miRNA-based therapy is their delivery, as these modulators must leave the circulatory system to get into the target tissue and should be able to cross blood-retina barrier. The other important issue is their circulatory half-life. The advances in drug delivery techniques, however, could open up the use of miRNAs for diabetic retinopathy.
As mentioned above, epigenetic modifications are influenced by environmental and dietary factors; many natural compounds have been tested to evaluate their beneficial effects on such modifications [99, 100]. Polyphenols, including resveratrol, a natural compound found in the skin of red grapes and a constituent of red wine, are implicated in the regulation of histone deacetylases, Sirt1 [101]. Curcumin (diferuloylmethane), a natural compound commonly used as a curry spice, is shown to modulate a number of histone modifying enzymes and miRNAs [102, 103], and our previous work has shown that its curcumin ameliorates retinal metabolic abnormalities postulated to be important in the development of diabetic retinopathy [104]. Thus, natural compounds could have potential benefits in inhibiting the development of retinopathy in diabetic patients via modulating both metabolic abnormalities and epigenetic modifications.
The role of epigenetic modifications in the pathogenesis of diabetic retinopathy is an emerging area. Recent research has shown that the diabetes environment fosters epigenetic modifications of a number of genes implicated in the pathogenesis of diabetic retinopathy, and enzymes responsible for bringing in the epigenetic changes are altered. The use of the inhibitors and mimics is targeted to regulate the histone modification, DNA methylation and miRNAs are being targeted for a number of chronic diseases [105–107], and the recent advances in the drug delivery to the retina are now opening up the field for their future testing for this devastating disease which a diabetic patient fears the most.
References
- 1.Zheng Y, He M, Congdon N. The worldwide epidemic of diabetic retinopathy. Indian Journal of Ophthalmology. 2012;60:428–431. doi: 10.4103/0301-4738.100542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank RN. Diabetic Retinopathy. New England Journal of Medicine. 2004;350(1):48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- 3.Lim LS, Wong TY. Lipids and diabetic retinopathy. Expert Opinion on Biological Therapy. 2012;12(1):93–105. doi: 10.1517/14712598.2012.641531. [DOI] [PubMed] [Google Scholar]
- 4.Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. The Lancet. 2010;376(9735):124–136. doi: 10.1016/S0140-6736(09)62124-3. [DOI] [PubMed] [Google Scholar]
- 5.Zhang W, Liu H, Rojas M, Caldwell RW, Caldwell RB. Anti-inflammatory therapy for diabetic retinopathy. Immunotherapy. 2011;3(5):609–628. doi: 10.2217/imt.11.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitada M, Zhang Z, Mima A, King GL. Molecular mechanisms of diabetic vascular complications. Journal of Diabetes Investigation. 2010;1(3):77–89. doi: 10.1111/j.2040-1124.2010.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos JM, Mohammad G, Zhong Q, Kowluru RA. Diabetic retinopathy, superoxide damage and antioxidants. Current Pharmaceutical Biotechnology. 2011;12(3):352–361. doi: 10.2174/138920111794480507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowluru RA. Diabetes-induced elevations in retinal oxidative stress, protein kinase C and nitric oxide are interrelated. Acta Diabetologica. 2001;38(4):179–185. doi: 10.1007/s592-001-8076-6. [DOI] [PubMed] [Google Scholar]
- 9.Torres JM, Cox NJ, Philipson LH. Genome wide association studies for diabetes: perspective on results and challenges. Pediatric Diabetes. 2013;14:90–96. doi: 10.1111/pedi.12015. [DOI] [PubMed] [Google Scholar]
- 10.Donaghue KC, Margan SH, Chan AKF, et al. The association of aldose reductase gene (AKR1B1) polymorphisms with diabetic neuropathy in adolescents. Diabetic Medicine. 2005;22(10):1315–1320. doi: 10.1111/j.1464-5491.2005.01631.x. [DOI] [PubMed] [Google Scholar]
- 11.Ma J, Li Y, Zhou F, Xu X, Guo G, Qu Y. Meta-analysis of association between the Pro12Ala polymorphism of the peroxisome proliferator-activated receptor-γ2 gene and diabetic retinopathy in Caucasians and Asians. Molecular Vision. 2012;18:2352–2360. [PMC free article] [PubMed] [Google Scholar]
- 12.Bonnefond A, Saulnier PJ, Stathopoulou MG, Grarup N, Ndiaye NC, Roussel R, et al. What is the contribution of two genetic variants regulating VEGF levels to type 2 diabetes risk and to microvascular complications? PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0055921.e55921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tewari S, Santos JM, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxidants & Redox Signaling. 2012;17:492–504. doi: 10.1089/ars.2011.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villeneuve LM, Natarajan R. The role of epigenetics in the pathology of diabetic complications. American Journal of Physiology: Renal Physiology. 2010;299(1):F14–F25. doi: 10.1152/ajprenal.00200.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong Q, Kowluru RA. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes. 2011;60(4):1304–1313. doi: 10.2337/db10-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010;107(9):1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Yao H, Lin S, Zhu X, Shen Z, Lu G. Transcriptional and epigenetic regulation of human microRNAs. Cancer Letters. 2013;331:1–10. doi: 10.1016/j.canlet.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 19.McCarthy N. Epigenetics: histone modification. Nature Reviews Cancer. 2013;13, article 379 [Google Scholar]
- 20.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nature Structural & Molecular Biology. 2013;20:259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 21.Portela A, Esteller M. Epigenetic modifications and human disease. Nature Biotechnology. 2010;28(10):1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 22.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes and Development. 2011;25(10):1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams K, Christensen J, Pedersen MT, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473(7347):343–349. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8(11):1698–1710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu J-K. Active DNA demethylation mediated by DNA glycosylases. Annual Review of Genetics. 2009;43:143–166. doi: 10.1146/annurev-genet-102108-134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 28.Zhong Q, Kowluru RA. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: role of histone methylation. Investigative Ophthalmology & Visual Science. 2013;54:244–250. doi: 10.1167/iovs.12-10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong Q, Kowluru RA. Regulation of matrix metallopeptidase-9 by epigenetic modifications, and the development of diabetic retinopathy. Diabetes. 2013;62(7):2559–2568. doi: 10.2337/db12-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaissière T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutation Research. 2008;659(1-2):40–48. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 32.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26(37):5420–5432. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 33.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 34.Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell. 2002;109(7):801–806. doi: 10.1016/s0092-8674(02)00798-5. [DOI] [PubMed] [Google Scholar]
- 35.Kouzarides T. Histone methylation in transcriptional control. Current Opinion in Genetics and Development. 2002;12(2):198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 36.Kubicek S, Jenuwein T. A crack in histone lysine methylation. Cell. 2004;119(7):903–906. doi: 10.1016/j.cell.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 37.Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Current Opinion in Genetics and Development. 2005;15(2):163–176. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Nightingale KP, O’Neill LP, Turner BM. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Current Opinion in Genetics and Development. 2006;16(2):125–136. doi: 10.1016/j.gde.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Guo H-B, Guo H. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(21):8797–8802. doi: 10.1073/pnas.0702981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lachner M, O’Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. Journal of Cell Science. 2003;116(11):2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- 41.Forneris F, Binda C, Battaglioli E, Mattevi A. LSD1: oxidative chemistry for multifaceted functions in chromatin regulation. Trends in Biochemical Sciences. 2008;33(4):181–189. doi: 10.1016/j.tibs.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Musri MM, Carmona MC, Hanzu FA, Kaliman P, Gomis R, Párrizas M. Histone demethylase LSD1 regulates adipogenesis. Journal of Biological Chemistry. 2010;285(39):30034–30041. doi: 10.1074/jbc.M110.151209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garzon R, Calin GA, Croce CM. MicroRNAs in cancer. Annual Review of Medicine. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- 44.Dykxhoorn DM, Novina CD, Sharp PA. Killing the messenger: short RNAs that silence gene expression. Nature Reviews Molecular Cell Biology. 2003;4(6):457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 45.Baer C, Claus R, Plass C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Research. 2013;73:473–477. doi: 10.1158/0008-5472.CAN-12-3731. [DOI] [PubMed] [Google Scholar]
- 46.Lorenzen JM, Martino F, Thum T. Epigenetic modifications in cardiovascular disease. Basic Research in Cardiology. 2012;107(2, article 0245) doi: 10.1007/s00395-012-0245-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dwivedi RS, Herman JG, McCaffrey TA, Raj DSC. Beyond genetics: epigenetic code in chronic kidney disease. Kidney International. 2011;79(1):23–32. doi: 10.1038/ki.2010.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shanmugam MK, Sethi G. Role of epigenetics in inflammation-associated diseases. Subcellular Biochemistry. 2012;61:627–657. doi: 10.1007/978-94-007-4525-4_27. [DOI] [PubMed] [Google Scholar]
- 49.Pham TX, Lee J. Dietary regulation of histone acetylases and deacetylases for the prevention of metabolic diseases. Nutrients. 2012;4:1868–1886. doi: 10.3390/nu4121868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ding G-L, Wang F-F, Shu J, et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes. 2012;61(5):1133–1142. doi: 10.2337/db11-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ling C, Del Guerra S, Lupi R, et al. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia. 2008;51(4):615–622. doi: 10.1007/s00125-007-0916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tewari S, Zhong Q, Santos JM, Kowluru RA. Mitochondria DNA replication and DNA methylation in the metabolic memory associated with continued progression of diabetic retinopathy. Investigative Ophthalmology & Visual Science. 2012;53:4881–4888. doi: 10.1167/iovs.12-9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes. 2009;58(12):2718–2725. doi: 10.2337/db09-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Medical Genomics. 2010;3, article 33 doi: 10.1186/1755-8794-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akçay T, Dinçer Y, Celebi N, Ilkova H. O6-methylguanine DNA methyltransferase activity in diabetic patients. Diabetes Research and Clinical Practice. 2003;61:1–6. doi: 10.1016/s0168-8227(03)00063-9. [DOI] [PubMed] [Google Scholar]
- 56.Williams KT, Garrow TA, Schalinske KL. Type I diabetes leads to tissue-specific DNA hypomethylation in male rats. Journal of Nutrition. 2008;138(11):2064–2069. doi: 10.3945/jn.108.094144. [DOI] [PubMed] [Google Scholar]
- 57.Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(26):9047–9052. doi: 10.1073/pnas.0803623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhong Q, Kowluru RA. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. Journal of Cellular Biochemistry. 2010;110(6):1306–1313. doi: 10.1002/jcb.22644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olsen AS, Sarras MP, Jr., Leontovich A, Intine RV. Heritable transmission of diabetic metabolic memory in zebrafish correlates with DNA hypomethylation and aberrant gene expression. Diabetes. 2012;61(2):485–491. doi: 10.2337/db11-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson RF, Fazzari MJ, Niu H, Barzilai N, Simmons RA, Greally JM. Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. Journal of Biological Chemistry. 2010;285(20):15111–15118. doi: 10.1074/jbc.M109.095133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feng B, Chen S, George B, Feng Q, Chakrabarti S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes/Metabolism Research and Reviews. 2010;26(1):40–49. doi: 10.1002/dmrr.1054. [DOI] [PubMed] [Google Scholar]
- 62.Wang XH, Qian RZ, Zhang W, Chen SF, Jin HM, Hu RM. MicroRNA-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clinical and Experimental Pharmacology and Physiology. 2009;36(2):181–188. doi: 10.1111/j.1440-1681.2008.05057.x. [DOI] [PubMed] [Google Scholar]
- 63.Fu Y, Zhang Y, Wang Z, et al. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. American Journal of Nephrology. 2010;32(6):581–589. doi: 10.1159/000322105. [DOI] [PubMed] [Google Scholar]
- 64.Krupa A, Jenkins R, DongLuo D, Lewis A, Phillips A, Fraser D. Loss of microRNA-192 promotes fibrogenesis in diabetic nephropathy. Journal of the American Society of Nephrology. 2010;21(3):438–447. doi: 10.1681/ASN.2009050530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Madsen-Bouterse SA, Kowluru RA. Oxidative stress and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Reviews in Endocrine and Metabolic Disorders. 2008;9(4):315–327. doi: 10.1007/s11154-008-9090-4. [DOI] [PubMed] [Google Scholar]
- 66.Syreeni A, El-Osta A, Forsblom C, et al. Genetic examination of SETD7 and SUV39H1/H2 methyltransferases and the risk of diabetes complications in patients with type 1 diabetes. Diabetes. 2011;60(11):3073–3080. doi: 10.2337/db11-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kadiyala CS, Zheng L, Du Y, Yohannes E, Kao HY, Miyagi M. Acetylation of retinal histones in diabetes increases inflammatory proteins: effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC) Journal of Biological Chemistry. 2012;287:25869–25880. doi: 10.1074/jbc.M112.375204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kanwar M, Chan P-S, Kern TS, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Investigative Ophthalmology and Visual Science. 2007;48(8):3805–3811. doi: 10.1167/iovs.06-1280. [DOI] [PubMed] [Google Scholar]
- 69.Kowluru RA, Atasi L, Ho Y-S. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Investigative Ophthalmology & Visual Science. 2006;47(4):1594–1599. doi: 10.1167/iovs.05-1276. [DOI] [PubMed] [Google Scholar]
- 70.Kowluru RA, Kowluru V, Xiong Y, Ho Y-S. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radical Biology and Medicine. 2006;41(8):1191–1196. doi: 10.1016/j.freeradbiomed.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 71.Madsen-Bouterse SA, Zhong Q, Mohammad G, Ho Y-S, Kowluru RA. Oxidative damage of mitochondrial DNA in diabetes and its protection by manganese superoxide dismutase. Free Radical Research. 2010;44(3):313–321. doi: 10.3109/10715760903494168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kowluru RA. Role of matrix metalloproteinase-9 in the development of diabetic retinopathy and its regulation by H-Ras. Investigative Ophthalmology & Visual Science. 2010;51(8):4320–4326. doi: 10.1167/iovs.09-4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kowluru RA, Mohammad G, Dos Santos JM, Zhong Q. Abrogation of MMP-9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes. 2011;60(11):3023–3033. doi: 10.2337/db11-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perrone L, Devi TS, Hosoya K-I, Terasaki T, Singh LP. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. Journal of Cellular Physiology. 2009;221(1):262–272. doi: 10.1002/jcp.21852. [DOI] [PubMed] [Google Scholar]
- 75.McArthur K, Feng B, Wu Y, Chen S, Chakrabarti S. MicroRNA-200b regulates vascular endothelial growth factor-mediated alterations in diabetic retinopathy. Diabetes. 2011;60(4):1314–1323. doi: 10.2337/db10-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kovacs B, Lumayag S, Cowan C, Xu S. MicroRNAs in early diabetic retinopathy in streptozotocin-induced diabetic rats. Investigative Ophthalmology & Visual Science. 2011;52(7):4402–4409. doi: 10.1167/iovs.10-6879. [DOI] [PubMed] [Google Scholar]
- 77.Silva VAO, Polesskaya A, Sousa TA, et al. Expression and cellular localization of microRNA-29b and RAX, an activator of the RNA-dependent protein kinase (PKR), in the retina of streptozotocin-induced diabetic rats. Molecular Vision. 2011;17:2228–2240. [PMC free article] [PubMed] [Google Scholar]
- 78.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 79.Valko M, Morris H, Cronin MTD. Metals, toxicity and oxidative stress. Current Medicinal Chemistry. 2005;12(10):1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 80.Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. International Journal of Biochemistry and Cell Biology. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 81.Wang J, Wu Z, Li D, Li N, Dindot SV, Satterfield MC. Nutrition, epigenetics, and metabolic syndrome. Antioxidants & Redox Signaling. 2012;17:282–301. doi: 10.1089/ars.2011.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Valinluck V, Tsai H-H, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Research. 2004;32(14):4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chervona Y, Costa M. The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic Biol Med. 2012;53:1041–1047. doi: 10.1016/j.freeradbiomed.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3630–3635. doi: 10.1073/pnas.1012311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dzitoyeva S, Chen H, Manev H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiology of Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Madsen-Bouterse SA, Mohammad G, Kanwar M, Kowluru RA. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxidants and Redox Signaling. 2010;13(6):797–805. doi: 10.1089/ars.2009.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Santos JM, Tewari S, Goldberg AFX, Kowluru RA. Mitochondrial biogenesis and the development of diabetic retinopathy. Free Radical Biology and Medicine. 2011;51(10):1849–1860. doi: 10.1016/j.freeradbiomed.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ohgaki K, Kanki T, Fukuoh A, et al. The C-terminal tail of mitochondrial transcription factor A markedly strengthens its general binding to DNA. Journal of Biochemistry. 2007;141(2):201–211. doi: 10.1093/jb/mvm020. [DOI] [PubMed] [Google Scholar]
- 89.Santos JM, Tewari S, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radical Biology and Medicine. 2012;53:1729–1737. doi: 10.1016/j.freeradbiomed.2012.08.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Santos JM, Kowluru RA. Impaired transport of mitochondrial transcription factor A (TFAM) and the metabolic memory phenomenon associated with the progression of diabetic retinopathy. Diabetes/Metabolism Research and Reviews. 2013;29:204–213. doi: 10.1002/dmrr.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eglen RM, Reisine T. Screening for compounds that modulate epigenetic regulation of the transcriptome: an overview. Journal of Biomolecular Screening. 2011;16(10):1137–1152. doi: 10.1177/1087057111417871. [DOI] [PubMed] [Google Scholar]
- 92.Champion C, Guianvarc’h D, Sénamaud-Beaufort C, et al. Mechanistic insights on the inhibition of C5 DNA methyltransferases by zebularine. PLoS ONE. 2010;5(8) doi: 10.1371/journal.pone.0012388.e12388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim J, Hwang J, Jeong H, et al. Promoter methylation status of VEGF receptor genes-a possible epigenetic biomarker to anticipate the efficacy of intracellular-acting vegf-targeted drugs in cancer cells. Epigenetics. 2012;7(2):191–200. doi: 10.4161/epi.7.2.18973. [DOI] [PubMed] [Google Scholar]
- 94.Choi K-C, Myung GJ, Lee Y-H, et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Research. 2009;69(2):583–592. doi: 10.1158/0008-5472.CAN-08-2442. [DOI] [PubMed] [Google Scholar]
- 95.Peart MJ, Tainton KM, Ruefli AA, et al. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Research. 2003;63(15):4460–4471. [PubMed] [Google Scholar]
- 96.Khabele D, Son D-S, Parl AK, et al. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: implications for therapy. Cancer Biology and Therapy. 2007;6(5):795–801. doi: 10.4161/cbt.6.5.4007. [DOI] [PubMed] [Google Scholar]
- 97.Glass E, Viale PH. Histone deacetylase inhibitors: novel agents in cancer treatment. Clinical Journal of Oncology Nursing. 2013;17:34–40. doi: 10.1188/13.CJON.34-40. [DOI] [PubMed] [Google Scholar]
- 98.Caroli A, Cardillo MT, Galea R, Biasucci LM. Potential therapeutic role of microRNAs in ischemic heart disease. Journal of Cardiology. 2013;61:315–320. doi: 10.1016/j.jjcc.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 99.Khan SI, Aumsuwan P, Khan IA, Walker LA, Dasmahapatra AK. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chemical Research in Toxicology. 2012;25(1):61–73. doi: 10.1021/tx200378c. [DOI] [PubMed] [Google Scholar]
- 100.Reuter S, Gupta SC, Park B, Goel A, Aggarwal BB. Epigenetic changes induced by curcumin and other natural compounds. Genes and Nutrition. 2011;6(2):93–108. doi: 10.1007/s12263-011-0222-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chung S, Yao H, Caito S, Hwang J-W, Arunachalam G, Rahman I. Regulation of SIRT1 in cellular functions: role of polyphenols. Archives of Biochemistry and Biophysics. 2010;501(1):79–90. doi: 10.1016/j.abb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bora-Tatar G, Dayangaç-Erden D, Demir AS, Dalkara S, Yelekçi K, Erdem-Yurter H. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: activity and docking studies. Bioorganic and Medicinal Chemistry. 2009;17(14):5219–5228. doi: 10.1016/j.bmc.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 103.Howell JC, Chun E, Farrell AN. Global microRNA expression profiling: curcumin (diferuloylmethane) alters oxidative stress-responsive microRNAs in human ARPE-19 cells. Molecular Vision. 2013;19:544–560. [PMC free article] [PubMed] [Google Scholar]
- 104.Kowluru RA, Kanwar M. Effects of curcumin on retinal oxidative stress and inflammation in diabetes. Nutrition and Metabolism. 2007;4, article 8 doi: 10.1186/1743-7075-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu Q, Quinn AM, Patel MP, Semus SF, Graves AP, Bandyopadhyay D. Perspectives on the discovery of small-molecule modulators for epigenetic processes. Journal of Biomolecular Screening. 2012;17:555–571. doi: 10.1177/1087057112437763. [DOI] [PubMed] [Google Scholar]
- 106.Nana-Sinkam SP, Croce CM. Clinical applications for microRNAs in cancer. Clinical Pharmacology & Therapeutics. 2013;93:98–104. doi: 10.1038/clpt.2012.192. [DOI] [PubMed] [Google Scholar]
- 107.Oki Y, Issa J-PJ. Review: recent clinical trials in epigenetic therapy. Reviews on Recent Clinical Trials. 2006;1(2):169–182. doi: 10.2174/157488706776876490. [DOI] [PubMed] [Google Scholar]