

Abstract
While there have been significant advances in our understanding of the autoimmune responses and the molecular nature of the target autoantigens in primary biliary cirrhosis (PBC), unfortunately these data have yet to be translated into new therapeutic agents. We have taken advantage of a unique murine model of autoimmune cholangitis in which mice expressing a dominant negative form of transforming growth factor β receptor II (dnTGFβRII), under the control of the CD4 promoter, develop an intense autoimmune cholangitis associated with serological features similar to human PBC. CD40-CD40 ligand (CD40L) is a major receptor–ligand pair that provides key signals between cells of the adaptive immune system, prompting us to determine the therapeutic potential of treating autoimmune cholangitis with anti-CD40L antibody (anti-CD40L; MR-1). Four-week-old dnTGFβRII mice were injected intraperitoneally with either anti-CD40L or control immunoglobulin (Ig)G at days 0, 2, 4 and 7 and then weekly until 12 or 24 weeks of age and monitored for the progress of serological and histological features of PBC, including rigorous definition of liver cellular infiltrates and cytokine production. Administration of anti-CD40L reduced liver inflammation significantly to 12 weeks of age. In addition, anti-CD40L initially lowered the levels of anti-mitochondrial autoantibodies (AMA), but these reductions were not sustained. These data indicate that anti-CD40L delays autoimmune cholangitis, but the effect wanes over time. Further dissection of the mechanisms involved, and defining the events that lead to the reduction in therapeutic effectiveness will be critical to determining whether such efforts can be applied to PBC.
Keywords: anti-CD40 ligand antibody, autoimmunity, cholangitis, primary biliary cirrhosis
Introduction
Primary biliary cirrhosis (PBC) is a unique chronic, fibrotic liver disease predominantly affecting women and characterized by an immune-mediated specific destruction of the small intrahepatic bile ducts [1–3]. Although the aetiology is unknown, results from a series of our previous study suggest that the pathogenesis of human PBC is attributed primarily to autoreactive T cells [4–6]. Recently, we have described several murine models of PBC allowing dissection of the immune response and preclinical testing of potential novel therapies. Notably, mice expressing a dominant negative form of transforming growth factor-β receptor type II (dnTGFβRII) under the control of the CD4 promoter develop anti-mitochondrial antibodies and an autoimmune cholangitis. In these mice, adoptive transfer of CD8+ T cells recapitulates the cholangitis [7] while depletion of B cells exacerbates the disease [8], indicating the interconnected roles between autoreactive T and B cells in this model.
CD40–CD40 ligand (CD40L) is a receptor–ligand pair that provides key communication signals between cells of the adaptive immune system. CD40L is required for generating optimal CD4+ and CD8+ T cell responses through activation of dendritic cells [9]. Numerous studies have documented the key role played by the interaction between CD40 and CD40L in priming immune responses, expanding antigen primed T cells, up-regulating the expression of other co-stimulatory molecules and promoting the release of cytokines and chemokines by immune cells [10–13]. Dysregulation in the CD40–CD40L co-stimulation pathway are featured prominently in systemic autoimmune and tissue-specific autoimmune disease [14–17]. Levels of CD40L are correlated positively with hepatic immunoglobulin (Ig) production [18]. Further, apoptotic intrahepatic biliary epithelial cells frequently express increased levels of CD40 and are associated with CD40L-expressing T cells and macrophages [19]. More recently, our group demonstrated that IgM levels inversely correlate with CD40L promoter methylation [20]. These findings prompted us to explore the therapeutic use of a monoclonal antibody with specificity for CD40L.
Materials and methods
Animals
dnTGFβRII mice were bred onto a C57BL/6J (B6) strain background at the animal facilities of the University of California at Davis. All mice were genotyped at 3–4 weeks of age to confirm the dnTGF-βRII transgene [21]. Mice were fed a sterile rodent helicobacter medicated dosing system (three-drug combination) diet (Bio-Serv, Frenchtown, NJ, USA) and maintained in individually ventilated cages under specific pathogen-free conditions. Sulfatrim (Hi-tech Pharmacal, Amityville, NY, USA) was delivered through drinking water. All protocols were approved by the University of California Animal Care and Use Committee.
Anti-CD40L antibody
Anti-CD40L (anti-CD40L; MR-1) (BioXCell, MR-1; BE0017-1) was utilized for the studies herein at a concentration of 2·5 mg/ml. Four-week-old dnTGFβRII mice (n = 10–12 per group) were injected intraperitoneally with 25 mg/kg body weight of either anti-CD40L or control IgG on days 0, 2, 4 and 7 and thence weekly until 12 or 24 weeks of age.
Flow cytometry analysis
Liver and spleen mononuclear cells (MNCs) were isolated as described previously [7,21]; 1×106 cells were resuspended in staining buffer [0·2% bovine serum albumin (BSA), 0·04% ethylenediamine tetraacetic acid (EDTA) and 0·05% sodium azide in phosphate-buffered saline (PBS)], divided into 25-μl aliquots, and incubated first with anti-mouse FcR blocking reagent (BioLegend, San Diego, CA, USA) for 15 min at 4°C and stained for 30 min at 4°C with cocktails containing combinations of various fluorochrome-conjugated monoclonal antibodies (mAb) for CD4, CD8α, CD44, CD62L, NK1·1, CD69, CD19 (Biolegend) and T cell receptor (TCR)-β (eBioscience, San Diego, CA, USA). For intracellular cytokine staining, cells were resuspended in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS) (RPMI-FBI) and stimulated at 37°C for 4 h with leucocyte activation cocktail in the presence of BD GolgiPlug (BD Pharmingen, San Diego, CA, USA). The cells were stained for surface CD4, CD8, NK1·1 and TCR-β, fixed and permeabilized with BD Cytofix/Cytoperm Solution (BD Biosciences), then stained for intracellular interferon-γ (IFN-γ) (BioLegend) [22]. A fluorescence activated cell sorter (FACS)can flow cytometer (BD Immunocytometry Systems, San Jose, CA, USA), upgraded for detection of five colours by Cytek Development (Fremont, CA, USA), was used to acquire data, analysed with Cellquest Pro software.
Anti-mitochondrial antibodies (AMA)
AMAs were detected using our recombinant PDC-E2-based enzyme-linked immunosorbent assay (ELISA) as described previously, using our standardized protocol [21,23,24].
Histopathology
Livers were collected and fixed in 4% paraformaldehyde, embedded in paraffin, cut into 4-μm sections, deparaffinized and stained with haematoxylin and eosin (H&E) [25]. Portal inflammation and bile duct damage were evaluated by a ‘blinded’ pathologist. Portal inflammation were scored as follows: 1, normal liver histology; 2, minimal inflammation; 3, mild inflammation; 4, moderate inflammation; and 5, severe inflammation. The bile duct damage was graded as: 1, no significant changes of bile duct; 2, mild to moderate cell infiltrate with bile duct destruction; 3, moderate to severe cell infiltrate with bile duct loss.
Cytokine analysis
Total protein was extracted from 30 mg of frozen liver tissues by homogenization in T-Per® Tissue Protein Extraction buffer (Thermo, Rockford, IL, USA) containing a protease inhibitor cocktail (Roche, Indianapolis, IN, USA). The homogenized tissue suspension was centrifuged at 12 000 g for 20 min at 4°C and supernatant fluid stored at −80°C. Serum and tissue lysate levels of IFN-γ, tumour necrosis factor (TNF)-α, IL-6, IL-17A, IL-4, IL-2 and IL-10 were measured with a cytokine bead array assay using the mouse T helper type 1 (Th1)/Th2/Th17 cytokine kit (BD Biosciences). The protein concentration of liver extracts was measured using the BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA)
Statistical analysis
A two-tailed unpaired Mann–Whitney test and a χ2 analysis were used to analyse the data; P-values < 0·05 were considered statistically significant.
Results
Anti-CD40L suppresses peripheral T cell activation in dnTGFβRII mice
We first examined the effect of anti-CD40L administration on the frequencies of activated T cells in peripheral blood after 2 weeks of treatment. As shown in Fig. 1a, anti-CD40L treatment did not significantly affect the frequencies of total CD4 and CD8 T cells in peripheral blood. However, for both CD4+ and CD8+ populations, naive (CD44+CD62L+) T cells were increased and effector (CD44−CD62L−) and memory (CD44+CD62L+) T cells were decreased significantly by anti-CD40L (Fig. 1a). We next evaluated whether Th1/Th2/Th17-related cytokines were also affected by anti-CD40L. After 8 weeks of treatment, serum levels of IFN-γ, IL-4 and IL-17A were significantly lower in the anti-CD40L-treated mice compared with the control mice (Fig. 1b). Similarly, the level of other inflammatory cytokines such as IL-2, TNF-α, IL-6 and IL-10 were also lower in serum from the treatment group (data not shown).
Fig. 1.
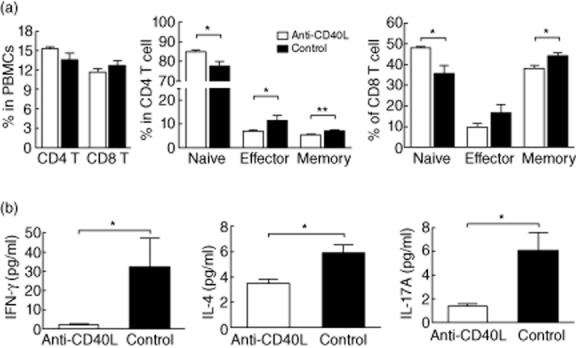
Suppression of T cell activation by anti-CD40L treatment. (a) The frequency of CD4, CD8 T cells and their activated subsets in peripheral blood mononuclear cells (MNCs) after 2 weeks of anti-CD40L. (b) Serum levels of interferon (IFN)-γ, interleukin (IL)-4 and IL-17A after 8 weeks of anti-CD40L of immunoglobulin (Ig)G. Data are expressed as mean ± standard error of the mean. *P < 0·05; two-tailed unpaired Mann–Whitney U-test.
Anti-CD40L ameliorates cholangitis in dnTGFβRII mice
dnTGFβRII mice treated with anti-CD40L demonstrated a significant decrease in portal pathology (Fig. 2). Of note, although some mice exhibited minimal portal inflammation, interlobular biliary cell damage was not observed in any treated mice (Fig. 2b). In contrast, at 12 weeks of age, control dnTGFβRII mice developed readily recognized cholangitis highlighted by the presence of lymphocyte infiltrates and bile duct damage (Fig. 2).
Fig. 2.
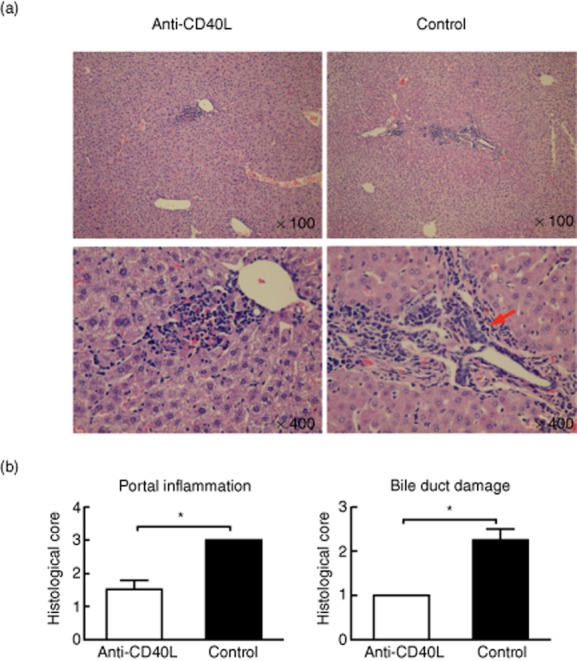
Eight weeks of anti-CD40L treatment suppresses portal inflammation and cholangitis. (a) Representative haematoxylin and eosin (H&E)-stained liver sections from dominant negative form of transforming growth factor beta receptor type II (dnTGFβRII) mice 8 weeks after treatment with anti-CD40L or control immunoglobulin (Ig)G. Arrows point to the interlobular bile duct destruction. (b) Portal inflammation and bile duct destruction scores. *P < 0·05; χ2 test.
Analysis of intrahepatic MNCs revealed a decrease in the frequency of natural killer (NK) T cells with anti-CD40L treatment, while other subsets, including CD4+ T, CD8+ T, NK and B cells, were not significantly different (Fig. 3a). However, the frequencies of effector (CD44+CD62L− population) CD4+ and CD8+ T cells were decreased significantly in liver and spleen with anti-CD40L treatment (Fig. 3b). The percentage of CD69+ cells was decreased significantly among both CD4+ and CD8+ T cells in anti-CD40L treated mice (Fig. 3c). Of note, intracellular staining data demonstrated that the percentages of IFN-γ-producing CD4 T cells in liver were decreased markedly in mice after anti-CD40L treatment (Fig. 3d).
Fig. 3.

Eight weeks of anti-CD40L treatment suppresses CD4+ and CD8 T+ cell activation. (a) Absolute number of intrahepatic immune cell subpopulations. (b) Frequency of naive (CD44−CD62L+), effector (CD44+CD62L−), and memory (CD44+CD62L+) subpopulations in CD4+ and CD8+ T cell subsets in spleen and liver. (c) CD69+ and (d) interferon (IFN)-γ-expressing cell in intrahepatic CD4+ and CD8+ T cells in anti-CD40L and immunoglobulin (Ig)G (control)-treated mice. Data are expressed as the mean ± standard error of the mean. *P < 0·05; two-tailed unpaired Mann–Whitney U-test.
Loss of anti-CD40L effects in dnTGFβRII mice after 20 weeks
In contrast to the 8-week treatment group, livers from mice treated for 20 weeks demonstrated no significant differences in the severity of cholangitis or bile duct damage between anti-CD40L-treated mice and control mice (Fig. 4a,b). However, there was a persistent decrease in hepatic and splenic effector (CD44+CD62L−) CD8+ T cells (Fig. 5a). In contrast, neither intrahepatic nor splenic CD4+ T cell subsets differed significantly between treated and control groups. Further, the absolute number of effector CD8 T cells in liver was not significantly different between anti-CD40L-treated and control mice (Fig. 5b). Intracellular cytokine staining showed that IFN-γ-producing CD4+ and CD8+ T cells did not exhibit differences between treated and control mice (Fig. 5c). In addition, the levels of the various proinflammatory cytokines IFN-γ, IL-17A, TNF-α, IL-4, IL-6 and IL-10 were not significantly different in liver protein extracts from anti-CD40L treated compared with control mice (Fig. 5d and data not shown).
Fig. 4.
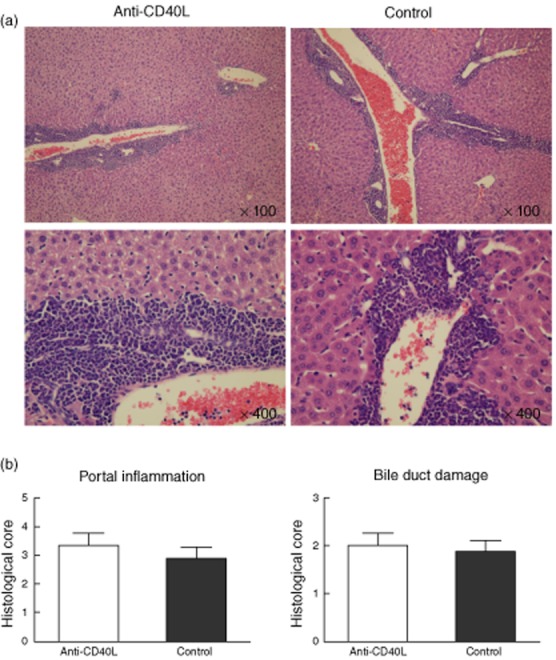
The effects of anti-CD40L following 20 weeks of treatment (24 weeks of age). Note that there were no significant differences between treated and control mice, unlike analysis at 12 weeks of age. (a) Representative haematoxylin and eosin (H&E)-stained liver sections from dominant negative form of transforming growth factor beta receptor type II (dnTGFβRII) mice treated with anti-CD40L or control immunoglobulin (Ig)G. (b) Histological scores of portal inflammation and bile duct destruction.
Fig. 5.
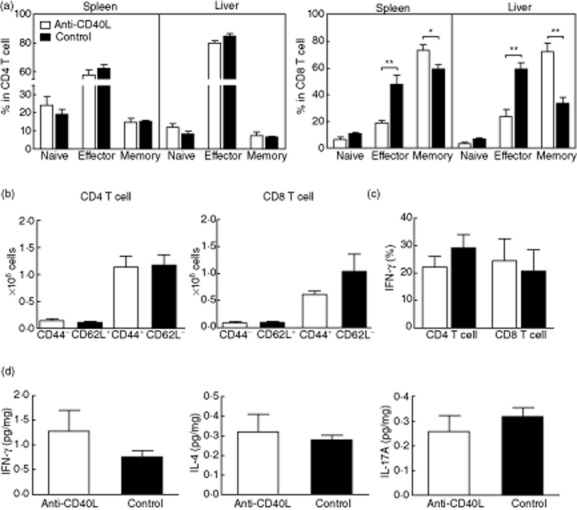
T cell activation after 20 weeks of anti-CD40L treatment. (a) The frequency of naive (CD44−CD62L+), effector (CD44+CD62L−), and memory (CD44+CD62L+) subpopulations in CD4+ and CD8+ T cell subsets in spleen and liver. (b) The absolute numbers of effector (CD44+CD62L−) CD4+ and CD8+ T cells in liver of anti-CD40L and control immunoglobulin (Ig)G-treated mice. (c) Frequency of interferon (IFN)-γ producing CD4+ and CD8+ T cells in liver. (d) Levels of IFN-γ, interferon (IL)-4 and IL17A in liver lysates from mice treated for 20 weeks. Data are expressed as the mean ± standard error of the mean. *P < 0·05; **P < 0·01; two-tailed unpaired Mann–Whitney U-test.
AMA production in dnTGFβRII mice
Compared to control mice, antibodies to PDC-E2 in CD40L-treated mice were reduced significantly following 8 weeks of treatment (12 weeks of age) [optical density (OD) value, anti-CD40L, 0·4548 ± 0·0499 versus control group, 1·1214 ± 0·2491]. No significant differences in AMA levels were found between the anti-CD40L-treated and the control group at 24 weeks of age.
Discussion
Beneficial effects of blocking CD40L have been observed in preclinical models of multiple sclerosis, arthritis, systemic lupus erythematosus and Alzheimer's disease [26–30]. We thus took advantage of our murine model of autoimmune cholangitis, the dnTGFβRII mice, to investigate the potential therapeutic effects of anti-CD40L in PBC. Given that our previous studies suggested that the pathogenesis of PBC is primarily secondary to autoreactive T cells, and in particular CD8+ T cells [4–7], we reasoned that by suppressing CD8+ T cells by blockade of the CD40–CD40L pathway we would reduce autoimmune cholangitis. Our results demonstrate herein that the use of anti-CD40L to 12 weeks of age reduces portal inflammation significantly. However, this effect was lost at 24 weeks of age. Hence, the use of anti-CD40L significantly delays, but does not prevent, the development of autoimmune cholangitis.
A key factor in our study is the time of treatment initiation. We chose to start treatment at 4 weeks of age for several reasons. At 4 weeks of age, mice have normal thymic cellularity and subset distribution and no changes in the distribution of cellular subsets in the periphery. However, at 4 weeks of age, there are already the beginnings of a change in the percentage of cells with a naive CD62highCD44low phenotype and an increase of T cells with an activated/memory phenotype [31]. We demonstrate that even after 2 weeks of treatment with anti-CD40L, there was a marked decrease of effector and memory T cell frequencies. In addition, after 8 weeks of treatment, CD4+ and CD8+ effector T cells, as well as NK T cells, in liver were reduced significantly. Further, intra-hepatic IFN-γ-producing CD4+ T cells were suppressed significantly by anti-CD40L treatment. These data are consistent with our hypothesis that autoactivation of T cells in these mice is CD40–CD40L-dependent, and suggest that CD40L blockade would be an effective strategy for the treatment of PBC.
In addition to the effects of anti-CD40L on T cells, there was also suppressed production of prototypical anti-mitochondrial antibodies [32], reflecting a secondary effect on B cells due to the inhibition of CD4+ T cells. Given that CD8+ T cells are the primary contributors to autoimmune cholangitis in this model based upon our adoptive transfer experiments [7], anti-CD40L might inhibit B cell-mediated liver infiltration of CD8+ T cells from extrahepatic lymphoid organs [24]. Alternatively, blocking the CD40/CD40L signalling pathway may suppress development of PDC-E2 primed CD8+ T cells due to the lower level of AMA, which may be responsible for cross-priming CD8+ T cells, a process in which antigen-presenting cells bind PDC–E2–Ig complexes on their Fc receptors and cross-prime CD8+ T cells into activated PDC–E2-reactive phenotypes [5]. However, we emphasize the lack of a durable effect after 24 weeks of treatment with anti-CD40L, data consistent with the immunohistology.
There are potential explanations for the lack of a robust and continued efficacy of treatment. We considered the possibility that there were neutralizing antibodies against anti-CD40L. The chronic administration of exogenous antibodies often causes the development of anti-drug antibodies (ADA) which can limit the efficiency of antibodies, as well as hypersensitivity reactions, immunotoxicological reactions and other adverse events [33–35]. However, we were unable to detect ADA in serum of the long-term anti-CD40L-treated dnTGFβRII mice (data not shown). There are other possible explanations for the reduction in efficacy. First, it is possible that over time there is binding to target antigens in tissues other than necessary for the therapeutic effect. Secondly, either non-specific binding or cross-reactivity with non-target antigens may occur. Either of these would lead to an underdosing of anti-CD40L for maintenance of remission and loss of efficacy [33]. Clearly, the treatment of PBC has been difficult, and therapeutic efforts with other biologicals have also had shortcomings [24], despite major advances in the immunobiology of PBC [20,36–39]. Thirdly, it is also possible that further study of the pharmacokinetics of anti-CD40L might lead to more optimal results and long-term therapy, as reflected by similar studies in other experimental models [40,41]. Finally, we should emphasize that in the work herein we studied a hamster anti-mouse antibody and we note that a fully murine sequence might reduce any anti-hamster reactivity. There is a major need to develop therapies in human PBC, and the use of animal models is a logical first step for determining whether the newer biologicals can be applied to patients.
Acknowledgments
The authors thank Yugo Ando, Jinjun Wang, Jun Zhang, Chen-yen Yang, Weici Zhang, Kerstien Padgett and Tom P. Kenny for technical support in this experiment. We also thank Nikki Phipps for support in preparing this article. Financial support was provided by National Institutes of Health grant DK090019.
Author contributions
H. T. performed the experiments. G.-X. Y. performed the experiments and interpreted the data. N. I. interpreted the data and contributed to the writing of the manuscript. S. J. K. designed the experiment and interpreted the data. K. K. performed the experiments. K. T. performed the experiments and interpreted the data. P. L. designed the experiments, interpreted the data and wrote the manuscript. R. L. C. designed the experiments, interpreted the data and wrote the manuscript. A. A. A. designed the experiments, interpreted the data and wrote the manuscript. T. J. interpreted the data and edited the manuscript. C. B. designed the experiments, interpreted the data and wrote the manuscript. M. E. G. designed the experiments, interpreted the data and wrote the manuscript.
Disclosures
There are no financial, professional or personal conflicts for any authors.
References
- 1.Gershwin ME, Ansari AA, Mackay IR, et al. Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev. 2000;174:210–225. doi: 10.1034/j.1600-0528.2002.017402.x. [DOI] [PubMed] [Google Scholar]
- 2.Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: convenient and inconvenient truths. Hepatology. 2008;47:737–745. doi: 10.1002/hep.22042. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka H, Leung PS, Kenny TP, Gershwin ME, Bowlus CL. Immunological orchestration of liver fibrosis. Clin Rev Allergy Immunol. 2012;43:220–229. doi: 10.1007/s12016-012-8323-1. [DOI] [PubMed] [Google Scholar]
- 4.Shimoda S, Van de Water J, Ansari A, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest. 1998;102:1831–1840. doi: 10.1172/JCI4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kita H, Lian ZX, Van de Water J, et al. Identification of HLA-A2-restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J Exp Med. 2002;195:113–123. doi: 10.1084/jem.20010956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kita H, Matsumura S, He XS, et al. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002;109:1231–1240. doi: 10.1172/JCI14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang GX, Lian ZX, Chuang YH, et al. Adoptive transfer of CD8(+) T cells from transforming growth factor beta receptor type II (dominant negative form) induces autoimmune cholangitis in mice. Hepatology. 2008;47:1974–1982. doi: 10.1002/hep.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moritoki Y, Zhang W, Tsuneyama K, et al. B cells suppress the inflammatory response in a mouse model of primary biliary cirrhosis. Gastroenterology. 2009;136:1037–1047. doi: 10.1053/j.gastro.2008.11.035. [DOI] [PubMed] [Google Scholar]
- 9.Feau S, Arens R, Togher S, Schoenberger SP. Autocrine IL-2 is required for secondary population expansion of CD8(+) memory T cells. Nat Immunol. 2011;12:908–913. doi: 10.1038/ni.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Song G. The role of CD40–CD154 interaction in cell immunoregulation. J Biomed Sci. 2004;11:426–438. doi: 10.1007/BF02256091. [DOI] [PubMed] [Google Scholar]
- 12.Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol. 2009;21:293–300. doi: 10.1016/j.smim.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 14.Ciferska H, Horak P, Hermanova Z, et al. The levels of sCD30 and of sCD40L in a group of patients with systemic lupus erythematodes and their diagnostic value. Clin Rheumatol. 2007;26:723–728. doi: 10.1007/s10067-006-0389-9. [DOI] [PubMed] [Google Scholar]
- 15.Goules A, Tzioufas AG, Manousakis MN, Kirou KA, Crow MK, Routsias JG. Elevated levels of soluble CD40 ligand (sCD40L) in serum of patients with systemic autoimmune diseases. J Autoimmun. 2006;26:165–171. doi: 10.1016/j.jaut.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Vakkalanka RK, Woo C, Kirou KA, Koshy M, Berger D, Crow MK. Elevated levels and functional capacity of soluble CD40 ligand in systemic lupus erythematosus sera. Arthritis Rheum. 1999;42:871–881. doi: 10.1002/1529-0131(199905)42:5<871::AID-ANR5>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 17.Ludwiczek O, Kaser A, Tilg H. Plasma levels of soluble CD40 ligand are elevated in inflammatory bowel diseases. Int J Colorect Dis. 2003;18:142–147. doi: 10.1007/s00384-002-0425-4. [DOI] [PubMed] [Google Scholar]
- 18.Mayo MJ, Mosby JM, Jeyarajah R, et al. The relationship between hepatic immunoglobulin production and CD154 expression in chronic liver diseases. Liver Int. 2006;26:187–196. doi: 10.1111/j.1478-3231.2005.01211.x. [DOI] [PubMed] [Google Scholar]
- 19.Afford SC, Ahmed-Choudhury J, Randhawa S, et al. CD40 activation-induced, Fas-dependent apoptosis and NF-kappaB/AP-1 signaling in human intrahepatic biliary epithelial cells. FASEB J. 2001;15:2345–2354. doi: 10.1096/fj.01-0088com. [DOI] [PubMed] [Google Scholar]
- 20.Lleo A, Liao J, Invernizzi P, et al. Immunoglobulin M levels inversely correlate with CD40 ligand promoter methylation in patients with primary biliary cirrhosis. Hepatology. 2012;55:153–160. doi: 10.1002/hep.24630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oertelt S, Lian ZX, Cheng CM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655–1660. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 22.Tsuda M, Zhang W, Yang GX, et al. Deletion of interleukin (IL)-12p35 induces liver fibrosis in dominant-negative TGFbeta receptor type II mice. Hepatology. 2013;57:806–816. doi: 10.1002/hep.25829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wakabayashi K, Lian ZX, Leung PS, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48:531–540. doi: 10.1002/hep.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moritoki Y, Lian ZX, Lindor K, et al. B-cell depletion with anti-CD20 ameliorates autoimmune cholangitis but exacerbates colitis in transforming growth factor-beta receptor II dominant negative mice. Hepatology. 2009;50:1893–1903. doi: 10.1002/hep.23238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida K, Yang GX, Zhang W, et al. Deletion of interleukin-12p40 suppresses autoimmune cholangitis in dominant negative transforming growth factor beta receptor type II mice. Hepatology. 2009;50:1494–1500. doi: 10.1002/hep.23132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerritse K, Laman JD, Noelle RJ, et al. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci USA. 1996;93:2499–2504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durie FH, Fava RA, Foy TM, Aruffo A, Ledbetter JA, Noelle RJ. Prevention of collagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science. 1993;261:1328–1330. doi: 10.1126/science.7689748. [DOI] [PubMed] [Google Scholar]
- 28.Mohan C, Shi Y, Laman JD, Datta SK. Interaction between CD40 and its ligand gp39 in the development of murine lupus nephritis. J Immunol. 1995;154:1470–1480. [PubMed] [Google Scholar]
- 29.Ma J, Xu J, Madaio MP, et al. Autoimmune lpr/lpr mice deficient in CD40 ligand: spontaneous Ig class switching with dichotomy of autoantibody responses. J Immunol. 1996;157:417–426. [PubMed] [Google Scholar]
- 30.Tan J, Town T, Crawford F, et al. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- 31.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 32.Leung PS, Iwayama T, Coppel RL, Gershwin ME. Site-directed mutagenesis of lysine within the immunodominant autoepitope of PDC-E2. Hepatology. 1990;12:1321–1328. doi: 10.1002/hep.1840120612. [DOI] [PubMed] [Google Scholar]
- 33.Brennan FR, Morton LD, Spindeldreher S, et al. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. mAbs. 2010;2:233–255. doi: 10.4161/mabs.2.3.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358:1109–1117. doi: 10.1056/NEJMoa074943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emi Aikawa N, de Carvalho JF, Artur Almeida Silva C, Bonfa E. Immunogenicity of anti-TNF-alpha agents in autoimmune diseases. Clin Rev Allergy Immunol. 2010;38:82–89. doi: 10.1007/s12016-009-8140-3. [DOI] [PubMed] [Google Scholar]
- 36.Selmi C, Meroni PL, Gershwin ME. Primary biliary cirrhosis and Sjogren's syndrome: autoimmune epithelitis. J Autoimmun. 2012;39:34–42. doi: 10.1016/j.jaut.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang W, Ono Y, Miyamura Y, Bowlus CL, Gershwin ME, Maverakis E. T cell clonal expansions detected in patients with primary biliary cirrhosis express CX3CR1. J Autoimmun. 2011;37:71–78. doi: 10.1016/j.jaut.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q, Selmi C, Zhou X, et al. Epigenetic considerations and the clinical reevaluation of the overlap syndrome between primary biliary cirrhosis and autoimmune hepatitis. J Autoimmun. 2013;41:140–145. doi: 10.1016/j.jaut.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 39.You Z, Wang Q, Bian Z, et al. The immunopathology of liver granulomas in primary biliary cirrhosis. J Autoimmun. 2012;39:216–221. doi: 10.1016/j.jaut.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Early GS, Zhao W, Burns CM. Anti-CD40 ligand antibody treatment prevents the development of lupus-like nephritis in a subset of New Zealand black × New Zealand white mice. Response correlates with the absence of an anti-antibody response. J Immunol. 1996;157:3159–3164. [PubMed] [Google Scholar]
- 41.Quezada SA, Eckert M, Adeyi OA, Schned AR, Noelle RJ, Burns CM. Distinct mechanisms of action of anti-CD154 in early versus late treatment of murine lupus nephritis. Arthritis Rheum. 2003;48:2541–2554. doi: 10.1002/art.11230. [DOI] [PubMed] [Google Scholar]