

Abstract
Endothelial cell (EC) apoptosis seems to play an important role in the pathophysiology of pulmonary arterial hypertension (PAH). We aimed to test the hypothesis that circulating anti-endothelial cell antibodies (AECA) of PAH patients induce EC apoptosis. Immunoglobulin (Ig)G was purified from sera of PAH patients (n = 26), patients with systemic lupus erythematosus (SLE) nephritis without PAH (n = 16), patients with systemic sclerosis (SSc) without PAH (n = 58) and healthy controls (n = 14). Human umbilical vein endothelial cells (HUVECs) were incubated with patient or healthy control IgG for 24 h. Thereafter, apoptosis was quantified by annexin A5 binding and hypoploid cell enumeration by flow cytometry. Furthermore, real-time cell electronic sensing (RT–CES™) technology was used to monitor the effects of purified IgG from patient and healthy control IgG on HUVECs. As demonstrated previously, IgG of AECA-positive SLE nephritis patients (n = 7) induced a higher percentage of apoptosis of HUVECs compared to IgG of AECA-negative SLE nephritis patients and healthy controls. Furthermore, IgG of AECA-positive SLE nephritis patients induced a marked decrease in cell index as assessed by RT–CES™ technology. IgG of AECA-positive PAH patients (n = 12) and SSc patients (n = 13) did not alter the percentage of HUVEC apoptosis or cell index compared to IgG of AECA-negative PAH and SSc patients and healthy controls. AECA-positive PAH patients, in contrast to SLE nephritis patients, do not have circulating IgG AECA that enhances apoptosis of HUVECs in vitro. Further studies should focus on other mechanisms by which AECA may enhance EC apoptosis in PAH, such as antibody-dependent cell-mediated cytotoxicity.
Keywords: anti-endothelial cell antibodies, apoptosis, endothelial cells, pulmonary arterial hypertension, real-time cell electronic sensing
Introduction
Pulmonary arterial hypertension (PAH) is an orphan disease associated with great impact on patients' morbidity and mortality [1,2]. PAH is incurable and the prognosis remains poor, despite improved treatment options [3]. Therefore, a better understanding of its pathophysiology is essential for designing novel therapeutic approaches. Pulmonary vascular remodelling involving intimal, medial and adventitial layers is one of the hallmarks of PAH [4]. The mechanisms causing and propagating vascular changes in PAH remain unclear; however, pulmonary endothelial cell (EC) dysfunction is considered a key player in this process [5]. It has been postulated that injury to the pulmonary endothelium leads to EC apoptosis resulting in destabilization of the pulmonary vascular intima and uncontrolled proliferation of ECs [5,6]. In-vitro studies with human pulmonary microvascular ECs demonstrated that hyper-proliferative and apoptosis-resistant ECs could be generated after the induction of EC apoptosis by vascular endothelial growth factor (VEGF) receptor blockade in combination with high fluid shear stress [6]. Moreover, studies in animal models of PAH also support the importance of EC apoptosis in the early stages of PAH [7–9]. Thus, both in-vitro and in-vivo experiments suggest a link between EC apoptosis and the concomitant development of the angioproliferative lesions as found in PAH [10].
Autoimmune factors are believed to play a role in PAH pathophysiology [11,12]. Anti-endothelial cell antibodies (AECA) are found in the majority of connective tissue disease (CTD)-associated PAH and idiopathic PAH (IPAH) patients [13,14]. AECA are a heterogeneous group of autoantibodies capable of reacting with different EC-related antigenic structures [15]. AECA are present in a variety of systemic autoimmune diseases, including systemic sclerosis (SSc), systemic lupus erythematosus (SLE) and vasculitis [16]. Functional capacities of AECA include activation of ECs and/or induction of EC apoptosis [15,17]. Previously, our group demonstrated the capacity of purified immunoglobulin (Ig)G from AECA-positive patients with SLE nephritis to induce EC apoptosis directly in vitro [18]. The functional capacity of AECA in PAH regarding EC apoptosis is unknown.
Therefore, we investigated the capacity of purified IgG from AECA-positive PAH patients to induce apoptosis of human umbilical vein endothelial cells (HUVECs) in vitro. Apoptosis was quantified by means of annexin A5 binding and hypoploid cell enumeration. Furthermore, we monitored the effects of purified IgG of AECA-positive PAH patients on HUVECs by real-time cell electronic sensing (RT–CES™) technology. This system is a quantitative, non-invasive and real-time assay for monitoring cellular health and behaviour in culture [19]. However, the RT–CES™ system can also be utilized as a cytotoxicity assay to test cytotoxic compounds, as reported by Kirstein et al. [19].
AECA-positive SSc and SLE nephritis patients without PAH were included as disease control cohorts. AECA-negative PAH, SSc and SLE patients, as well as healthy controls, were included as negative control cohorts.
Materials and methods
Patients and controls
A total of 114 participants categorized in four cohorts were included. SLE and diffuse cutaneous SSc patients met the diagnostic criteria of The American College of Rheumatology [20,21]. Patients with limited cutaneous SSc fulfilled the criteria of LeRoy and Medsger [22].
PAH
This cohort encompassed 14 IPAH and 12 SSc-associated PAH patients, all of whom were seen consecutively in our hospital. All the SSc-associated PAH patients were diagnosed with the limited cutaneous form of SSc. PAH was confirmed by right heart catheterization and defined as a mean pulmonary arterial pressure greater than 25 mm Hg at rest with a capillary wedge pressure lower than 15 mm Hg. The diagnosis IPAH was established if further clinical assessment, laboratory investigation, high-resolution computed tomography, ventilation/perfusion lung scan and complete lung function did not show any underlying disease resulting in pulmonary hypertension [23].
SSc without PAH
This cohort encompassed 58 patients, 49 with the limited and nine with the diffuse cutaneous form. Echocardiographically, none of these patients had signs of PAH (estimated right ventricular pressure less than 40 mm Hg).
The PAH and SSc cohorts were recruited consecutively by physicians from the multi-disciplinary PAH team of the Maastricht University Medical Centre.
SLE without PAH
This cohort consisted of 16 consecutive SLE patients with biopsy-proven SLE nephritis [18]. Echocardiographically, none of them had signs of PAH. Sera from these patients were obtained at time of renal biopsy.
Healthy controls
This cohort comprised 14 healthy individuals, who are retired co-workers of the Maastricht University Medical Centre. All subjects gave their informed consent prior to participation.
IgG purification
IgG purification from sera was achieved by affinity chromatography, as described previously [18].
Isolation and culture of HUVECs
HUVECs were isolated from normal term umbilical cord veins and cultured according to the method described previously [18].
Detection of IgG AECA by an EC-based enzyme-linked immunosorbent assay (ELISA)
A modified cyto-ELISA with unfixed HUVECs in their third passage was performed to detect IgG AECA specifically targeting EC surface antigens, as described previously [18]. All experiments were performed at 4°C to preserve the viability of the ECs, unless stated otherwise. Briefly, confluent EC monolayers were washed and incubated with medium [RPMI-1640 containing 1% heat-inactivated fetal calf serum (FCS) adjusted at pH 6·0] for 45 min. Thereafter, ECs were incubated in triplicate with 100 μl/well of either patient or control sera diluted 1 : 100 in medium for 1·5 h. Following three washing steps, the bound IgG AECA were detected with a 1-h incubation of alkaline phosphatase-conjugated goat F(ab)2 anti-human IgG (American Qualex Manufacturers, San Clemente, CA, USA), respectively. Cells were subsequently washed and incubated for 1 h with p-nitrophenyl phosphate (Sigma, St Louis, MO, USA) at room temperature. After stopping the reaction with 5N NaOH (pH 11·0), the optical density (OD) at 405 nm was measured in a Biorad 550 microplate reader (Bio-Rad Laboratories, Veenendaal, the Netherlands).
Cut-off points based on the OD values from the PAH cohort compared to the healthy controls were calculated using a receiver operator characteristics (ROC) curve analysis [13].
Measurement of apoptosis as induced by purified IgG
HUVEC monolayers were trypsinized with trypsin/ethylenediamine tetraacetic acid (EDTA) (0·25%/0·2%). Detached cells were resuspended in culture medium consisting of RPMI-1640 with Glutamax-1 (Gibco, Breda, the Netherlands) supplemented with 10% heat-inactivated FCS (iFCS) (Integro BV, Lelystad, the Netherlands) and centrifuged. Cell pellets were subsequently resuspended in culture medium and incubated in separate wells of precoated 12-well plates (Costar Corning, Bornem, Belgium) with 160 μg/ml of IgG from each individual patient and control in a final concentration of 5·105 cells/ml at 37°C under 5% CO2. The optimal IgG concentration was determined by a concentration–response curve using IgG from several SLE patients (data not shown). HUVECs in separate wells were incubated with either culture medium containing 10% iFCS, culture medium without iFCS (cell starvation) or culture medium containing 10% iFCS and 5 nmol/ml staurosporine as internal negative and positive controls, respectively, for apoptosis. Staurosporine, a widely used apoptogenic agent, has been shown to induce EC apoptosis via focal adhesion kinase dephosphorylation and focal adhesion disassembly independent of focal adhesion kinase proteolysis [24].
After 24 h incubation, supernatants were collected while attached cells were washed in phosphate-buffered saline (PBS; containing 0·15 mol/l NaCl, 0·01 mol/l phosphate, pH 7·4), trypsinized, and collected. All collected supernatants, washing fluids and trypsinized cells were combined and divided subsequently into two Falcon tubes (BD Biosciences, Bedford, MA, USA), washed with PBS and centrifuged. One sample was used to measure annexin V binding, while the other sample was used for the enumeration of hypoploid cells, respectively. Experiments were repeated three times on three different HUVEC isolates.
Binding of annexin A5
Cell pellets were resuspended in annexin A5 buffer (10 mM Hepes/NaOH, pH 7·4, 150 mM NaCl, 5 mM KCl, 2·5 mM CaCl2·H2O, 1 mM MgCl2) and centrifuged. Subsequently, the cells were incubated in 300 μl of the same buffer containing 250 ng/ml fluorescein isothiocyanate (FITC)-conjugated annexin A5 (from Dr C. P. M. Reutelingsperger) for 10 min at room temperature in the dark. Propidium iodide (PI) (Calbiochem®; EMD Chemicals, Inc., Gibbstown, NJ, USA) was added to exclude dead cells, diluted to a final concentration of 10 μg/ml. All cell preparations were examined with a fluorescence activated cell sorter (FACS)Canto II (BD Biosciences, San Jose, CA, USA) using the diva software (BD Biosciences) for analysis. The percentage of annexin A5 single-positive cells (early apoptotic cells) was calculated within the viable population of cells.
Enumeration of hypoploid cells
Enumeration of hypoploid cells was carried out as described previously [25,26]. Briefly, cell pellets were resuspended and fixed with 70% ethanol for 2 h at −20°C. Subsequently, cells were centrifuged and resuspended in PI incubation buffer (45 mM Na2HPO4, 2·5 mM citric acid and 0·1% Triton X-100) for 20 min at 37°C. PI was added to a final concentration of 10 μg/ml. All cell preparations were examined with a FACSCanto II (BD Biosciences) using the diva software (BD Biosciences) for analysis. Doublets were ‘gated-out’ by making use of a two-parameter measurement scheme in which a plot of pulse peak height versus area (integral) PI signal allowed for identification and exclusion of doublets.
HUVECs monitoring by the RT–CES™ system
The principles and components of RT–CES™ (ACEA Biosciences Inc., San Diego, CA, USA) technology have been described previously [27–29]. Briefly, the RT–CES system allows for non-invasive monitoring of target cells by using impedance sensor technology. Electrode impedance, which is displayed and recorded as cell index (CI) values, reflect the biological status of monitored cells, including the cell number, cell viability, morphology and adhesion quality.
We monitored the effects of purified IgG from a subgroup of PAH (n = 16), SSc (n = 12) and SLE nephritis (n = 6) patients and healthy controls (n = 6) on HUVECs with the RT–CES™ system. We performed three experiments with the RT–CES™ system, each experiment with different HUVEC batches but with the same purified IgG from the above-mentioned subgroups. HUVECs were seeded at a density of 4500 cells per well on 96-well plates integrated with microelectrodes at the bottom of the wells (E-plates™; ACEA Biosciences Inc.). Briefly, cells were trypsinized, centrifuged and resuspended in culture medium consisting of RPMI-1640 with Glutamax-1 (Gibco) supplemented with 10% iFCS (Integro BV) and counted. Background measurements were taken after adding 50 μl of the culture medium to the wells of the E-Plate™. Cells were adjusted to the appropriate concentration, and 100 μl of the cell suspension was added to the E-plate™ wells. Thereafter, cell attachment, spreading and proliferation were monitored every 15 min using the RT–CES system. The cells were in the log growth phase after approximately 2–3 h after seeding, depending on the HUVEC batch used in the respective experiment. At this point, being similar within each HUVEC batch, the cells were treated with 160 μg/ml patient or control IgG in triplicate and monitored continuously for 48 h. HUVECs incubated in culture medium without iFCS (cell starvation) and HUVEC treated with 5 nmol/ml staurosporine in 10% iFCS served as internal positive controls for apoptosis.
Statistical analyses
Data were analysed with spss statistical software version 15·0 for Windows. For comparison among the five cohorts for normal distributed variables we used one-way analysis of variance (anova). To compare two groups for non-parametric and normal distributed variables we used the Mann–Whitney U-test and Student's t-test, respectively. For comparison among nominal variables between groups we used the χ2 test. A Bonferroni correction was applied to the comparative tests used in our statistical analyses. Data are presented as median and interquartile ranges, with P < 0·05 indicating statistical significance.
Results
Demographic characteristics of the study cohorts
Participants in all four study cohorts did not differ significantly with respect to gender (P = 0·690) (Table 1). The age of the healthy controls, PAH and SSc patients did not differ significantly from each other. However, the SLE nephritis cohort encompassed younger participants (P < 0·0001).
Table 1.
Demographic characteristics of study cohorts
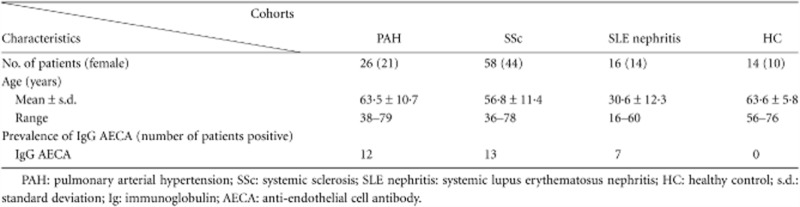 |
The prevalence of IgG AECA specifically targeting surface antigens on HUVECs in the different cohorts is presented in Table 1. IgG AECA prevalence in the PAH, SSc and SLE nephritis cohorts was significantly higher compared to the healthy controls (P = 0·002, P = 0·05 and P = 0·005, respectively). IgG AECA prevalence in the IPAH (n = 14) and SSc-associated PAH (n = 12) patients was 42·9 and 50·0%, respectively. The occurrence of IgG AECA in the SSc-associated PAH patients was significantly higher in comparison to SSc patients without PAH (P = 0·05).
IPAH patients were not using corticosteroids or immunosuppressive medication at the time of blood sampling, whereas two of the SSc-associated PAH patients used low-dose corticosteroid treatment (5 mg p.o.). In the SSc cohort, 22 of the 58 patients used low-dose corticosteroids in combination with immunosuppressive medication. In nine of the 16 SLE patients corticosteroids and immunosuppressive treatment was initiated some days before the renal biopsy was obtained.
No significant difference was observed between the IPAH and SSc-associated PAH patients with respect to the different parameters of disease severity, as presented in Table 2.
Table 2.
Parameters of disease severity in IPAH and SSc-associated PAH
 |
Induction of EC apoptosis by purified IgG
Assessment by binding of annexin A5
Levels of spontaneous apoptosis in HUVEC control cultures varied between 7·50 and 9·75%. The mean percentage of EC apoptosis induced by cell starvation and staurosporine was 45·95 and 57·40%, respectively. As demonstrated previously, purified IgG from the AECA-positive SLE patients induced a significantly higher percentage of apoptosis of HUVECs in comparison to AECA-negative SLE patients (P = 0·001) and healthy controls (P = 0·001) (Fig. 1). Purified IgG from the AECA-positive PAH patients did not induce a significantly higher percentage of apoptosis of HUVECs compared to the AECA-negative PAH patients (P = 0·94) and healthy controls (P = 0·80), as assessed by binding of annexin A5 (Fig. 1). Similarly, no induction of apoptosis was observed in the SSc cohort (Fig. 1). When analysing late apoptotic cells, defined as annexin A5/propidium iodide double-positive, similar results were obtained (data not shown). Further analysis of the PAH cohort demonstrated that IgG from the AECA-positive IPAH patients did not induce a significantly higher percentage of apoptosis of HUVECs compared to the AECA-negative PAH patients (P = 0·96) and healthy controls (P = 0·82) as assessed by binding of annexin A5.
Fig. 1.
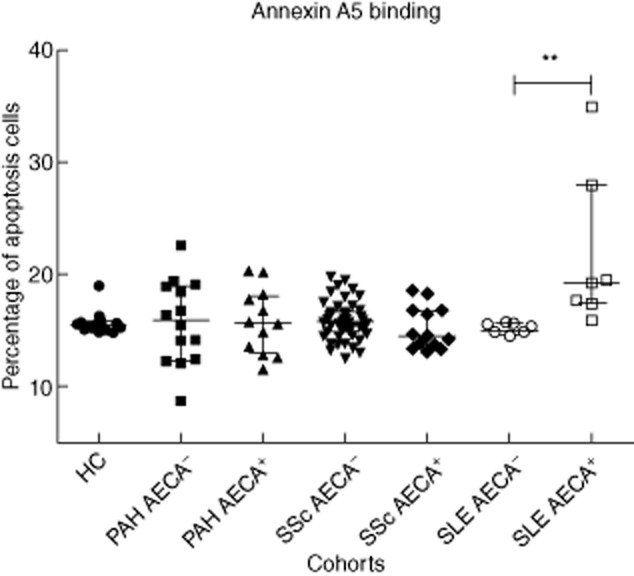
Apoptosis induction by purified immunoglobulin (Ig)G from either patients or healthy controls as assessed by binding of annexin V. Results are reported in the percentage of apoptotic cells defined by the percentage of annexin V-positive cells calculated within the viable population of cells. The lines represent median and interquartile ranges of the different study cohorts. **P = 0·001.
Assessment by enumeration of hypoploid cells
Levels of spontaneous apoptosis in HUVEC control cultures varied between 10·00 and 12·50%. The mean percentage of EC apoptosis induced by cell starvation and staurosporine was 55·46 and 66·80%, respectively. As demonstrated by the enumeration of hypoploid cells, purified IgG from the AECA-positive SLE patients induced a significantly higher percentage of apoptosis of HUVECs in comparison to AECA-negative SLE patients (P = 0·001) and healthy controls (P < 0·0001) (Fig. 2).
Fig. 2.
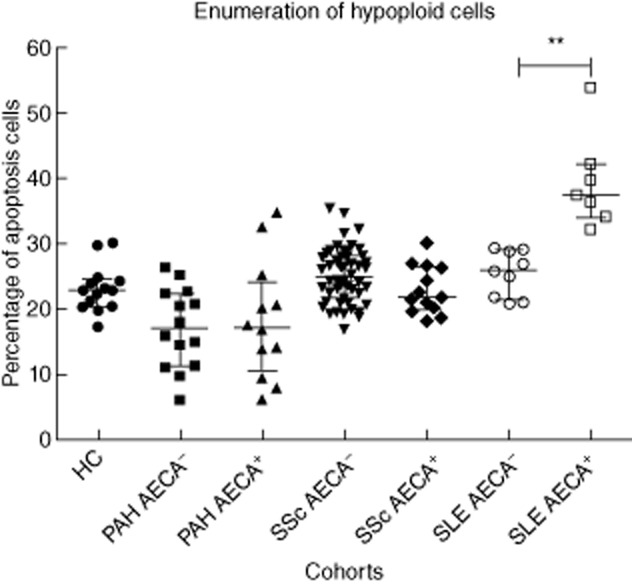
Apoptosis induction by purified immunoglobulin (Ig)G from either patients or healthy controls as assessed by enumeration of hypoploid cells. Results are reported in the percentage of apoptotic cells. The lines represent median and interquartile ranges of the different study cohorts. **P = 0·001.
Purified IgG from the AECA-positive PAH patients did not induce a higher percentage of apoptosis of HUVECs compared to the AECA-negative PAH patients (P = 0·92) and healthy controls (P = 0·08), as assessed by the enumeration of hypoploid cells (Fig. 2). Also in the SSc cohort, no induction of apoptosis was observed (Fig. 2). Further analysis of the PAH cohort demonstrated that IgG from the AECA-positive IPAH patients did not induce a significantly higher percentage of apoptosis of HUVECs compared to the AECA-negative PAH patients (P = 0·94) and healthy controls (P = 0·09), as assessed by the enumeration of hypoploid cells.
Effects of purified IgG on HUVECs cellular status assessed by the RT–CES system
Incubation with IgG from AECA-positive SLE (n = 3) patients induced a significant decrease in the CI value compared to IgG from AECA-negative SLE (n = 3) patients (P = 0·050) and healthy controls (P = 0·020) (Fig. 3). In fact, IgG from AECA-positive SLE patients induced a decrease in CI value of 79%, which was comparable with the decrease in CI values induced by cell starvation (82%) and incubation with 5 nmol/ml staurosporine (93%). Incubation of HUVECs with IgG from the AECA-positive PAH (n = 8) and SSc (n = 6) patients, however, did not alter the CI value significantly compared to IgG from the AECA-negative PAH (n = 8) and SSc (n = 6) patients (P = 0·248 and P = 0·749, respectively) and healthy controls (P = 0·121 and P = 0·337, respectively).
Fig. 3.
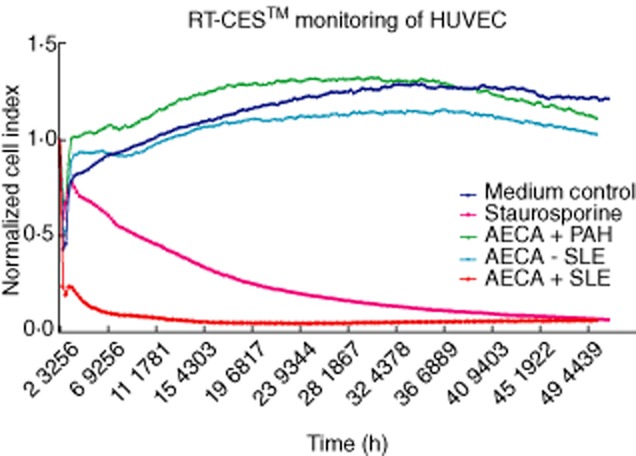
Results of real-time cell electronic sensing (RT–CES) monitoring of human umbilical vein endothelial cells (HUVECs) incubated with purified immunoglobulin (Ig)G from one representative anti-endothelial cell antibody (AECA)-positive and one AECA-negative systemic lupus erythematosus (SLE) patient. Confidence intervals (CI) values from the medium control, one representative AECA-positive pulmonary arterial hypertension (PAH) patient and incubation with 5 nmol/ml staurosporine are also shown. Results are reported in CI values of the HUVECs monitored dynamically every 15 min using the RT–CES™ system.
Discussion
The aetiology of PAH is still poorly understood, and it is postulated that dysfunction of pulmonary ECs plays an important role in the pathophysiology of PAH [5]. EC dysfunction may lead to pulmonary vascular remodelling and ultimately to the development of PAH [4,5]. Mounting evidence suggests an important role for EC apoptosis in this process. Taraseviciene-Stewart et al. demonstrated that selective blockade of the vascular endothelial growth factor receptor 2 (VEGFR-2) resulted in severe irreversible pulmonary hypertension associated with precapillary arterial endothelial cell proliferation in chronically hypoxic rats [7]. EC apoptosis following VEGFR-2 blockade was a prerequisite for endothelial proliferation, because caspase inhibition throughout the course of chronic hypoxia and VEGFR-2 blockade prevented EC proliferation and the development of severe pulmonary hypertension [7]. The paradox that apoptosis fosters emergence of apoptosis resistant and hyperproliferative cells is not novel [30].
The factors that trigger EC apoptosis in PAH remain unclear. Autoimmune factors may be among them [12,30]. Recently, we reported that the majority of PAH patients have circulating AECA specifically targeting cell surface antigens of ECs [13]. To study the specificity of AECA towards ECs in our study we determined the reactivity of our patients' sera towards human fibroblasts by means of a cyto-ELISA with unfixed normal human dermal fibroblast (NHDF). The sera of the AECA-positive PAH patients did not show any reactivity towards NHDF compared to the sera of the healthy controls (data not shown).
We also demonstrated that IgG from AECA-positive patients with SLE nephritis induce EC apoptosis in vitro by a mechanism as yet unknown [18]. In avian SSc, AECA have been shown to induce EC apoptosis, which is considered a primary pathogenic event in SSc [31]. However, conflicting data have been published concerning the mechanisms by which AECA exert EC apoptosis in human SSc [17,32]. AECA in SSc have been shown to directly induce apoptosis [17]. Alternatively, EC apoptosis may be induced by antibody-dependent cell-mediated cytotoxicity (ADCC) [32]. Irrespective of the mechanism, AECA have been shown to exert pro-apoptotic activity on ECs. Hence we hypothesized that AECA could be the trigger leading to the development of PAH by inducing EC apoptosis which subsequently activates a cascade culminating in EC proliferation. In the present study we demonstrate, surprisingly, that in contrast to IgG from AECA-positive SLE patients the IgG from AECA-positive PAH patients do not induce apoptosis of EC. We confirmed this finding by employing three different methods, of which the RT–CES™ technology is a new method, to measure cell viability by high-throughput screening [28].
The lack of apoptosis-inducing activity of purified IgG from AECA-positive PAH patients suggests that other circulating factors may trigger EC apoptosis. Kahaleh et al. suggested serum-mediated endothelial injury and demonstrated the presence of granular enzymes (granzymes) in sera of SSc patients [33]. Granzymes gain access to the cells following cellular membrane damage by perforin [34]. We tested sera from PAH patients on their ability to induce EC apoptosis in vitro to assess whether serum factors other than IgG could induce EC apoptosis. However, none of the tested sera from AECA-positive PAH expressed EC apoptosis-inducing activity (data not shown).
ADCC is another proposed mechanism of EC apoptosis in SSc [32]. This mechanism of EC apoptosis requires antibodies and appropriate effector cells. Sgonc et al. found activated natural killer (NK) cells to be absolutely necessary for the AECA-dependent apoptosis induction in EC cultures [32]. In the present study we did not address this mechanism of EC apoptosis in PAH.
In conclusion, we report a discrepancy between IgG from AECA-positive PAH patients and IgG from AECA-positive SLE patients, with the latter expressing EC apoptosis inducing activity and the former lacking this activity. We were unable to find circulating pro-apoptotic factors in PAH patients that would support the EC apoptosis hypothesis of PAH. It is important to mention that we used HUVECs in our study and that, ideally, patients' own pulmonary ECs should be used to study pro-apoptotic activities of circulating IgG. Nevertheless, our study demonstrates that circulating IgG from AECA-positive patients differ bioactively between diseases and cannot, therefore, be incorporated in a general cause–consequence relationship solely on the basis of their shared feature of binding to EC.
Acknowledgments
Special thanks go to Drs B. Broers (cardiologist) from the Orbis Medisch Centrum in Sittard-Geleen, the Netherlands, for recruitment of PAH patients. The authors also thank N. Deckers from the Cardiovascular Research Institute Maastricht (CARIM), the Netherlands, for his excellent technical assistance and advice with regard to the RT–CES™ assays. This research was supported by financially Actelion Pharmaceuticals Nederland BV (Woerden, the Netherlands).
Disclosure
The authors declare that they have no conflict of interests.
References
- 1.Rubin L. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–117. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 2.McLaughlin V, Presberg K, Doyle R, et al. Prognosis of pulmonary arterial hypertension*: ACCP evidence-based practice guidelines. Chest. 2004;126:78S–92S. doi: 10.1378/chest.126.1_suppl.78S. [DOI] [PubMed] [Google Scholar]
- 3.Matura L, Carroll D. Human responses to pulmonary arterial hypertension. J Cardiovasc Nurs. 2010;25:420–427. doi: 10.1097/JCN.0b013e3181d25458. [DOI] [PubMed] [Google Scholar]
- 4.Tuder R, Marecki J, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28:23–42. doi: 10.1016/j.ccm.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Budhiraja R, Tuder R, Hassoun P. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 6.Sakoa S, Taraseviciene-Stewart L, Lee J, Wood K, Cool C, Voelkel N. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cell. FASEB J. 2005;19:1178–1180. doi: 10.1096/fj.04-3261fje. [DOI] [PubMed] [Google Scholar]
- 7.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, McMahon G, Waltenberger J. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 8.Thomas H, Lame M, Dunston S, Segall H, Wilson D. Monocrotaline pyrrole induces apoptosis in pulmonary artery endothelial cells. Toxicol Appl Pharmacol. 1998;151:236–244. doi: 10.1006/taap.1998.8458. [DOI] [PubMed] [Google Scholar]
- 9.Voelkel N, Tuder R. Hypoxia-induced pulmonary vascular remodeling: a model for what human disease? J Clin Invest. 2000;106:733–738. doi: 10.1172/JCI11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jurasz P, Courtman D, Babaie S, Stewart D. Role of apoptosis in pulmonary hypertension: from experimental models to clinical trials. Pharmacol Ther. 2010;126:1–8. doi: 10.1016/j.pharmthera.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Mouthon L, Guillevin L, Humbert M. Pulmonary arterial hypertension: an autoimmune disease? Eur Respir J. 2005;26:986–988. doi: 10.1183/09031936.05.00112105. [DOI] [PubMed] [Google Scholar]
- 12.Nicolls M, Taraseviciene-Stewart L, Rai P, Badesch D, Voelkel N. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26:1110–1118. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- 13.Arends S, Damoiseaux J, Duijvestijn A, et al. Prevalence of anti-endothelial cell antibodies in idiopathic pulmonary arterial hypertension. Eur Respir J. 2010;35:923–925. doi: 10.1183/09031936.00164209. [DOI] [PubMed] [Google Scholar]
- 14.Tamby M, Chanseaud Y, Humbert M, et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax. 2005;60:765–772. doi: 10.1136/thx.2004.029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belizna C, Duijvestijn A, Hamidou M, Cohen Tervaert J. Antiendothelial cell antibodies in vasculitis and connective tissue disease. Ann Rheum Dis. 2006;65:1545–1550. doi: 10.1136/ard.2005.035295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belizna C, Cohen Tervaert J. Specificity, pathogenecity, and clinical value of antiendothelial cell antibodies. Semin Arthritis Rheum. 1997;27:98–109. doi: 10.1016/s0049-0172(97)80010-8. [DOI] [PubMed] [Google Scholar]
- 17.Bordron A, Dueymes M, Levy Y, et al. The binding of some human antiendothelial cell antibodies induces endothelial cell apoptosis. J Clin Invest. 1998;101:2029–2035. doi: 10.1172/JCI2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Paassen P, Duijvestijn A, Debrus-Palmans L, Damoiseaux J, Vroomen M, Cohen Tervaert J. Induction of endothelial cell apoptosis by IgG antibodies from SLE patients with nephropathy. A potential role for anti-endothelial cell antibodies. Ann NY Acad Sci. 2007;1108:147–156. doi: 10.1196/annals.1422.017. [DOI] [PubMed] [Google Scholar]
- 19.Kirstein S, Atienza J, Xi B, et al. Live cell quality control and utility of real-time cell electronic sensing for assay development. Assay Drug Dev Technol. 2006;4:545–553. doi: 10.1089/adt.2006.4.545. [DOI] [PubMed] [Google Scholar]
- 20.Tan E, Cohen A, Fries J, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 21.Masi A, Rodnan G, Medsger T. Preliminary cirteria for the classification of systemic sclerosis (scleroderma) Arthritis Rheum. 1980;23:581–590. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 22.LeRoy E, Medsger TJ. Criteria for the classification of early systemic sclerosis. J Rheumatol. 2001;28:1573–1576. [PubMed] [Google Scholar]
- 23.Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(Suppl. 12):40S–47. doi: 10.1016/j.jacc.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 24.Kabir J, Lobo M, Zachary I. Staurosporine induces endothelial cell apoptosis via focal adhesion kinase dephosphorylation and focal adhesion disassembly independent of focal adhesion kinase proteolysis. Biochem J. 2002;367:145–155. doi: 10.1042/BJ20020665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicoletti I, Migliorati G, Pagliacci M, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 26.Relou I, Damen C, van der Schaft D, Groenewegen G, Griffioen A. Effect of culture conditions on endothelial cell growth and responsiveness. Tissue Cell. 1998;30:525–530. doi: 10.1016/s0040-8166(98)80032-3. [DOI] [PubMed] [Google Scholar]
- 27.Solly K, Wang X, Xu X, Strulovici B, Zheng W. Application of real-time cell electronic sensing (RT–CES) technology to cell-based assays. Assay Drug Dev Technol. 2004;2:363–372. doi: 10.1089/adt.2004.2.363. [DOI] [PubMed] [Google Scholar]
- 28.Zhu J, Wang X, Xu X, Abassi Y. Dynamic and label-free monitoring of natural killer cell cytotoxic activity using electronic cell sensor arrays. J Immunol Methods. 2006;309:25–33. doi: 10.1016/j.jim.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 29.Abassi Y, Jackson J, Zhu J, O'Connell J, Wang X, Xu X. Label-free, real-time monitoring of IgE-mediated mast cell activation on microelectronic cell sensor arrays. J Immunol Methods. 2004;292:195–205. doi: 10.1016/j.jim.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 30.Sakao S, Tatsumi K, Voelkel N. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res. 2009;10:95–103. doi: 10.1186/1465-9921-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sgonc R, Gruschwitz M, Dietrich H, Recheis H, Gershwin M, Wick G. Endothelial cell apoptosis is a primary pathogenic event underlying skin lesions in avian and human scleroderma. J Clin Invest. 1996;98:785–792. doi: 10.1172/JCI118851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sgonc R, Gruschwitz M, Boeck G, Sepp N, Gruber J, Wick G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum. 2000;43:2550–2562. doi: 10.1002/1529-0131(200011)43:11<2550::AID-ANR24>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 33.Kahaleh B, Fan P. Mechanism of serum-mediated endothelial injury in scleroderma: identification of a granular enzyme in scleroderma skin and sera. Clin Immunol Immunopathol. 1997;83:32–40. doi: 10.1006/clin.1996.4322. [DOI] [PubMed] [Google Scholar]
- 34.Kahaleh B. The microvascular endothelium in scleroderma. Rheumatology. 2008;47:V14–15. doi: 10.1093/rheumatology/ken279. [DOI] [PubMed] [Google Scholar]