

Abstract
A green one-pot approach is used to synthesize surface morphology controlled hollow silica spheres in aqueous solution under ambient conditions. Using cationic polystyrene particles as a template and aminopropyltriethoxysilane (APTES) as a structural directing agent, the surface roughness and wall thickness could be easily controlled by varying the concentration of tetraethoxysilane (TEOS). Smooth surface of hollow silica spheres with different wall thickness was obtained using anionic polystyrene particles. The resulted hollow silica spheres with rough surface had better performance in carrying and releasing a model drug.
Introduction
Monodisperse hollow silica spheres have a variety of applications such as encapsulation, drug-delivery, ultra-sound imaging, chromatography, waste removal, separation and catalysis, and controlled-release of drugs.1–6 Silica spheres with hierarchical surface morphologies and well-defined structures gained much attention over the last few years due to their light weight, high surface area and roughness with interesting potential applications. A variety of silica spheres with novel surface morphologies have been synthesized including raspberry-like, sunflower-like, snowman-like, petal-like, daisy-shaped and multipod-like.7–10 For example, raspberry-like silica spheres have been used as antifogging coatings or superhydrophobic films that mimic the surface topology of self-cleaning lotus leaves.11, 12 Among the methods for the synthesis of hollow silica spheres, in situ deposition of silica against polystyrene (PS) particles still retains a facile and effective approach since it is relatively inexpensive and can achieve well-defined morphology with controllable size. As a polymer, the PS template can be easily removed by either calcination or decomposition upon exposure to organic solvents.13 Thus, they are well-suited to serve as sacrificial templates and widely adopted for the fabrication of hollow spheres. An ammonia-catalysed sol-gel process is commonly used to synthesize silica spheres using original or modified PS spheres as templates. This method usually results in smooth and homogeneous silica shells with variable thickness. Hollow silica spheres with increased surface roughness can be synthesized by specially modified PS spheres or co-assembly of pre-synthesized small silica particles and large PS spheres.14–16 The preparation processes are time-consuming as they usually need multiple steps or special equipment and often require harsh conditions, such as elevated temperature (≥200 °C), non-physiological pH values, and the use of either large amount of surfactants and/or toxic organic solvents. Therefore, a facile, energy-efficient and environmentally friendly method is required for actual applications.
In nature, the positively charged diatoms and sponges can nucleate silica under ambient conditions in aqueous solution.17 Inspired by nature, a template with amino groups has been developed to facilitate the synthesis of silica nanocomposites by biomimetic approach.18, 19 Such approach is environmentally benign and economic and thus become more attractive. Here, we introduce a one-pot synthesis of hollow silica spheres with controlled surface nanostructures under ambient conditions in aqueous solution. Namely, the synthesis of silica spheres and dissolution of PS spheres are performed in one reaction. By this approach, the resultant silica hollow spheres have uniform size and their surface morphologies can be easily tuned by varying concentrations of tetraethoxysilane (TEOS) (Scheme 1). To the best of our knowledge, the synthesis of hollow silica spheres with controlled surface morphology by biomimetic approach has never been reported.
Scheme 1.

Schematic diagram showing the one-pot morphology-controlled synthesis of hollow silica spheres. Hydrolyzed APTES is absorbed onto the surface of amino-PS spheres forming silica nuclei. Less polycondensation of TEOS on the nuclei results in relatively smooth silica shell. More polycondensation of TEOS on the nuclei leads to the growth of the nuclei in spherical manner, which in turn increases the surface roughness of silica spheres. Finally, the adjacent silica particles meet and merge together. The PS core is removed by solvent simultaneously during synthesis of silica spheres.
Experimental
Synthesis of hollow silica spheres
The method follows our previous report with some modifications.11 The amino-PS or carboxyl-PS particles (2.5% w/v in water) (Spherotech Inc.) were diluted 20 times by water. Aminopropyltriethoxysilane (APTES) (2×10−3 mmol) was mixed with 500 μl PS particles and agitated by a vortex mixer. The mixture was then incubated at room temperature for 2–3 min. TEOS (0.5 μl, 1 μl, 2 μl, 4 μl) was added to the mixture while stirring was continued for at least 3 min. The mixed solution was left for ~30 min and 0.1 vol % of CH2Cl2 was added to the solution. The mixture was then aged at room temperature for 4 h.
Characterization of silica spheres
Transmission electron microscopy (TEM) and Scanning electron microscopy (SEM) were used to observe the morphologies of the obtained silica spheres. The synthesized silica spheres were diluted with water and ultrasonicated for 15 min. The morphologies of the hollow silica spheres were then characterized using a TEM (Zeiss 10) and SEM (JEOL 880).
TGA and nitrogen adsorption/desorption analysis
About 5 mg of the original PS particles, hollow silica spheres or silica spheres with PS (No CH2Cl2 added) were dried at 60 °C overnight before examination. All the powders were heated in air from room temperature to 800 °C at a scan rate of 20 °C min−1. The surface area was determined according to the Brunauer-Emmett-Teller (BET) method through nitrogen adsorption/desorption analysis.
Model peptide loading and release
The hollow silica spheres (3 mg) were mixed with 0.1 mg of model peptide labelled with FITC (VTAMEPGQCG-Lys (FITC) (Invitrogen) in 2 ml of phosphate buffered saline (PBS) buffer solution (pH 7.2) and stirred for 48 h at 4 °C. Afterwards, the drug-loaded hollow silica spheres were washed two times by PBS. The peptide-loaded hollow silica spheres were agitated at room temperature. At each checking point, the solution was centrifuged at 7000 rpm for 15 min, and 0.1 ml of the solution was taken and measured using a spectrophotometric microplate reader (Biotek). Another 0.1 ml of the fresh PBS buffer was added to keep a constant volume. The morphology of silica spheres were characterized by a fluorescence microscope (Nikon).
Results and discussion
The amino-PS spheres were diluted 20 times by water and then mixed with the APTES. After vortex and stayed for 2–3 minutes, TEOS was added to the solution under stirring. The mixture was then left at room temperature and aged for 4 h. SEM images of the hollow silica spheres obtained at varied TEOS concentrations are illustrated in Fig. 1. All of the silica spheres maintained a spherical morphology with a narrow size distribution. Relatively smooth surface of silica spheres but occasionally decorated with a few small particles were observed when the volume of TEOS is 0.5 μl (Fig. 1a). The diameter of silica spheres is slightly larger than that of the original PS spheres. With increased volume of TEOS (1 μl), the surface roughness of the hollow silica spheres highly increased by the presence of silica nanoparticles with a dimension of about 15 nm, which is ideal for the fabrication of superhydrophobic or superhydrophilic films (Fig. 1b). Multipod-like silica spheres were obtained when the volume of TEOS increased to 2 μl. The dimension of the small silica nanoparticles was increased up to ~35 nm on the surface of silica spheres (Fig. 1c). As the volume of TEOS was increased to 4 μl, the silica spheres exhibited coarse surface morphology (Fig. 1d), arising from the growth of nucleated silica particles due to further polycondensation of TEOS until adjacent silica particles meet and fuse together. Meanwhile, some silica spheres with smaller diameters but very smooth surface morphology appeared in the solution. Low magnification of the silica spheres was shown in Fig. S1.
Fig. 1.

SEM images of the hollow silica spheres obtained at various amounts of TEOS using amino-PS spheres as templates. (a) Relatively smooth surface morphology of silica spheres was obtained using 0.5 μl of TEOS. (b) Highly increased surface roughness of silica spheres exhibited raspberry-like structures using 1 μl of TEOS. (c) Multipod-like surface morphology was formed when using 2 μl of TEOS. (d) 4 μl of TEOS resulted in excessive silica formation. Some irregular silica spheres were nucleated with large silica particles. At the same time, some smaller silica particles with smooth surfaces were observed.
Transmission electron microscopy (TEM) images of hollow silica spheres with different surface morphologies were shown in Fig. 2. Dissolution of PS spheres was carried out via a one-step process during the synthesis of silica spheres by a small amount of CH2Cl2 (0.1% v/v). When 1.5 ml PS spheres were mixed with 0.5 μl TEOS, smooth hollow silica spheres were obtained (Fig. 2a). The shell thickness is about 5 nm. The dimension and cavity of the hollow spheres are almost equal to that of the PS spheres, clearly indicating they are transcribed from the PS templates. When the volume of TEOS was increased to 1 μl, the surface roughness of the silica spheres was highly increased with protruding surface morphologies. As shown in Fig. 2b, the as-synthesized silica spheres presented raspberry-like structures. The diameter of hollow silica spheres is 207±10 nm and wall thickness is increased to about 8 nm. Except for increased surface roughness of hollow silica spheres, no major changes in the dimension and wall thickness of the silica spheres were observed when the volume of TEOS was further increased to 2 μl (Fig. 2c). As observed by SEM, TEM images also showed large solid silica particles nucleated on the surface of hollow silica spheres. However, the silica spheres with smaller diameters and smooth surface morphology are solid (Fig. 2d). Small silica particles are induced in the aqueous solution by excessive TEOS. The PS core particles are unable to capture the rapidly forming silica particles completely and equably, thus resulted in the formation of extra free silica particles.20 The silica spheres without the dissolution of PS core by CH2Cl2 showed PS/SiO2 hybrid core-shell structures (Fig. S2).
Fig. 2.
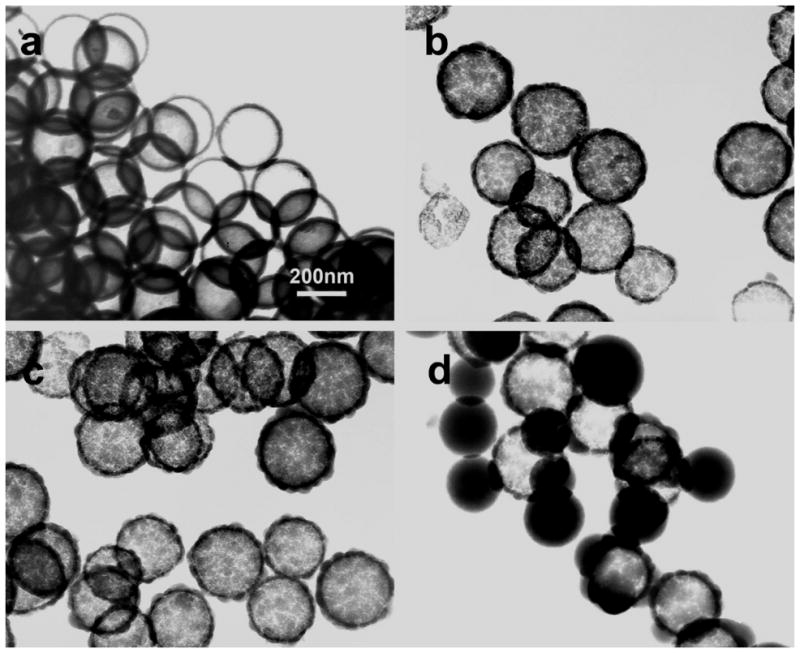
TEM images of the well-defined hollow silica spheres produced at various amounts of TEOS using amino-PS spheres as template. (a) Hollow silica spheres with smooth surface morphology were obtained using 0.5 μL of TEOS after the removal of PS cores by CH2Cl2. (b) Hollow silica spheres with thicker silica coating but rough surface morphology were observed using 1 μl of TEOS. (c) The outer surface of silica spheres became rougher using 2 μl of TEOS. (d) Some free solid silica particles were formed in the solution using 4 μl of TEOS.
Recently, we found that APTES could serve as a structure-directing agent in biotemplated synthesis of silica nanostructures.21 Namely, APTES can be pre-absorbed onto the biotemplate surface and hydrolyzed to form spherical silica nuclei. Then the polycondensation of TEOS on these nuclei leads to the growth of silica on the biotemplate surface, generating a pearl-necklace-like morphology of silica nanotubes with rough surface. A possible explanation is that the transcription is based on a so-called “surface mechanism”, which means that the initial nucleation of silica does not start simultaneously all over the nanotubes but begins on some specific points, followed by the growth of the nuclei in a spherical manner until the adjacent spheres meet and fuse together.22 APTES is hydrolytically unstable in an aqueous solution due to the rapid self-hydrolysis. In this work, the amino-groups on the PS surface may further catalyse the hydrolysis of APTES. The aminopropyl groups of APTES keep intact but the ethoxy groups are hydrolysed forming transient silanol groups. These transient groups are then condensed to yield aminopropyl-functionalized silica particles with hydroxyl and amino groups on their surface. Finally, these silica particles are attached to the amino-PS surface via hydrogen bonding to serve as nuclei (Scheme S1). After TEOS was added to the solution and hydrolyzed, polycondensation of TEOS follows on the surfaces of these nuclei. Only a thin layer of tiny silica particles is coated on PS template to achieve a smooth surface morphology when the concentration of TEOS is low. Further condensation of TEOS will proceed on the surfaces of these silica particles leading to the growth of the nuclei in spherical manner. The surface roughness of PS spheres is increased due to the protruding silica particles from their surfaces, generating raspberry-like morphology. After that, there will be a net growth of silica on the coated silica surface until the meeting and consequent fusion of neighbouring particles. Silica encapsulation and dissolution of the PS spheres can be done in one pot simultaneously in situ when a small amount of CH2Cl2 is added to dissolve PS cores (Scheme 1).
In order to better understand the effects of surface charges on TEOS hydrolysis and condensation, in a control experiment, hollow silica spheres were synthesized using negatively charged carboxyl-PS spheres using the same method on amino-PS spheres. Interestingly, higher concentrations of TEOS do not increase the surface roughness but only the wall thickness of hollow silica spheres (Fig. S3). The wall thickness of hollow silica spheres was further increased with the increase in the concentrations of TEOS. However, the outer surface of silica shells remained smooth. The negatively charged groups on the PS surface enhance the absorption of APTES to the surface by electrostatic interactions and the strong nucleophilic residues catalyze the hydrolysis of APTES to form silica nuclei on the surface of carboxyl-PS spheres.21, 23 Then the subsequent polycondensation of TEOS proceed on the layer of nuclei, resulting in the formation of silica shells with smooth surface (Scheme S2).
The importance of APTES and surface charge of PS spheres were further confirmed by the direct addition of TEOS without the prior addition of APTES. After 20 μl of TEOS was mixed with 1.5 ml amino-PS spheres for 7 days, a uniform thin layer of silica shell was formed on the PS spheres (Fig. S4a). After the PS cores were removed by CH2Cl2, some of the hollow spheres were deformed, probably because their silica shells were too thin to endure the electron beam (Fig. S4b). This indicates that the positively charged amino residues can catalyse the hydrolysis and polycondensation of TEOS directly just like the silica formation induced by diatoms and sponges in nature. However, when TEOS was mixed with carboxyl-PS spheres, no silica condensation was observed on their surfaces, but some solid silica particles were formed in the solution (data not shown). The negatively charged silica sols cannot interact directly with carboxyl-functionalized PS spheres. These control experiments show that the synthesis of hollow silica spheres with controlled surface morphology can only be produced using both amino-PS spheres as templates and APTES as a structure-directing agent (Schemes S1 and 1). We have consistently found that APTES could serve as a structure-directing agent for promoting silica nucleation on biological templates such as phage and flagella.21, 24, 25 Furthermore, when a higher concentration of APTES (10−2 mmol) was mixed with 500 μl PS particles first and then mixed with 1 μl TEOS, rougher surfaces of silica spheres were observed. At the same time, some free silica particles were formed and attached onto the surface of spheres (Fig. S5).
The samples were further analyzed by thermal gravimetric analysis (TGA) to verify the presence of PS cores and evaluate their removal (Fig. 3). The weight-loss below 300 °C is attributed to the evaporation of physically adsorbed water and residual solvent. The decomposition of PS cores is located in the region from 300 to 560 °C (Fig. 3b), according to the TGA curve of pure PS core (Fig. 3a). The decomposition of silica bonded groups such as –OH and/or unhydrolyzed –OR occurs at the region from 420 to 600 °C (Fig. 3c).20, 26 The BET surface areas of raspberry-like and smooth hollow spheres were determined to be 439.53 and 311.42 m2/g respectively.
Fig. 3.
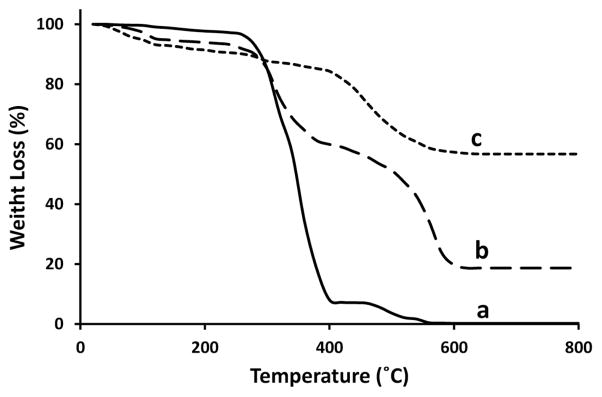
TGA curves of (a) cationic PS spheres, (b) PS/SiO2 hybrid core-shell particles, and (c) hollow silica (samples of Fig. 1b).
To demonstrate the application of the unique hollow spheres in drug delivery, a peptide labelled with FITC (VTAMEPGQCG-Lys-FITC) was used as a model drug and the release profiles conducted in PBS buffer solutions were displayed in Fig. 4. The loading capacities27 of raspberry-like and smooth hollow spheres were 0.024 mg and 0.0146 mg model drug per mg of spheres, respectively. The drug release profiles (Fig. 4a) showed raspberry-like hollow silica spheres (shown in Fig. 1b) released more drugs than smooth surface of hollow silica spheres (shown in Fig. 1a) due to their larger surface area. The loading of drugs on the hollow silica spheres does not need further surface modification. The peptide can be absorbed on the surface of silica spheres by electrostatic interactions and hydrogen bonding. Because APTES has been incorporated in the silica coating on PS core and it has aminopropyl groups, the surface of the synthesized silica spheres exhibits functional hydroxyl- and amino-groups. After the raspberry-like hollow silica spheres were loaded with the FITC labeled peptide, they were also examined using a fluorescence microscope (Fig. 4b). The green fluorescence dots confirmed the success of drug loading. Some larger dots should be aggregation of several silica spheres in the aqueous solution.
Fig. 4.

Release profiles of model peptide drug from raspberry-like and smooth hollow silica spheres in the PBS buffer (pH 7.2) (Left). Fluorescence microscopic image of drug-loaded raspberry-like hollow silica spheres (Right), showing the successful loading of fluorescently labelled model drug.
Conclusions
In summary, the monodisperse hollow silica spheres with controlled surface morphology are prepared via one-pot polymerization under mild condition in aqueous solution. The PS spheres are subsequently, even synchronously, ‘dissolved’ during condensation of TEOS without thermal treatment. This biomimetic “green” process is facile, cost-efficient, environmentally friendly and energy saving. It is very useful for large-scale production especially in industry applications. The effects of surface charge on the polycondensation of TEOS shed light on the nucleation mechanism of silica and give more information for the preparation of silica based materials in the future. The enhanced drug loading and release of raspberry-like hollow silica spheres indicate that they may have promising biomedical applications such as drug delivery.
Supplementary Material
Acknowledgments
We would like to thank the National Science Foundation (DMR-0847758, CBET-0854465, CBET-0854414 and CMMI-1234957), National institute of Health (4R03AR056848-03, 1R21EB015190-02 and 5R01HL092526-02), Department of Defense Peer Reviewed Medical Research Program (W81XWH-12-1-0384), Oklahoma Center for the Advancement of Science and Technology (HR11-006) and Oklahoma Center for Adult Stem Cell Research (434003) for their generous financial support.
Footnotes
Electronic Supplementary Information (ESI) available: [Nucleation mechanism of silica on anionic and cationic PS particles, TEM and SEM images of silica spheres using anionic and cationic PS particles as templates, TEM images of silica spheres using TEOS only]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Yang J, Lee J, Kang J, Lee K, Suh JS, Yoon HG, Huh YM, Haam S. Langmuir. 2008;24:3417–3421. doi: 10.1021/la701688t. [DOI] [PubMed] [Google Scholar]
- 2.Hu H, Zhou H, Du J, Wang Z, An L, Yang H, Li F, Wu H, Yang S. Journal of Materials Chemistry. 2011;21:6576–6583. [Google Scholar]
- 3.Ma Y, Qi L, Ma J, Wu Y, Liu O, Cheng H. Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2003;229:1–8. [Google Scholar]
- 4.Bae E, Chah S, Yi J. Journal of Colloid and Interface Science. 2000;230:367–376. doi: 10.1006/jcis.2000.7064. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Y, Shi J, Shen W, Dong X, Feng J, Ruan M, Li Y. Angewandte Chemie International Edition. 2005;44:5083–5087. doi: 10.1002/anie.200501500. [DOI] [PubMed] [Google Scholar]
- 6.Seo JS, Whang D, Lee H, Jun SI, Oh J, Jeon YJ, Kim K. Nature. 2000;404:982–986. doi: 10.1038/35010088. [DOI] [PubMed] [Google Scholar]
- 7.Yu M, Wang H, Zhou X, Yuan P, Yu C. Journal of the American Chemical Society. 2007;129:14576–14577. doi: 10.1021/ja077110r. [DOI] [PubMed] [Google Scholar]
- 8.Yang X, Dai T, Lu Y. Polymer. 2006;47:441–447. [Google Scholar]
- 9.Perro A, Reculusa S, Bourgeat-Lami E, Duguet E, Ravaine S. Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2006;284–285:78–83. [Google Scholar]
- 10.Reculusa S, Mingotaud C, Bourgeat-Lami E, Duguet E, Ravaine S. Nano Letters. 2004;4:1677–1682. [Google Scholar]
- 11.Du X, Liu X, Chen H, He J. The Journal of Physical Chemistry C. 2009;113:9063–9070. [Google Scholar]
- 12.Ming W, Wu D, van Benthem R, de With G. Nano Letters. 2005;5:2298–2301. doi: 10.1021/nl0517363. [DOI] [PubMed] [Google Scholar]
- 13.Lu Y, McLellan J, Xia Y. Langmuir. 2004;20:3464–3470. [PubMed] [Google Scholar]
- 14.Qian Z, Zhang Z, Song L, Liu H. Journal of Materials Chemistry. 2009;19:1297–1304. [Google Scholar]
- 15.Zhang Y, Chen H, Shu X, Zou Q, Chen M. Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2009;350:26–32. [Google Scholar]
- 16.Wu X, Tian Y, Cui Y, Wei L, Wang Q, Chen Y. The Journal of Physical Chemistry C. 2007;111:9704–9708. [Google Scholar]
- 17.van Bommel KJC, Friggeri A, Shinkai S. Angewandte Chemie International Edition. 2003;42:980–999. doi: 10.1002/anie.200390284. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Lind JU, Trogler WC. Chemistry of Materials. 2008;20:2875–2877. [Google Scholar]
- 19.Pi M, Yang T, Yuan J, Fujii S, Kakigi Y, Nakamura Y, Cheng S. Colloids and Surfaces B: Biointerfaces. 2010;78:193–199. doi: 10.1016/j.colsurfb.2010.02.031. [DOI] [PubMed] [Google Scholar]
- 20.Chen M, Wu L, Zhou S, You B. Advanced Materials. 2006;18:801–806. [Google Scholar]
- 21.Wang F, Li D, Mao C. Advanced Functional Materials. 2008;19:4007–4013. [Google Scholar]
- 22.van Bommel KJC, Shinkai S. Langmuir. 2002;18:4544–4548. [Google Scholar]
- 23.Brinker CJ. Journal of Non-Crystalline Solids. 1988;100:31–50. [Google Scholar]
- 24.Mao C, Wang F, Cao B. Angewandte Chemie International Edition. 2012;51:6411–6415. doi: 10.1002/anie.201107824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li D, Qu X, Newton SMC, Klebba PE, Mao C. Journal of Materials Chemistry. 2012;22:15702–15709. doi: 10.1039/C2JM31034A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao S, Jin X, Yuan X, Wu W, Hu J, Sheng W. Journal of Polymer Science Part A: Polymer Chemistry. 2010;48:1332–1338. [Google Scholar]
- 27.Zhao W, Chen H, Li Y, Li L, Lang M, Shi J. Advanced Functional Materials. 2008;18:2780–2788. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.