

Abstract
The developmental brain gene NPAS3 stands out as a hot spot in human evolution because it contains the largest number of human-specific, fast-evolving, conserved, non-coding elements. In this paper we studied 2xHAR142, one of these elements that is located in the fifth intron of NPAS3. Using transgenic mice, we show that the mouse and chimp 2xHAR142 orthologues behave as transcriptional enhancers driving expression of the reporter gene lacZ to a similar NPAS3 expression subdomain in the mouse central nervous system. Interestingly, the human 2xHAR142 orthologue drives lacZ expression to an extended expression pattern in the nervous system. Thus, molecular evolution of 2xHAR142 provides the first documented example of human-specific heterotopy in the forebrain promoted by a transcriptional enhancer and suggests that it may have contributed to assemble the unique properties of the human brain.
Keywords: NPAS3, brain, evolution, schizophrenia, exaptation, MIR
1. Introduction
A major goal of current evolutionary studies is the identification of the genetic basis underlying the phenotypic differences that characterize Homo sapiens. In the last 6 Myr of human lineage evolution, the genes that contributed to peculiar human traits have probably acquired novel mutations in their coding or regulatory sequence, leading to the origin of differential protein functional domains or changes in their spatial or temporal expression, respectively. The identification of the genetic bases underlying the evolution of H. sapiens has been fuelled in the past decade by the availability of the human, several other primate and mammalian genomes. By comparing sequences from orthologue loci of several vertebrate genomes, including the human and chimpanzee, it is possible to identify brain genes and genomic regions showing accelerated evolution in the human lineage as candidates to reveal the molecular basis of the particular configuration of the human brain. Several studies have identified signatures of positive selection in the human lineage in the coding regions of brain genes, suggesting that they might have played a role during recent human evolution [1–6]. In addition, as part of genome-wide study to identify non-protein-coding accelerated regions in the human genome, Pollard et al. [7] identified the most dramatic human accelerated regions, HAR1, that is part of a novel RNA gene (HAR1F) that is expressed in Cajal–Retzius neurons in the developing human neocortex. Furthermore, the authors showed that HAR1F is co-expressed with reelin, an extracellular matrix glycoprotein secreted by Cajal–Retzius neurons that is of fundamental importance in specifying the six-layer structure of the human cortex [7]. More recently, the analyses of duplications and deletions in the human lineage have led to the identification of several genes related to brain function that underwent human-specific copy number variation [8]. Among them, the gene SRGAP2 is noteworthy, as it has at least one fixed human-specific partial duplication in the human genome. SRGAP2 is a negative regulator of neuronal migration and promotes neurite outgrowth. It has been hypothesized that the partially duplicated protein dimerizes with the full-length SRGAP2 protein, and thus it acts as a dominant partial inhibitor, presumably leading to neotenous changes, including increased density of longer neurite spines [9,10].
Taking advantage of four recent independent genome-wide studies performed to identify accelerated conserved non-protein coding regions in the human genome [7,11–14] that altogether detected approximately 1800 human accelerated elements (HAEs), we have recently identified the gene NPAS3 (neuronal PAS domain-containing protein 3) as an exceptional case in human evolution as this gene contains the largest cluster of 14 HAEs in the human genome [15]. NPAS3 encodes a transcription factor of the bHLH-PAS family [16] and is mainly expressed in the developing central nervous system of mice and humans as well as in the adult brain [16,17]. Analysis of NPAS3-deficient mice showed that this gene plays an important role during brain development and the control of neurosignalling pathways [18,19]. In addition, NPAS3 was identified as a candidate gene for schizophrenia in family [20–23] and association studies [24,25].
Using an enhancer assay in transgenic zebrafish, we have recently shown that 11 out of the 14 HAEs present in NPAS3 drive reporter gene expression to the developing central nervous system, suggesting that the accelerated evolution of the these regulatory sequences could have had an impact on the expression of NPAS3 in humans [15].
To explore the possibility that the human-specific genetic changes present in NPAS3 played a role in brain evolution, we aim to study in more detail the expression of NPAS3-HAE elements in a mammalian model. In this paper, we studied 2xHAR142, one of the 14 NPAS3-HAEs, that is located in the fifth intron of NPAS3. Using transgenic mice, we show that the mouse and chimp 2xHAR142 orthologues behave as transcriptional enhancers, driving expression of the reporter gene lacZ to a similar NPAS3 expression subdomain in the mouse central nervous system. Interestingly, the human 2xHAR142 orthologue drives lacZ expression to an extended expression pattern in the nervous system. In addition, we report here that 2xHAR142 enhancer contains sequences derived from a short interspersed element (SINE) retroposon of the MIR family. The high level of conservation of the SINE-derived sequences suggests that the 2xHAR142 enhancer originated at least in part from an exaptation event in the lineage leading to placental mammals.
2. Results
(a). The accelerated non-coding region 2xHAR142 behaves as a developmental enhancer
We have recently performed a meta-analysis combining the data of four recent independent genome-wide studies designed to identify human genomic accelerated regions through different comparisons and algorithms [7,11–14] that identified 1800 non-overlapping human accelerated elements (HAEs) in the human genome [15]. In that work, we identified the transcriptional units and genomic 1-megabase (1-Mb) intervals of the entire human genome carrying the highest number of HAEs. We found that the brain developmental transcription factor NPAS3 contains the largest cluster of non-coding accelerated regions in the human genome, with up to 14 elements that are highly conserved in mammals, including primates, but carry human-specific nucleotide substitutions [15]. Among these 14 HAEs, we decided to study in detail the function and evolution of the region 2xHAR142 that comprises 165 bp in the human genome (chr14: 33 118 402–33 118 566; figure 1). In order to study the function of the 2xHAR142 accelerated element, we cloned 502 bp containing the PhastCons conserved region containing the human 2xHAR142 (2xHAR142-Hs) sequence cloned upstream of the HSP68 minimal promoter fused to the reporter gene lacZ and generated transgenic mouse lines (figure 1). We generated five independent transgenic mouse lines (see electronic supplementary material, figure S1) and studied the expression pattern of the reporter gene lacZ at three mouse-developmental stages. Although we observed line-to-line variation, most probably owing to variable numbers and position of transgene insertions in the genome (see electronic supplementary material, figure S1), we observed that the 2xHAR142-Hs transgene behaves as a consistent developmental enhancer driving the expression of lacZ to the central nervous system, including the brain and the spinal cord at the three stages analysed (figure 1; electronic supplementary material, figure S1). At E10.5 and E12.5, we observed expression of lacZ in regions of the hindbrain, the midbrain and the forebrain. At E12.5 and E14.5 in the forebrain, the expression is mainly located at the developing cortex (figure 1e,f), in the hippocampal neuroepithelium bed nucleus of the stria terminalis, ventral thalamus and the hypothalamus (figure 1e,f). In the midbrain, we observed expression at the tegmentum and the cerebral peduncles. In the hindbrain, we observed extensive expression, including the cerebellar neuroepithelium, the pontine reticular nucleus, the fasciculus, etc. Outside the nervous system, we observed lacZ expression in the limb buds, the eyes, developing inner ear, vertebral body primordia, etc. (figure 1; electronic supplementary material, figure S1).
Figure 1.

Location of exapted repeats and HAEs at the NPAS3 locus in the human genome. (a) Location of the NPAS3 gene in NCBI build 36.1 of the human genome assembly showing exapted elements [26,27] and HAEs [15]. (b) Conservation of the 2xHAR142 sequence of the region according to the UCSC Genome Browser (www.genome.ucsc.edu). (c) Scheme of the transgene containing the 2xHAR142 sequence ligated to the mouse minimal Hsp68 promoter followed by the Escherichia coli lacZ reporter gene. Below, expression pattern of lacZ driven by 2xHAR142-Hs at E10.5 (d), E12.5 (e) and E14.5 (f) whole mount embryos and histologically selected sections of a representative 2xHAR142-Hs transgenic mouse line. Forebrain (fb); midbrain (mb); hindbrain (hb); spinal cord (sc); lateral ventricle (lv); preoptic area (pa); olfactory bulb (ob); ganglionic eminence (ge); thalamus (th); hypothalamus (hy); hippocampus (hi).
To determine whether the accelerated evolution led to changes in the expression pattern of this developmental enhancer, we generated and analysed transgenic mice carrying the 2xHAR142 mouse (2xHAR142-Mm) and chimpanzee (2xHAR142-Pt) orthologues. We found that three independent lines of 2xHAR142-Mm mice expressed consistently the reporter gene lacZ at a reduced expression domain that includes a subdomain of the 2xHAR142-Hs expression pattern. We observed expression in the spinal cord, in the hindbrain and low expression in the midbrain. At the forebrain, we observed expression in the preoptic area, but notably these transgenic mice did not show any expression in the developing cortex (figure 2; electronic supplementary material, figure S1).
Figure 2.
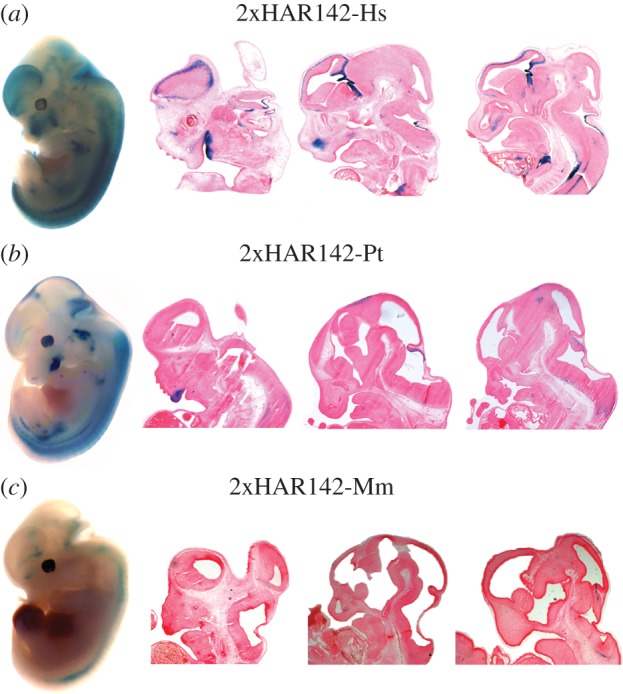
Comparative expression analysis of human, chimpanzee and mouse 2xHAR142 orthologue sequences in transgenic mice. Whole mount and selected sagital cryostat sections of F1 transgenic mouse embryos showing lacZ expression patterns obtained with each transgene in a (a) human, (b) chimpanzee and (c) mouse representative transgenic line at E12.5.
To address whether the differences in expression were only driven by the human accelerated region and not present in other primates, we generated three transgenic mouse lines carrying the chimpanzee 2xHAR142 sequence (2xHAR142-Pt). We analysed the expression pattern of the reporter gene driven by the 2xHAR142-Pt sequence and observed that the expression of lacZ in the developing nervous system is more similar to the one driven by the 2xHAR142-Mm than to the 2xHAR142-Hs (figure 2; electronic supplementary material, figure S1). In fact, expression domains include the spinal cord and the hindbrain, and low expression was observed in the midbrain. In the forebrain, we only observed expression in the preoptic area, whereas we did not observe expression in the developing cortex driven by the 2xHAR142-Pt sequence, in high contrast to the strong expression in this region driven by the human orthologue (figure 2; electronic supplementary material, figure S1). In addition, we noted that the 2xHAR142-Pt sequence drives expression to the eyes, limb buds, developing inner ear, vertebral body primordia, etc. (see electronic supplementary material, figure S1). The expression in the limb buds and vertebral body primordia seems to be a characteristic of the two primate enhancers but is absent in the mouse enhancer. More work will be necessary to assess whether these expression domains were gained in the primate lineage or lost in the mouse lineage. We observed that the 2xHAR142-Hs sequence shows 98% identity with the chimpanzee sequence (seven human-specific substitutions on the PhastCons region; electronic supplementary material, figure S3) and 85% identity with the mouse sequence harbouring 37 substitutions across the PhastCons region. These data suggest that the changes acquired after the split of the human and chimpanzee lineages could be responsible for the acquisition of new expression territories driven by the human sequence in the developing nervous system.
(b). The 2xHAR142 sequence drives the expression of the lacZ reporter gene to an overlapping expression domain of the gene NPAS3
In order to determine whether the enhancer sequence 2xHAR142 behaves as an NPAS3 regulatory sequence, we aimed to compare the expression of the 2xHAR142 sequence with the NPAS3 gene expression pattern. The expression pattern of the gene NPAS3 during mouse central nervous system development has only been partially described [16], consequently we also aimed to analyse in detail expression domains of NPAS3 during mouse development. Owing to the difficulty in getting antibodies to detect NPAS3 during mouse development, we studied the expression pattern of NPAS3 using in situ hybridization (ISH). We performed ISH studies at the three developmental stages that we had analysed in transgenic mice. In addition, to study the cellular types where NPAS3 is expressed during mouse development, we performed fluorescent ISH studies to detect NPAS3 in combination with different cellular markers in the nervous system.
At E10.5, we observed that the NPAS3 is expressed all across the neuroepithelium in the hindbrain, midbrain and forebrain (figure 3a–j). Expression of NPAS3 greatly overlaps with cells expressing the undifferentiated neural precursor cell (NPC) marker SOX2. We observed numerous cells expressing both SOX2 and NPAS3 (figure 3b–e and g–j). On the other hand, we detected less density of cells expressing NPAS3 in the external layers of the neuroepithelium where cells express the early neuronal markers DCX and β III-tubulin or TUJ1 (figure 3a,f).
Figure 3.

NPAS3 expression in the developing mouse brain. The expression of NPAS3 is shown through ISH in combination with immunohistochemical detection of the early neuronal markers DCX and β III-tubulin (TUJ1) and the undifferentiated NPC marker SOX2 in cryostat sections of E10.5 and E12.5 mouse embryos. Expression of NPAS3 at E10.5 is shown in the hindbrain (a–e) and the forebrain (f–j) in combination with DCX (a,f) and SOX2 (b–e and g–j). At E12.5 expression is shown in the spinal cord (k–o), the hindbrain (p–y and aa–ee) and the forebrain (ff–jj). In the spinal cord, NPAS3 is shown in combination with SOX2 (k) and TUJ1 at low (l) and high magnification (m–o). In the hindbrain, expression is shown in combination with DCX (p–t), SOX2 (u–y) and TUJ1 (aa–ee). In the forebrain, the expression is shown in combination with TUJ1 at low (ff,gg) and high magnification (hh–jj).
At E12.5, we observed expression of NPAS3 in the developing central nervous system as well as other areas including the inner ear, the eye and the olfactory epithelium (not shown). In the central nervous system, we detected expression in the forebrain, midbrain, hindbrain and spinal cord (figure 3). In the hindbrain, NPAS3 is expressed all across the neuroepithelium, and especially high expression is observed in the ventricular zone. This area close to the fourth ventricle is populated by SOX2-expressing cells and we observed that a very high proportion of these cells also express NPAS3 (figure 3u–y). Additionally, numerous NPAS3-expressing cells away from the ventricle also expressed DCX (figure 3p–t) and fewer expressed TUJ1 (figure 3aa–ee). In the forebrain, at the developing cortex, we observed a similar pattern of high density of NPAS3-SOX2-expressing cells in the ventricular zone (not shown) and cells in the cortical plate expressing NPAS3 and TUJ1.
There are technical difficulties in simultaneously detecting NPAS3 and β-galactosidase. Thus, we used the detection of β-galactosidase in combination with the cellular markers that we had used in conjunction with NPAS3-ISH to infer whether the expression of the reporter gene lacZ and the NPAS3 gene overlap. We observed that the expression of β-galactosidase driven by the 2xHAR142-Pt and 2xHAR142-Mm sequence co-localizes mainly with DCX- and TUJ1-expressing cells, whereas minimum overlap was observed in SOX2-expressing cells in the hindbrain and spinal cord (figure 4; electronic supplementary material, figure S2).
Figure 4.

β-Galactosidase expression pattern at E12.5 in different cellular types in the developing brain. (a) Schematic of an E12.5 whole embryo where a red line indicates the approximate location of the histological section located below. (b) Low-resolution image of a histological section of a transgenic mice expressing β-galactosidase under the control of the 2XHAR142-Pt sequence in the hindbrain and spinal cord. White boxes over the image indicate approximate location of the high-resolution images shown on the right. High-resolution confocal photographs at the right show the expression pattern of the reporter gene lacZ protein product β-galactosidase (bgal) at different levels of the spinal cord (c,d,g,h,k,l,o,p) and the hindbrain (s–z and aa–bb). The expression of β-galactosidase is shown in combination with the expression of the early neuronal markers DCX and β III-tubulin (TUJ1) and the undifferentiated NPC marker SOX2 in the developing central nervous system. The expression of NPAS3 in combination with the same cellular type markers is shown for some cases at approximate similar levels in the spinal cord (e,f,i,j,m,n) and the hindbrain (cc,dd).
However, we had observed that the expression pattern of NPAS3 at E12.5 includes the complete neuroepithelium at the spinal cord and the hindbrain (figures 3 and 4e,f,i,j,m,n,dd), and NPAS3 is highly expressed in SOX2-positive cells. Altogether, our results indicate that the enhancer sequences 2xHAR142-Pt and 2xHAR142-Mm drive the expression of the reporter gene lacZ to a subdomain of the NPAS3 expression pattern and suggest that this enhancer participates in the expression regulation of NPAS3.
(c). The accelerated non-coding region 2xHAR142 contains sequences derived from an MIR retroposon
We observed that 30 nucleotides of the 2xHAR142 region overlap with the exapted element exap2775 as described by Lowe et al. [26] (figure 1; electronic supplementary material, figure S3). The term ‘exaptation’ was suggested in 1992 by Brosius & Gould [28] to name the examples of transposable elements (TEs) acquiring novel functions in the genome. Since then, many examples of TE exaptation as putative or demonstrated transcriptional regulatory regions have accumulated (for a review, see [29]); however, only in a few cases it was possible to demonstrate functionally that regulatory innovation was mediated by the TE exaptation event [29]. The exap2775 region comprises 73 bp that are included in the PhastCons conserved sequence (280 bp) that contains the accelerated region 2xHAR142 (figure 1; electronic supplementary material, figure S3). As exap2775 does not overlap with a Repeat Masker annotated mobile element, we used several strategies in order to find out the identity of the mobile element present in the conserved region. In addition, we aimed to determine the time at which this element originated in the history of vertebrates.
In order to determine the type of mobile element from which exap2775 originated, we searched orthologue sequences of exap2775 in the human genome using BLAT. We found that significant hits of exap2775 in the human genome were located in annotated SINE retroposons of the MIR family. MIRs or mammalian-wide interspersed repeats are t-RNA-derived SINEs [30] from the CORE-SINE superfamily [31]. To identify the subfamily of MIRs from which the exap2775 element originated, we performed exhaustive sequence comparisons using BLAST among exap2775 and all MIR sequences present in the RepBase database. We found that the region of highest similarity correspond to the CORE region of MIRs. In addition, we found the highest similarity to members of the MIRb and MIRc subfamilies (see electronic supplementary material, figure S3). To confirm this finding, we also searched the exap2775 element using the P-clouds tool designed to detect TEs using a different strategy to that used by Repeat Masker [32]. We found that exap2775 overlaps with a P-clouds MIR annotated element in the human genome (see electronic supplementary material, figure S3).
The exapted region 2775 is conserved among placental mammals and not present in non-placental mammals and other vertebrates (figure 1; electronic supplementary material, figure S3). In order to trace the origin of this sequence, we performed BLAST searches using the exap2775 region as query against all sequenced non-placental mammal vertebrate genomes. BLAST searches in the Monodelphis domestica genome showed that all significant hits were located in MIR-derived sequences, although none of them was located in the NPAS3 locus. We observed similar results searching the platypus (Ornitorhynchus anatinus) genome where all significant hits were found in CORE-SINEs-derived sequences of the type Mon1, closely related to MIRs. On the other hand, searches in the chicken genome (Gallus gallus) resulted in a few low-score hits that where not located in identified mobile elements. Re-BLAST of the identified sequences of the chicken genome indicated that they were not originated by mobile elements but unique sequences in the genome. Similarly, the highest scoring hits in the zebrafish genome where not in mobile-elements-derived sequences. Altogether, our analyses indicated that the mobile element from which the sequence exap2775 originated, was derived from a SINE retroposon, most probably of the MIR family. In addition, our data showed that although SINE elements of the MIR family are present in non-placental mammalian species, the exaptation of the MIR element from which exap2775 originated occurred in the lineage leading to placental mammals. Even though full-length MIR retroposons are around 250 bp, only a smaller portion of the consensus element was recognized as the repeat derived element exap2775 (73 bp). In order to test whether additional signatures of the original full-length MIR retroposon are still recognizable in the enhancer sequence tested, we have aligned mammalian orthologues of the sequence containing the 2xHAR142 region with full-length MIR consensus sequences from RepBase (MIR, MIRb and MIRc) and particular MIR instances from the genome that were retrieved using exap2775 as a query (see electronic supplementary material, figure S3). We found that additional signatures of the original MIR are still recognizable outside the exap2775 region, suggesting that a full-length MIR element inserted in the region in the lineage leading to placental mammals and accumulated mutations before it started to be under purifying selection (see electronic supplementary material, figure S3). Although a complete proof of the functional relevance of the MIR-derived sequences would require further deletional studies, the high level of conservation (see electronic supplementary material, figure S3) suggest that the MIR-derived sequences were probably exapted into cis-regulatory sequences and maintained under purifying selection all throughout the evolution of placental mammals.
(d). Acceleration and exaptation
Afterwards, we aimed to analyse whether exap2775 was the only non-coding conserved sequence derived from a mobile element in the NPAS3 gene genomic region. We found that 10 exapted repeats had already been identified by Lowe et al. [26] in the NPAS3 region and only one of them (exap2775) overlaps an HAE (figure 1). Our genome-wide analysis found that from the 1800 HAEs only 31 overlap exapted repeats from the 10 402 described by Lowe et al. [26] covering 8743 bases of the human genome. Twenty eight of the 31 HAEs originated from exapted mobile elements correspond to HACNS and three to 2xHARs.
A second database of exapted repeat elements has been more recently described [12,27] containing a total of 284 857 regions. Using this database that we call exapted repeats 2011, we mapped 138 exapted repeats in the NPAS3 region, ranging from 5 to 238 bp in length. Three of these newly described elements overlapped with the previous database (exap2772 and exap2778). In addition, we found no overlap between exapted repeats 2011 and HAEs in the NPAS3 genomic region, whereas at the whole-genome level, we found that 133 exapted repeats 2011 overlap with HAEs. To analyse whether NPAS3 shows an exceptional amount of exapted element in the genome, we analysed the distribution of exapted repeats by gene. For that purpose, we obtained a unique non-overlapping database of exapted elements from the 2007 and the 2011 databases and intersected this with a database of gene transcriptional units, defined as the genome interval delimited by the largest reported RefSeq transcript for each gene. This analysis showed that the NPAS3 region accumulates a total of 145 exapted repeats (figure 1; electronic supplementary material, table S1). We observed that even though some exapted repeats were reported as individual exaptation events, they are clustered very closely and overlap the same mobile element, suggesting that they constitute a unique exaptation event (see electronic supplementary material, table S1). We discovered that from a total of 19 897 annotated human RefSeq genes (human genome assembly 2006; NCBI36/hg18), 10 754 carry at least one exapted repeat in their transcriptional unit. We found that 10 707 genes contained between 1 and 144 exapted repeats, whereas only 39 genes display more than 145 exapted repeats. Surprisingly, we observed that there are several genes harbouring more than 300 exapted repeats in their transcriptional unit (see electronic supplementary material, table S2). We found that, as expected, genes with long transcriptional units accumulate large amounts of exapted elements (see electronic supplementary material, table S2). In order to take into account the gene-length factor, we also analysed the accumulation of exapted element by fixed length units of 1 Mb of the human genome. We found that two out of three of the 1-Mb genomic regions that accumulate the largest amounts of exapted elements (more than 400) are the ones that contain the top harbouring exapted element genes PTPRT and CSMD2 (see electronic supplementary material, table S2). More work will be necessary to clarify why exapted elements accumulate in particular genes or genomic regions.
Subsequently, to characterize in more detail and to date the exaptation events at the NPAS3 region, we analysed the type of mobile element from which the exapted regions originated. We found that 137 elements overlapped Repeat Masker annotations (Repeat Masker v. 3.2.7), facilitating its identification. Out of these 137 elements, 50 were identified as SINEs, 70 as long interspersed elements (LINEs) and 10 as DNA transposon-derived elements (see electronic supplementary material, table S1). Our data indicate that the types of mobile element from which the exapted regions originated in the NPAS3 region do not differ from the general trend that has been reported at the genome level [26,27]. Additionally, using BLAST searches, we identified the mobile elements from which the exapted repeats originated that do not overlap with Repeat Masker annotations in the genome (see electronic supplementary material, table S1).
Thereafter, to date all the exaptation events at the NPAS3 locus, we analysed the conservation of the exapted elements using sequence alignments of all available vertebrate genomes for each element. The majority of the NPAS3 exapted elements seem to have been co-opted in the lineage leading to placental mammals (85; 61.6%). In addition, 28.26% and 10.15% were exapted in the lineage leading to mammals and vertebrates, respectively (see electronic supplementary material, table S1).
3. Discussion
In this paper, we show that the human accelerated region 2xHAR142 is a mammalian novelty that seems to have originated, at least partially, from the exaptation of a SINE retroposon of the MIR family in the lineage leading to placental mammals. In addition, we show that the 2xHAR142 sequence is not the only conserved non-coding sequence in the NPAS3 locus containing mobile element-derived sequences, because the transcriptional unit of this gene accumulates 145 exapted elements as previously defined [12,26,27]. Moreover, we show that of the 19 897 annotated human RefSeq genes in the human genome, 10 754 carry at least one exapted repeat. We found that 10 707 genes contained between 1 and 144 exapted repeats, whereas only 47 genes display more than 145 exapted repeats, indicating that the locus containing NPAS3 underwent extensive co-option of mobile elements into non-coding conserved sequences throughout the evolution of vertebrates. Our findings add another example to the growing list of functionally characterized cases of transcriptional enhancers containing mobile-element-derived sequences throughout the evolution of living forms [33–38].
In this work, we also demonstrated that 2xHAR142 is an enhancer that drives the expression of the reporter gene lacZ to a subdomain of the NPAS3 expression pattern. We found that the accelerated evolution process that this sequence underwent in the human lineage has probably led to a shift in its expression pattern. In fact, we show here that transgenic mice carrying the human version of 2xHAR142 display an extended expression pattern compared with the chimpanzee or the mouse orthologues.
It has been hypothesized that distinctive human traits such as the use of a complex language, long-term planning and exceptional learning capacities are the result of molecular changes that led to modifications in the developmental programme of our brain that occurred during the past 6 Myr of human lineage evolution.
Almost 40 years ago, King & Wilson [39], on the basis of the Ohno hypothesis about the evolution of regulatory systems [40], proposed the idea that evolution of non-coding regulatory sequences played a major role in shaping the unique features of the human brain. However, a proof of concept of this idea is still pending. As protein-coding regions account for only one-third of all conserved sequences in the human genome and several studies performed in other species showed that lineage-specific evolution of regulatory sequences appeared to have driven phenotypic changes (see for reviews [41,42]), detection of human-specific accelerated conserved non-coding regions provides the raw material to identify those human-specific molecular changes that led to phenotypic evolution.
Recently, four independent genome-wide studies were performed to identify accelerated conserved non-protein coding regions in the human genome [7,11–14] that together detected approximately 1800 genomic human fast-evolving regions. Although these studies searched for conserved genomic regions with accelerated substitution rates specific for the human branch, each of them yielded partially overlapping output sequences, probably because they used different types and numbers of compared species and different algorithms to assess acceleration. As gene loci densely populated with HAEs are more likely to have contributed to human-specific novelties, we have recently performed a meta-analysis of these four public databases to identify clusters of human genomic accelerated elements. Our study revealed that most transcriptional units and 1 Mb intervals carry none, 1, 2 or 3 HAEs, and only very limited cases showed a concentration of HAEs greater than seven [15].
It has been more difficult to assess the functional impact of the evolutionary process than to identify signatures of accelerated evolution and positive selection in the human lineage. It has been shown that the most dramatic human accelerated region, HAR1, is part of a novel RNA gene (HAR1F) that is expressed in Cajal–Retzius neurons in the developing human neocortex from seven to 19 gestational weeks, a crucial period for cortical neuron specification and migration. HAR1F is co-expressed with reelin, a product of Cajal–Retzius neurons that is of fundamental importance in specifying the six-layer structure of the human cortex [7]. However, the HAR1F expression pattern in rhesus macaque is very similar to that observed in the human developing brain at comparable gestation stages. These results indicate that despite extensive sequence changes, the expression pattern of HAR1F in the developing cortex has been highly conserved since the divergence of hominoids and Old World monkeys around 25 Myr ago [7].
In addition, it has been reported that the conserved non-coding sequence HACNS1 that evolved extremely rapidly in humans acts as an enhancer of gene expression in transgenic mice. Moreover, it has been shown that the HACNS1 human orthologue gained a strong gene expression domain in the limbs compared with the chimpanzee and rhesus macaque orthologue elements [43]. However, the potential impact of human-specific changes in the expression of neighbour genes (CENTG2 and GBX2) and on limb development remains to be explored [43].
It has been shown that two amino acid substitutions in the transcription factor FOXP2 have been positively selected during human evolution [1,44]. In order to assess the functional impact of these evolutionary changes, these two amino acids were replaced in the mouse orthologue locus in order to create ‘humanized’ FOXP2 mice [45]. Extensive phenotypic analysis of these mice indicated that they have qualitatively different ultrasonic vocalizations, decreased exploratory behaviour and decreased dopamine concentrations in the brain, suggesting that the humanized Foxp2 allele affects basal ganglia. As humans who carry one non-functional FOXP2 allele show some alterations in the basal ganglia, the authors speculate that alterations in cortico-basal ganglia circuits as a result of the evolution of FOXP2 might have been important for the evolution of speech and language in humans. However, more research will be necessary to make a stronger link between FOXP2 and the evolution of speech in humans.
More recent work has identified the locus AUTS2 which contains the most significantly accelerated genomic region differentiating humans from Neanderthals. Using a combination of enhancer assays in transgenic zebrafish and mice, the authors characterized regulatory regions responsible for the expression of the gene in the brain as well as brain enhancers that overlap an autism spectrum disorder-associated deletion and brain enhancers that reside in regions implicated in human evolution [46].
We have recently shown that in addition to 2xHAR142, the gene NPAS3 accumulates 14 HAEs in its transcriptional unit, indicating that this gene is a hot spot in human evolution [15]. Using a transgenic zebrafish enhancer assay, we have shown that 11 out of the 14 HAEs present in NPAS3 drive reporter gene expression to the developing central nervous system, suggesting that the accelerated evolution of these regulatory sequences could have had an impact on the expression of NPAS3 in humans [15].
In this paper, we show that 2xHAR142 behaves as a transcriptional enhancer during mouse development, driving the expression of the reporter gene lacZ to a subdomain of the NPAS3 expression pattern. These results suggest that 2xHAR142 participates in the regulation of NPAS3 expression. In addition, our data also suggest that the overall expression of NPAS3 is probably controlled by other enhancer sequences. We have already shown that at least 11 NPAS3-HAEs behave as transcriptional enhancers in transgenic zebrafish [15]. Moreover, the large locus that the gene NPAS3 occupies in the genome means that it contains additional non-coding conserved sequences which could also participate in the regulation of its expression and which have not been characterized so far. Together these data suggest that the control of expression of the gene NPAS3 is very complex and further work needs to be done in order to understand it completely. So far, we have shown two cases of NPAS3-HAEs where accelerated evolution of non-coding regulatory sequences led to functional changes [15]. These data strongly suggest that the dramatic accelerated evolution that this gene underwent affecting 14 regulatory regions could have resulted in significant modifications of NPAS3 expression in humans.
Finally, the gain of function displayed by the 2xHAR142 human enhancer shown here provides the first documented example of human-specific heterotopy in the forebrain promoted by a transcriptional enhancer and suggests that it may have contributed to shape the expression of NPAS3 and the unique properties of the human brain.
4. Material and methods
(a). Sequences and databases
In order to obtain a unique non-overlapping database of exapted repeats, we intersect in the Table Browser of the UCSC Genome Browser (www.genome.ucsc.edu) the exapted repeats track [26] and a custom track of exapted repeats 2011 [12,27] obtained from the 29 mammals project website (http://www.broadinstitute.org/scientific-community/science/projects/mammals-models/29-mammals-project-supplementary-info). To obtain the number of exapted repeats for a gene transcriptional unit, we used the non-overlapping exapted repeats database with a database of gene transcriptional units, defined as the genome interval delimited by the largest reported RefSeq transcript for each gene or four datasets of 1 Mb-length genomic intervals that overlapped every 250 kb, using Galaxy tools (http://main.g2.bx.psu.edu/). All the analyses were performed in the human NCBI36/hg18 genome assembly. Identification of mobile elements was performed using the Repeat Masker track v. 3.2.7, and the repeat masking search was performed in the Repeat Masker web site (http://www.repeatmasker.org). We downloaded mobile element consensus sequences from Repbase (http://www.girinst.org/repbase/) and performed BLAST searches locally and in the BLAST web site (http://blast.ncbi.nlm.nih.gov). We also analysed the exap2775 sequence using P-clouds [32]. We used the MIR-specific custom tracks annotated in the human genome (available from [32]).
(b). Transgenes and transgenic mice production
Sequences 2xHAR142-Hs and 2xHAR142-Pt were amplified by proofreading PCR from human and chimpanzee genomic samples (chimpanzee samples were kindly donated by Adrian Sestelo). The resulting PCR fragments were directionally cloned into the vector pENTR/D-TOPO (Invitrogen, Carlsbad, CA, USA) through the CCAC sites added to the forward primer. Sequence identities were confirmed by sequencing. Fragments cloned in the pENTR/D-TOPO vector were transferred by site-specific recombination using the LR Clonase Enzyme mix (Invitrogen) to the pGW-Hsp68-lacZ vector which had previously been engineered to introduce recombination recognition sites (kindly donated by Dr Marcelo Nobrega, see reference [47]). Transgene 2xHAR142-Mm was amplified by proofreading PCR from a mouse genomic sample. The resulting PCR fragment was directionally cloned into the vector pZErO-2 (Invitrogen). Sequence identity was confirmed by sequencing. The plasmid pZErO-2/2xHAR142-Mm was digested with SalI and XmaI, and a fragment was subcloned into pHsp68-lacZ. Primer pairs used to amplify TG-2xHAR142-Hs and TG-2xHAR142-Pt were TG-2xHAR142-Hs–F: 5′-caccCAACCAACACCAGCTAGAAAG-3′, TG-2xHAR142-Pt: 5′-caccGCAACCAACACCAGCTTGAAAG-3′ and TG-2xHAR142-Hs/Pt-R: 5′-CAAGAATTCAATTAAATGCACAACTGAACAGG-3′. Primer pairs used to amplify TG-2xHAR142-Mm were TG-2xHAR142-Mm-F: 5′-ACGTCGACCTTTTGAGCCAGGTCCACTC-3′ and TG-2xHAR142-Mm-R: 5′-ACCCCGGGACGAGGGGGAAAAGATCACT-3′. Prior to microinjection, each pGW-Hsp68-lacZ-Hs/Pt vector was digested with SalI and pHsp68-LacZ-Mm was digested with SalI and Not I to liberate the transgenes. Then transgenes were eluted from an agarose gel using QIAquick Gel Extraction Kit (Qiagen, Valencia, CA, USA) and purified with Elutip-D columns (Schleicher & Schuell, Keene, NH, USA). After precipitating with NaOAc (3 M, pH 5.2) and ethanol, the DNA was washed with ethanol 70% and resuspended with microinjection buffer (filter sterile Tris–HCl 5 mM, pH 7.4; EDTA 0.1 mM). Transgenic mice were generated by pronuclear microinjection of B6CBF2 or FVB zygotes as described previously [34,36]. Microinjected zygotes were transferred to the oviduct of B6CBF2 pseudo-pregnant females. Transgenic pups were identified by PCR on ear punches genomic DNA with a combination of a forward transgene-specific primers and a reverse lacZ-specific primer. Primer pairs used were TG2xHAR142-Mm_F: 5′-TTCGGAATAAATGGGCTCCT-3′, TG2xHAR142-Hs/Pt_HSP_GW_F: 5′- TCCCTGTTCAGTTGTGCATT-3′ and LacZ_R1: 5′-AAGGGGGATGTGCTGCAAGGCG-3′. Mice were kept in a ventilated rack (Thoren Caging Systems, Hazleton, PA, USA) under a 12 L : 12 D cycle and 20–22°C room temperature (RT).
(c). X-Gal histochemistry
Transgene expression was determined in F1 mouse embryos of all independently generated pedigrees. Timed pregnant dams were obtained by mating B6CBF1 females with F0 or F1 transgenic males from all transgenic pedigrees. After killing the pregnant dam at defined post coitum days (dpc), embryos were removed immediately, washed with 1× phosphate-buffered saline (PBS; 0.1 M, pH 7.4) and fixed with 4% paraformaldehyde in 1× PBS during 30 min (for E9.5 to E12.5 dpc embryos) and 1 h (E14.5 dpc embryos). β-Galactosidase activity was determined in whole-mount embryos with 1 mg ml−1 of 5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid (X-Gal) in 1× PBS pH 7.3 containing 2.12 mg ml−1 of potassium ferrocyanide, 1.64 mg ml−1 of potassium ferricyanide, 2 mM MgCl2, 0.01% sodium deoxycholate and 0.02% NP-40 overnight at 37°C. To analyse β-galactosidase activity in sagittal sections, whole-mount embryos were first stained with X-Gal as described above. Then the specimens were rinsed in 1× PBS overnight at 4°C, cryoprotected in 10% sucrose solution in 1× PBS and embedded in 10% gelatin with 10% sucrose in the same buffer. The blocks were frozen for 1 min in isopentane cooled to −55°C in dry ice and were stored at −80°C. Serial 20 µm thick (10.5 dpc embryos) and 30 µm thick (12.5 and 14.5 dpc embryos) sections were cut in a cryostat (Leica CM1850, Germany), collected on Super-Frost Plus slides (Fisher Scientific, USA) and stored at −80°C until use. On the day of use, sections were air dried at RT for 2 h, post fixed with cold 4% paraformaldehyde in 1× PBS for 10 min and then washed with 1× PBS twice for 10 min each. X-Gal re-staining was performed at 37°C overnight. After X-Gal staining was completed, slides were washed three times for 5 min each in 1× PBS, once in water for 5 min and then incubated for 1 h in filtered 0.5% eosin (Sigma-Aldrich, USA) in a glacial acetic acid solution (0.16%, pH 6.0), then washed in distilled water for 15 s and dehydrated through ethanol series. Finally, slides were placed in xylene for 1 min and slip-covered using DPX (Fluka, Germany). Images were acquired using a Leica MZFLIII microscope coupled to a Leica DFC320 camera.
(d). In situ hybridization
The embryos were fixed by immersion in 4% paraformaldehyde solution in 1× PBS at 4°C overnight. The specimens were rinsed in 1× PBS at 4°C overnight, cryoprotected in 10% sucrose solution in 1× PBS and embedded in 10% gelatin with 10% sucrose in the same buffer. The blocks were frozen as described above. Serial 16 μm thick (10.5 dpc embryos) or 20 µm thick (12.5 and 14.5 dpc embryos) sagittal sections were cut in a cryostat (Leica CM1850, Germany), collected as parallel series on Super-Frost Plus slides (Fisher Scientific) and stored at –80°C until use. A 460 bp NPAS3 probe previously characterized [16] was amplified by proofreading PCR from mouse adult brain cDNA samples using the following primer pair: NPAS3probe1_F: 5′-CGAATAACTGCCCAGCATC-3′ and NPAS3probe1_R: 5′TTTTCCTTCTTGATTTAGTGCAAA 3′. The resulting PCR fragment was cloned into pGEM_Teasy (Promega, Madison, WI, USA) and sequenced to confirm identity. Plasmids containing the probe were linearized using SpeI and transcribed using T7 RNA polymerase (Roche, Penzberg, Germany) to generate digoxigenin-labelled NPAS3 antisense riboprobe. We performed ISH on cryosections as previously described [48]. Briefly, the thawed sections were postfixed with 4% paraformaldehyde in 1× PBS for 10 min and then washed three times with 1× PBS for 10 min. They were subsequently permeabilized in 1% Triton X-100 in 1× PBS for 30 min. Thereafter, prehybridization occurred at RT for 4 h in a solution containing 50% formamide, 10% dextran sulfate, 5× Denhardt's solution and 250 mg ml−1 t-RNA. Hybridization was performed with 200–300 ng ml−1 of the probe in the same hybridization solution at 72°C for 16 h. After hybridization, the sections were washed with 0.2× sodium-saline citrate buffer (SSC; 0.3 M sodium chloride, 30 mM trisodium citrate, pH 7) at 72°C for 2 h, thereafter 15 min with 0.2× SSC plus 2% H2O2 and then twice with a solution containing 100 mM NaCl, 0.1% Triton X-100 and 100 mM Tris–HCl (pH 7.5). After blocking with 10% normal donkey serum (NDS) in the same solution for 2 h, the sections were incubated overnight with horseradish peroxidase-conjugated, anti-digoxigenin Fab fragments (Roche, Germany; 1 : 500). The sections were washed twice with the same buffer and then incubated in 1/100 cyanine-3 tyramide reagent in 1× Plus Amplification Diluent (PerkinElmer Life and Analytical Sciences, Shelton, CT, USA). The tyramide signal amplification reaction was allowed to proceed for 30 min protected from light. Thereafter, the slides were washed with 1× PBS overnight.
(e). Immunohistochemistry
The embryos were fixed by immersion in 4% paraformaldehyde solution in 1× PBS at 4°C for 4 h. The specimens were cryoprotected and frozen as described above. Serial 16 µm thick (10.5 dpc embryos) or 20 µm thick (12.5 and 14.5 dpc embryos) sagittal sections were cut in a cryostat (Leica CM1850, Germany), collected as parallel series on Super-Frost Plus slides (Fisher Scientific) and stored at –80°C until use. Sections were air dried at RT for 1 h and incubated for 2 h at RT in blocking solution (10% NDS, 0.1% Triton X-100, 1× PBS) followed by incubation with a mix of primary antibodies diluted in blocking solution overnight at 4°C. Sections were washed three times in 1× PBS and then incubated sequentially for 1 h at RT with each secondary antibody. Primary antibodies used were rabbit polyclonal anti-β-galactosidase (1/1000; Cappel, MP Biomedical, Santa Ana, CA, USA), goat polyclonal anti-SOX-2 (1/100; Santa Cruz Biotechnology, Santa Cruz, CA, USA), goat polyclonal anti-Doublecortin (1/100; Santa Cruz Biotechnology) and mouse monoclonal anti-Neuronal Class III β-Tubulin (1/100; Covance, Princeton, NJ, USA). Secondary antibodies used were donkey anti-rabbit-AF555 (1/1000; Molecular Probes, Invitrogen), donkey anti-goat-AF488 (1/1000; Molecular Probes, Invitrogen), donkey anti-chicken-AF488 and donkey anti-mouse Cy5 (1/1000; Jackson Inmuno Research, Baltimore Pike, PA). Finally, sections were washed three times in 1× PBS and mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA). Images were acquired using a Nikon C1 confocal microscope using the EZ-C1 2.20 software and PlanApo objectives.
Acknowledgements
We thank Marta Treimun, Juan Manuel Baamonde, Marisol Costa and Adriana Barriento at the Transgenic Mouse Facility at the Centro de Estudios Científicos and Jessica Unger and Irina Garcia Suarez at the IBYME-INGEBI mouse facility for excellent technical assistance in transgenic mouse production and handling. We thank Marcelo Rubinstein for his help with transgenic mice generation and José Luis Ferrán for technical advice with in situ experiments. We thank Adrian Sestelo for chimpanzee samples and Gustavo Dziewczapolski for his inestimable contribution to our work.
All mouse procedures followed the Guide for the Care and Use of Laboratory Animals and were approved by the local institutional animal care and use committee.
Funding statement
This work was supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica (PICT 2008–1071), the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET-Argentina; PIP 0299) to L.F.F. and Proyecto de Financiamiento Basal CONICYT to R.L.L. G.B.K. and J.R.L. received a doctoral fellowship from the CONICET and R.L.L. received a doctoral fellowship from CONICYT.
References
- 1.Enard W, Przeworski M, Fisher SE, Lai CSL, Wiebe V, Kitano T, Monaco AP, Pääbo S. 2002. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418, 869–872 (doi:10.1038/nature01025) [DOI] [PubMed] [Google Scholar]
- 2.Evans PD, Anderson JR, Vallender EJ, Choi SS, Lahn BT. 2004. Reconstructing the evolutionary history of microcephalin, a gene controlling human brain size. Hum. Mol. Genet. 13, 1139–1145 (doi:10.1093/hmg/ddh126) [DOI] [PubMed] [Google Scholar]
- 3.Evans PD, Anderson JR, Vallender EJ, Gilbert SL, Malcom CM, Dorus S, Lahn BT. 2004. Adaptive evolution of ASPM, a major determinant of cerebral cortical size in humans. Hum. Mol. Genet. 13, 489–494 (doi:10.1093/hmg/ddh055) [DOI] [PubMed] [Google Scholar]
- 4.Evans PD, Gilbert SL, Mekel-Bobrov N, Vallender EJ, Anderson JR, Vaez-Azizi LM, Tishkoff SA, Hudson RR, Lahn BT. 2005. Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science 309, 1717–1720 (doi:10.1126/science.1113722) [DOI] [PubMed] [Google Scholar]
- 5.Evans PD, Vallender EJ, Lahn BT. 2006. Molecular evolution of the brain size regulator genes CDK5RAP2 and CENPJ. Gene 375, 75–79 (doi:10.1016/j.gene.2006.02.019) [DOI] [PubMed] [Google Scholar]
- 6.Mekel-Bobrov N, Gilbert SL, Evans PD, Vallender EJ, Anderson JR, Hudson RR, Tishkoff SA, Lahn BT. 2005. Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science 309, 1720–1722 (doi:10.1126/science.1116815) [DOI] [PubMed] [Google Scholar]
- 7.Pollard KS, et al. 2006. An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, 167–172 (doi:10.1038/nature05113) [DOI] [PubMed] [Google Scholar]
- 8.Fortna A, et al. 2004. Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol. 2, E207 (doi:10.1371/journal.pbio.0020207) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charrier C, et al. 2012. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell 149, 923–935 (doi:10.1016/j.cell.2012.03.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dennis MY, et al. 2012. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 149, 912–922 (doi:10.1016/j.cell.2012.03.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bush EC, Lahn BT. 2008. A genome-wide screen for noncoding elements important in primate evolution. BMC Evol. Biol. 8, 17 (doi:10.1186/1471-2148-8-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindblad-Toh K, et al. 2011. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (doi:10.1038/nature10530) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pollard KS, et al. 2006. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2, e168 (doi:10.1371/journal.pgen.0020168) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prabhakar S, Noonan JP, Paabo S, Rubin EM. 2006. Accelerated evolution of conserved noncoding sequences in humans. Science 314, 786 (doi:10.1126/science.1130738) [DOI] [PubMed] [Google Scholar]
- 15.Kamm GB, Pisciottano F, Kliger R, Franchini LF. 2013. The developmental brain gene NPAS3 contains the largest number of accelerated regulatory sequences in the human genome. Mol. Biol. Evol. 30, 1088–1102 (doi:10.1093/molbev/mst023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunskill EW, Witte DP, Shreiner AB, Potter SS. 1999. Characterization of NPAS3, a novel basic helix-loop-helix PAS gene expressed in the developing mouse nervous system. Mech. Dev. 88, 237–41 (doi:10.1016/S0925-4773(99)00182-3) [DOI] [PubMed] [Google Scholar]
- 17.Gould P, Kamnasaran D. 2011. Immunohistochemical analyses of NPAS3 expression in the developing human fetal brain. Anat. Histol. Embryol. 40, 196–203 (doi:10.1111/j.1439-0264.2010.01059.x) [DOI] [PubMed] [Google Scholar]
- 18.Brunskill EW, et al. 2005. Abnormal neurodevelopment, neurosignaling and behaviour in NPAS3-deficient mice. Eur. J. Neurosci. 22, 1265–1276 (doi:10.1111/j.1460-9568.2005.04291.x) [DOI] [PubMed] [Google Scholar]
- 19.Erbel-Sieler C, et al. 2004. Behavioral and regulatory abnormalities in mice deficient in the NPAS1 and NPAS3 transcription factors. Proc. Natl Acad. Sci. USA 101, 13 648–13 653 (doi:10.1073/pnas.0405310101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamnasaran D, Muir WJ, Ferguson-Smith MA, Cox DW. 2003. Disruption of the neuronal PAS3 gene in a family affected with schizophrenia. J. Med. Genet. 40, 325–332 (doi:10.1136/jmg.40.5.325) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pickard BS, Malloy MP, Porteous DJ, Blackwood DH, Muir WJ. 2005. Disruption of a brain transcription factor, NPAS3, is associated with schizophrenia and learning disability. Am. J. Med. Genet. B Neuropsychiatr. Genet. 136, 26–32 [DOI] [PubMed] [Google Scholar]
- 22.Pickard BS, Pieper AA, Porteous DJ, Blackwood DH, Muir WJ. 2006. The NPAS3 gene: emerging evidence for a role in psychiatric illness. Ann. Med. 38, 439–448 (doi:10.1080/07853890600946500) [DOI] [PubMed] [Google Scholar]
- 23.Yu L, Arbez N, Nucifora LG, Sell GL, Delisi LE, Ross CA, Margolis RL, Nucifora FC., Jr In press A mutation in NPAS3 segregates with mental illness in a small family. Mol. Psychiatry. (doi:10.1038/mp.2012.192) [DOI] [PubMed] [Google Scholar]
- 24.Huang J, et al. 2010. Cross-disorder genomewide analysis of schizophrenia, bipolar disorder, and depression. Am. J. Psychiatry 167, 1254–1263 (doi:10.1176/appi.ajp.2010.09091335) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pickard BS, Christoforou A, Thomson PA, Fawkes A, Evans KL, Morris SW, Porteous DJ, Blackwood DH, Muir WJ. 2008. Interacting haplotypes at the NPAS3 locus alter risk of schizophrenia and bipolar disorder. Mol. Psychiatry 14, 874–884 (doi:10.1038/mp.2008.24) [DOI] [PubMed] [Google Scholar]
- 26.Lowe CB, Bejerano G, Haussler D. 2007. Thousands of human mobile element fragments undergo strong purifying selection near developmental genes. Proc. Natl Acad. Sci. USA 104, 8005–8010 (doi:10.1073/pnas.0611223104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lowe CB, Haussler D. 2012. 29 mammalian genomes reveal novel exaptations of mobile elements for likely regulatory functions in the human genome. PLoS ONE 7, e43128 (doi:10.1371/journal.pone.0043128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brosius J, Gould SJ. 1992. On ‘genomenclature’: a comprehensive (and respectful) taxonomy for pseudogenes and other ‘junk DNA’. Proc. Natl Acad. Sci. USA 89, 10 706–10 710 (doi:10.1073/pnas.89.22.10706) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Souza FS, Franchini LF, Rubinstein M. 2013. Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong? Mol. Biol. Evol. 30, 1239–1251 (doi:10.1093/molbev/mst045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smit AF, Riggs AD. 1995. MIRs are classic, tRNA-derived SINEs that amplified before the mammalian radiation. Nucleic Acids Res. 23, 98–102 (doi:10.1093/nar/23.1.98) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilbert N, Labuda D. 1999. CORE-SINEs: eukaryotic short interspersed retroposing elements with common sequence motifs. Proc. Natl Acad. Sci. USA 96, 2869–2874 (doi:10.1073/pnas.96.6.2869) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. 2011. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 7, e1002384 (doi:10.1371/journal.pgen.1002384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bejerano G, Lowe CB, Ahituv N, King B, Siepel A, Salama SR, Rubin EM, James Kent W, Haussler D. 2006. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 441, 87–90 (doi:10.1038/nature04696) [DOI] [PubMed] [Google Scholar]
- 34.Franchini LF, Lopez-Leal R, Nasif S, Beati P, Gelman DM, Low MJ, de Souza FJS, Rubinstein M. 2011. Convergent evolution of two mammalian neuronal enhancers by sequential exaptation of unrelated retroposons. Proc. Natl Acad. Sci. USA 108, 15 270–15 275 (doi:10.1073/pnas.1104997108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pi W, Zhu X, Wu M, Wang Y, Fulzele S, Eroglu A, Ling J, Tuan D. 2010. Long-range function of an intergenic retrotransposon. Proc. Natl Acad. Sci. USA 107, 12 992–12 997 (doi:10.1073/pnas.1004139107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santangelo AM, de Souza FS, Franchini LF, Bumaschny VF, Low MJ, Rubinstein M. 2007. Ancient exaptation of a CORE-SINE retroposon into a highly conserved mammalian neuronal enhancer of the proopiomelanocortin gene. PLoS Genet. 3, 1813–1826 (doi:10.1371/journal.pgen.0030166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki T, et al. 2008. Possible involvement of SINEs in mammalian-specific brain formation. Proc. Natl Acad. Sci. USA 105, 4220–4225 (doi:10.1073/pnas.0709398105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tashiro K, et al. 2011. A mammalian conserved element derived from SINE displays enhancer properties recapitulating Satb2 expression in early-born callosal projection neurons. PLoS ONE 6, e28497 (doi:10.1371/journal.pone.0028497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King MC, Wilson AC. 1975. Evolution at two levels in humans and chimpanzees. Science 188, 107–116 (doi:10.1126/science.1090005) [DOI] [PubMed] [Google Scholar]
- 40.Ohno S. 1972. An argument for the genetic simplicity of man and other mammals. J. Hum. Evol. 1, 651–662 (doi:10.1016/0047-2484(72)90011-5) [Google Scholar]
- 41.Carroll SB. 2005. Evolution at two levels: on genes and form. PLoS Biol. 3, e245 (doi:10.1371/journal.pbio.0030245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carroll SB. 2008. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134, 25–36 (doi:10.1016/j.cell.2008.06.030) [DOI] [PubMed] [Google Scholar]
- 43.Prabhakar S, et al. 2008. Human-specific gain of function in a developmental enhancer. Science 321, 1346–1350 (doi:10.1126/science.1159974) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J, Webb DM, Podlaha O. 2002. Accelerated protein evolution and origins of human-specific features: Foxp2 as an example. Genetics 162, 1825–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Enard W, et al. 2009. A humanized version of Foxp2 affects cortico-basal ganglia circuits in mice. Cell 137, 961–971 (doi:10.1016/j.cell.2009.03.041) [DOI] [PubMed] [Google Scholar]
- 46.Oksenberg N, Stevison L, Wall JD, Ahituv N. 2013. Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 9, e1003221 (doi:10.1371/journal.pgen.1003221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poulin F, Nobrega MA, Plajzer-Frick I, Holt A, Afzal V, Rubin EM, Pennacchio LA. 2005. In vivo characterization of a vertebrate ultraconserved enhancer. Genomics 85, 774–781 (doi:10.1016/j.ygeno.2005.03.003) [DOI] [PubMed] [Google Scholar]
- 48.Sanchez-Calderon H, Martin-Partido G, Hidalgo-Sanchez M. 2005. Pax2 expression patterns in the developing chick inner ear. Gene Expr. Patterns 5, 763–773 (doi:10.1016/j.modgep.2005.04.007) [DOI] [PubMed] [Google Scholar]