

Abstract
In this paper, we provide a historical account of the contribution of a single line of research to our current understanding of the structure of cis-regulatory regions and the genetic basis for morphological evolution. We revisit the experiments that shed light on the evolution of larval cuticular patterns within the genus Drosophila and the evolution and structure of the shavenbaby gene. We describe the experiments that led to the discovery that multiple genetic changes in the cis-regulatory region of shavenbaby caused the loss of dorsal cuticular hairs (quaternary trichomes) in first instar larvae of Drosophila sechellia. We also discuss the experiments that showed that the convergent loss of quaternary trichomes in D. sechellia and Drosophila ezoana was generated by parallel genetic changes in orthologous enhancers of shavenbaby. We discuss the observation that multiple shavenbaby enhancers drive overlapping patterns of expression in the embryo and that these apparently redundant enhancers ensure robust shavenbaby expression and trichome morphogenesis under stressful conditions. All together, these data, collected over 13 years, provide a fundamental case study in the fields of gene regulation and morphological evolution, and highlight the importance of prolonged, detailed studies of single genes.
Keywords: shavenbaby gene, morphological change, cis-regulatory evolution, cis-regulatory structure, evolution of enhancer function
1. Introduction
This is a historical account of the contribution of a single line of research to our current understanding of the structure of cis-regulatory regions and the genetic basis for morphological evolution. It is not a comprehensive review of these subjects. Instead, this paper will review and expand upon the more concise papers that we have published on the topic over the past 13 years.
At the end of the twentieth century, few studies had provided evidence that evolution of a single locus had contributed to a morphological difference between species. At that time, multiple studies had shown that the expression patterns of genes that regulate development often were correlated with morphological differences between species [1–3]. These studies were performed usually by comparisons of distantly related taxa that involved large changes in the body plan, such as the distribution of limb types along an animal's body axis [4]. Such studies suggested that changes in expression patterns of key developmental regulatory genes might contribute to macroevolutionary patterns of morphological change, but there was little direct evidence for how individual genes contributed to morphological evolution.
For several reasons, it was also far from clear how the relevant evidence might be gathered. Comparisons between distantly related taxa were suspected to involve many genetic changes, some of which might have long ago obscured the key genetic events leading to morphological divergence. Comparisons between closely related species provided an obvious escape from this problem, but such studies involved multiple barriers. First, closely related species display mainly quantitative differences in morphological features and few qualitative differences. Most such quantitative differences were believed to result from changes at multiple loci and quantitative trait locus mapping had long failed to provide insights into the contributions of individual loci. Second, most closely related species cannot be hybridized, which eliminates a genetic approach to allow unbiased discovery of evolutionarily relevant mutations that contribute to species differences. These barriers imposed significant limitations to the study of the genetic causes of evolutionary change.
After working on the trichomes (hair-like projections of epidermal cells, formally called microtrichia) produced on adult legs for some years [5] and inspired by a paper by Dickinson et al. [6], David Stern encouraged his first graduate student, Elio Sucena, to examine the trichomes on first-instar larvae of species of the Drosophila melanogaster subgroup, hoping that some quantitative differences might be found and that these differences might be tractable targets of interspecific genetic analysis. Elio quickly discovered that larvae of Drosophila sechellia lacked a large swathe of trichomes on the dorsal and lateral regions of all body segments, a region of the so-called quaternary cells (figure 1; [7]). All other species in the species group retained the D. melanogaster-like pattern of quaternary trichomes and, by examining the distribution of these phenotypes on a phylogeny for the group, we found that D. sechellia evolved loss of quaternary trichomes within about the past 500 000 years. That is where our story begins. We will tell the story largely in chronological order with a few temporal rearrangements to clarify the biology.
Figure 1.
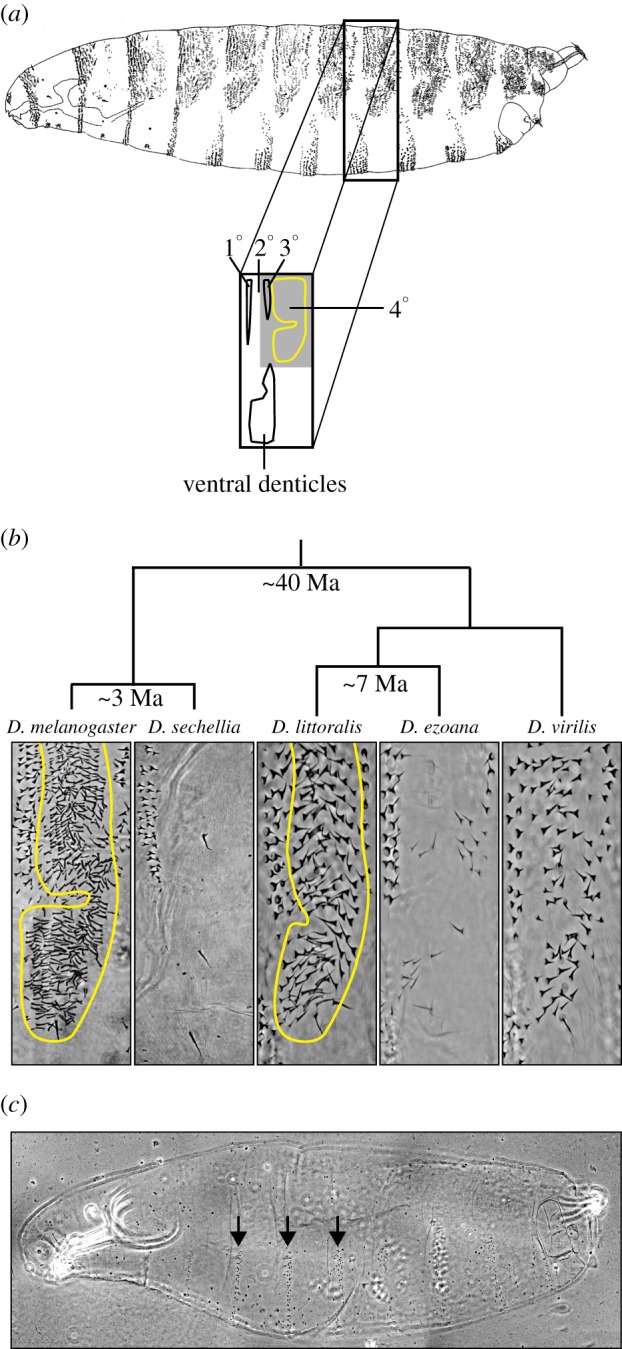
Trichome pattern variation in first-instar larvae of Drosophila species. (a) Drawing from the lateral perspective of a Drosohphila melanogaster first-instar larva. The black rectangle demarcates the fifth abdominal segment. On the dorsal cuticle, the primary (1°), tertiary (3°) and quaternary (4°) cells (light outline) differentiate trichomes, and the secondary (2°) cells differentiate naked cuticle. A group of stout trichomes (denticles) is present in the ventral cuticle. The grey area within the rectangle indicates the cuticle region shown in (b). (b) Detail of the dorsal cuticle in different species of the genus Drosophila. The quaternary cells of D. sechellia and D. ezoana produce ‘naked’ cuticle. By contrast, D. melanogaster, D. littoralis and D. virilis produce ‘hairy’ cuticles in the quaternary domain (light outline). (c) A svb null first-instar larva lacks dorsal and lateral trichomes and has fewer ventral denticles that are also reduced in size relative to wild-type ventral denticles (arrows). (Online version in colour.)
2. Trichome patterning differences between Drosophila sechellia and closely related species are due to changes in the transcriptional regulation of shavenbaby
A series of genetic crosses between Drosophila simulans and D. sechellia showed that the absence of quaternary trichomes segregated as a single Mendelian locus located on the X chromosome [8]. The D. sechellia allele (the ‘naked’ allele) was recessive to the ‘hairy’ alleles derived from D. simulans and from D. melanogaster. The Mendelian behaviour of this locus provided an unprecedented opportunity to study the genetic cause of morphological evolution. The fact that the D. sechellia allele was recessive to the D. melanogaster allele allowed us to localize the causal locus by performing deficiency mapping with existing D. melanogaster stocks carrying deletions of defined chromosomal regions. This analysis revealed that the causal locus was located in a region containing ten genes, only one of which, shavenbaby/ovo, was known to regulate trichome development. Further linkage analysis of the X chromosome using visible markers that could be scored in the larvae confirmed that the evolved locus co-segregated with the genomic region containing shavenbaby/ovo. Later, high-resolution genetic studies, described in §5, provided further genetic evidence that svb was the causal locus.
While D. sechellia had lost quaternary trichomes, svb null alleles caused loss of almost all trichomes on the first-instar larva (figure 1c); hence the gene name [9]. This observation was the first indication that the evolved svb allele represented a partial loss of function. In situ hybridization against svb mRNA in D. melanogaster and D. sechellia revealed that the expression pattern of svb closely matched the species-specific pattern of trichomes. This suggested that the evolved activity of the D. sechellia allele was caused by changes in the transcriptional regulation of svb. At this point in the story, it is worth taking a brief interlude to explain what is known about svb function, much of which has been revealed by the efforts of the laboratory of François Payre and Serge Plaza.
3. Shavenbaby: a master control gene that orchestrates the development of cuticular trichomes
The shavenbaby mRNA is one of three mRNAs produced by the ovo locus [10,11]. The other two mRNAs, ovoA and ovoB, are expressed only in the germline and are required for proper ovary development [10,11]. Svb is expressed in the epidermis of the embryo (figure 2a), and this expression is sufficient to induce cell-autonomous development of trichomes [12]. The activation of Svb protein requires the truncation of its N-terminus. The details of this post-translational step are not yet completely understood, but this modification is mediated by products of the polished rice (pri) gene, a polycistronic gene that encodes four short peptides [13]. Svb is required also for development of some of the trichomes on the adult cuticle [11] and for development of tarsal joints [14]. The complex expression pattern of svb suggests that this protein may have additional functions in tissues other than the epidermis.
Figure 2.

Shavenbaby embryonic expression is generated by the activity of multiple transcriptional enhancers. (a) Expression pattern of shavenbaby in the epidermis of a late stage Drosophila melanogaster embryo. (b) Schematic view of the structure of the svb/ovo locus. Black boxes indicate svb embryonic enhancers and grey boxes demarcate exons. The exons present in the svb mRNA are shown below this scheme. Svb protein contains a dominant repressor domain, an activator domain and a DNA-binding domain. The arrow marks the position where Svb is truncated, which converts Svb into a transcriptional activator. (c) Seven transcriptional enhancers generate the expression pattern of shavenbaby in the embryonic epidermis. The black regions within trichome domains schematize the expression patterns of enhancers DG2, DG3, Z, A, E3, E6 and 7. (Online version in colour.)
Genes upstream and downstream of svb have been identified. The embryonic expression pattern of svb is generated by the integration of many signalling pathways (Wnt, EFG-r, Hedgehog and Notch; [15]). In turn, Svb regulates directly or indirectly dozens of genes that participate in trichome morphogenesis [16,17], a complex cellular processes that involves the actin cytoskeleton [16]. The regulatory network governing trichome development thus resembles an ‘hourglass’ [11,18], with svb occupying a bottleneck, or master control position, in the flow of regulatory information. We have hypothesized that the position of svb in this regulatory network predisposes it to accumulate evolutionarily relevant mutations influencing trichome patterning [18].
Homologues of the ovo gene are found in vertebrates. In mice, ovo is required for hair development [19]. Is this a remarkable coincidence, or does this reflect an important evolutionary pattern? Mammalian hairs are obviously not homologues of insect trichomes. However, both insect trichomes and mammalian hairs are produced by ectodermal cells. To the best of our knowledge, nobody has tried to trace the evolutionary history of the expression patterns of svb/ovo across diverse ecdysozoans and deutorostomes, but it is possible that the involvement of svb/ovo in development of ectodermal structures reflects an ancestral pattern of svb/ovo expression in ectoderm and that this protein has been coopted into regulatory networks for development of diverse ectodermal structures in different lineages.
4. Regulation of svb embryonic epidermal expression
The embryonic expression pattern of svb (figure 2a) has been investigated almost entirely using reporter gene assays, where a segment of genomic DNA is tested for regulatory activity by attaching it to a minimal promoter and a reporter gene. We used this method to perform a comprehensive screen of the 90 kb upstream of the svb first exon. Previous analyses had shown that the large first intron of svb is devoid of regulatory elements that drive epidermal embryonic expression [20]. In summary, we discovered seven discrete regions that drive patterns of reporter gene expression that recapitulate part of the complete svb expression pattern and, all together, appear to recapitulate the complete svb expression pattern (figure 2b,c). These enhancers could not have been identified simply by comparing sequence conservation in the svb upstream region across Drosophila species, because enhancers and regions that do not appear to encode embryonic enhancers display similar levels of sequence conservation (figure 3).
Figure 3.

Pattern of sequence conservation in the shavenbaby cis-regulatory region. Scheme of a multiple sequence alignment of the shavenbaby upstream region in 12 Drosophila species (bottom). Mel, D. melanogaster; sech, D. sechellia; sim, D. simulans; yak, D. yakuba; ere, D. erecta; ana, D. ananassae; per, D. persimilis; pse, D. pseudoobscura; wil, D. willistoni; moj, D. mojavensis; vir, D. virilis; gri, D. grimshawii. Empty boxes indicate the position of svb embryonic enhancers and triangles indicate coding regions. Z678, A8 and 7H are the ‘minimal’ versions of enhancers Z, A and 7, respectively. The pattern of sequence conservation is shown in grey (top). Sequence identity was calculated in 100 bp windows.
We and our collaborators have dissected the five enhancers most proximal to the svb promoter to ‘minimal’ enhancer regions of approximately 500–1000 bp ([21]; G. K. Davis, A. P. McGregor, N. Frankel, F. Payre & D. Stern 2007–2013, unpublished data). The remaining two enhancers are delimited to regions of approximately 5 kb. For svb enhancers, the minimal fragment size to generate a coherent expression pattern is approximately 500 bp, because smaller fragments often show ectopic or very weak expression patterns ([21]; G. K. Davis, A. P. McGregor, N. Frankel, F. Payre & D. Stern 2007–2013, unpublished data). Nevertheless, even fragments of approximately 1000 bp may not contain all of the regulatory information required for proper regulation of expression. For example, D. melanogaster E6, a fragment of approximately 1000 bp, shows a correct expression onset when compared with the native gene, but continues to drive expression after the native gene has been shut off [21]. We do not know the source of this missing temporal regulation.
The expression patterns of the seven enhancers are largely complementary. For example, multiple enhancers drive expression in ventral cells, but each enhancer appears to drive in a different subset of cells (J. Crocker & D. Stern 2013, unpublished data). Likewise, multiple enhancers drive in largely complementary patterns in dorsal and lateral cells. Despite the fact that most enhancers drive expression in complementary patterns, we also found that many anatomical domains receive regulatory input from multiple enhancers. This is particularly pronounced for the quaternary cells, where multiple enhancers drive overlapping expression in the same cells ([22]; figure 2). We found this apparent redundancy of expression patterns puzzling and sought to explore the function of this redundancy using genetic approaches. Using reagents that allow targeted genomic modifications in D. melanogaster [23], we generated a precise deletion of 32 kb that removed enhancers Z, DG2 and DG3. Flies homozygous for this deletion were viable and fertile and, to our surprise, first-instar larvae displayed qualitatively normal patterns of trichomes in standard laboratory conditions. Nicolás Frankel hypothesized that these enhancers might function to confer robustness (i.e. canalization) on the phenotype in the face of environmental or genetic variability [24]. To test this idea, we reared embryos at temperatures at the extremes of what they might experience in the wild (17°C and 32°C), and, in a separate experiment, we reduced the levels of a svb activator called wingless [22]. Embryos carrying the deletion and experiencing extreme temperatures displayed a quantitative loss of trichomes in the quaternary domain where the Z and DG2 enhancers drive expression. Moreover, trichomes that were produced by these embryos were smaller and misshapen compared with those of the controls. Larvae carrying the deletion in a genetic background carrying one copy of the wingless gene also produced significantly fewer trichomes. By contrast, neither temperature stress nor the lack of one copy of wingless affected the number of trichomes in control flies. Hence, the genomic region containing the Z and DG2 enhancers is dispensable under optimal growth conditions but confers robustness under stressful conditions. We observed no loss of trichomes or any other obvious morphological changes in the ventral region, where the DG3 enhancer drives expression.
To confirm that the loss of canalization resulted from the loss of the Z and DG2 enhancers, and not from the removal of other DNA in the 32 kb deleted, we constructed a plasmid carrying the Z enhancer driving a svb cDNA and generated flies that carried this construct together with the 32 kb deletion. This construct rescued canalization only in the domain where the Z enhancer drives expression, providing compelling evidence that canalization results from expression driven at a sufficient level by multiple enhancers with overlapping expression patterns [22]. Some epidermal domains do not show overlapping expression patterns driven by multiple enhancers, and it is possible that the level of expression driven by single enhancers in these regions is sufficient to provide a robust phenotypic output under variable conditions.
This buffering mechanism is not unique to the shavenbaby gene. Expression of the Drosophila gene snail is also canalized through multiple enhancers [25]. Furthermore, the presence of enhancers with overlapping expression patterns seems to be a common cis-regulatory signature of developmental genes in both flies and mice [26]. Given the abundance of such enhancers in Drosophila and the mouse genome, it seems likely that apparently redundant enhancers confer canalization on gene expression and on the phenotype in diverse taxa.
5. The genetic changes underlying morphological evolution in Drosophila sechellia
We had shown previously that changes in transcriptional regulation of svb lead to production of naked cuticle, rather than quaternary trichomes, in D. sechellia [8]. Moreover, mapping evidence suggested that the causal genetic changes had occurred at the svb locus [8]. Therefore, one possible explanation for the loss of svb expression in D. sechellia was that all of the enhancers that drive expression in quaternary cells had lost their activity in the D. sechellia lineage. To test this hypothesis, we cloned DNA fragments from D. sechellia homologous to the five enhancers of D. melanogaster that drive expression in quaternary cells [22,27]. These species diverged about 3 Myr ago and their genomes are sufficiently similar that these homologous regions could be identified unambiguously. These DNA regions were tested in reporter gene assays within D. melanogaster. In each case, the D. sechellia orthologue showed remarkably lower levels of expression in quaternary cells than did the D. melanogaster enhancer [22,27]. This implied that the function of all five enhancers had evolved in the D. sechellia lineage.
The analysis of D. sechellia enhancers in D. melanogaster embryos alone could not rule out the possibility that additional linked loci had evolved and contributed to the absence of quaternary trichomes in D. sechellia. To test this hypothesis, we performed a high-resolution recombination mapping study of the svb locus [27]. We screened about 10 000 flies derived from a cross between D. sechellia and a strain of the ‘hairy’ species Drosophila mauritiana that carried phenotypic markers closely linked to svb, which allowed the identification of flies with recombination events within and around the svb locus. We isolated about 60 flies with recombination events within the svb locus. These recombinants demonstrated that genetic changes at the svb locus alone were sufficient to generate the phenotypic differences. In addition, larvae that carried the entire svb cis-regulatory region from D. sechellia produced no quaternary trichomes, even when they carried the entire coding region of svb from D. mauritiana. Finally, this experiment confirmed that multiple svb enhancers had evolved in D. sechellia to generate the loss of trichomes in different anatomical domains [27].
Hence, both reporter constructs and fine-scale recombination mapping supported the conclusion that multiple svb enhancers evolved to generate the naked cuticle phenotype in D. sechellia. The loss of quaternary trichomes is an example of a species difference controlled by variation at a single gene, yet multiple cis-regulatory changes have accumulated at this single locus [22,27].
6. Genetic changes in multiple enhancers and multiple changes within enhancers
Given the fact that multiple enhancers had evolved at svb, we then set out to characterize in further molecular detail how one of these enhancers had evolved. We focused on enhancer E6, which drives strong expression in quaternary cells in D. melanogaster (figure 4a) and in D. simulans, and which drives very weak expression in D. sechellia (figure 4a).
Figure 4.

Genetic changes underlying the evolution of enhancer E6 in D. sechellia. (a) Lateral view of the expression driven by enhancer E6 from D. melanogaster and D. sechellia in stage 15 embryos of D. melanogaster. (b) Selected regions of the full-length alignment of D. melanogaster (mel), D. sechellia (sec) and D. simulans (sim) E6 enhancer that include the seven clusters of sites uniquely derived in D. sechellia E6 (marked in bold). These sites were mutated in the rescue constructs depicted in (c). (c) Scheme of the constructs used to test the effects of mutations (above). Number of trichomes produced by E6 variants in a svb null background (graphs below). Number of trichomes rescued by the D. melanogaster (circles) and D. sechellia (triangles) constructs in the dorsal (left) and lateral (right) regions of the cuticle. Circles and triangles denote the mean number of trichomes. Vertical lines represent ± 1s.d. (n = 10). Larvae carrying D. sechellia rescue constructs produced zero trichomes in the lateral region and are not shown in the figure on the right. WT, wild-type construct. ALL, construct with all clusters mutated. Clusters highlighted in bold (x-axis) caused a significant change in the number of trichomes produced compared with the wild-type construct. (Online version in colour.)
Alignment of the E6 region from multiple species revealed 14 genetic changes that were uniquely derived in the D. sechellia sequence (figure 4b). Further sequence analysis showed that the D. sechellia region containing the 14 changes had evolved rapidly, consistent with the idea of positive selection in the D. sechellia lineage [21]. Some of these 14 sites were located very close to one other, and we therefore decided to test the effects of groups of these sites, resulting in seven candidate ‘clusters’ (figure 4b). We designed a series of ‘rescue constructs’ that allowed us to assay the phenotypic effects of genetic changes in E6. These constructs, which were all integrated into the same location in the D. melanogaster genome to control for position effects, carried the cDNA of svb downstream of different versions of E6 (figure 4c). To test the contribution of each of the clusters of evolved sites, we generated site-specific mutations in E6 within the context of a larger fragment that also included the E3 enhancer, located about 2 kb upstream of E6 (figure 4c). The E3 enhancer from all species in the D. melanogaster species group drives reporter gene expression in ventral cells and this ventral expression served as an internal control. Sites in the seven clusters were altered separately from the D. melanogaster state to the D. sechellia state in a D. melanogaster fragment, and vice versa for the D. sechellia fragment, and then two additional constructs were made that contained all of the 14 changes in the reciprocal directions. We then measured the activity of these rescue constructs and quantified the number of trichomes produced in a svb null background (figure 4c).
All constructs produced similar numbers of ventral trichomes, indicating that E3 (the internal positive control) generated consistent results despite changes in E6. In addition, the D. melanogaster wild-type rescue construct recovered many, but not all, quaternary trichomes. This was expected, because multiple enhancers are required to produce the complete pattern of quaternary trichomes [22,27]. The D. sechellia wild-type construct produced a small number of trichomes, consistent with the very low levels of reporter gene expression driven by the D. sechellia E6 enhancer (the residual expression from the D. sechellia E6 enhancer is likely to be an artefact of the reporter construct, because svb mRNA is not detected in quaternary cells of D. sechellia and larvae of this species do not produce quaternary trichomes). The D. melanogaster construct carrying all of the D. sechellia-specific changes caused a large decrease in the number of trichomes compared with the construct carrying the wild-type D. melanogaster E6. Likewise, the reciprocal changes in the D. sechellia construct caused a large increase in the number of trichomes produced. In both cases, however, the 14 changes did not recapitulate the number of trichomes produced by the wild-type E6 construct from the other species (figure 4c). This observation indicates that at least one of the clusters affects the activity of E6 and that at least one other change (other than the 14 targeted) contributes to the function of the E6 region. Four of the seven clusters caused small changes in trichome numbers (figure 4c), ranging from 5% to 30% of the total effect [21]. The sum of the effect sizes of individual clusters did not equal the effect size of the construct with all 14 changes, indicating the existence of epistatic interactions between sites. It is possible that transcription factors that recognize different sites interact to generate this epistasis.
In conclusion, at least five changes with small phenotypic effects evolved in the D. sechellia E6 enhancer to influence trichome patterning. In total, we have so far documented that at least nine mutations in the svb cis-regulatory region generated what, at first sight, seemed like a simple morphological difference between species. It is striking that all substitutions affected the phenotype in the direction consistent with the species differences, providing weak evidence that these substitutions evolved under positive selection [28].
7. Morphological convergence caused by parallel cis-regulatory changes at the shavenbaby locus
The pattern of quaternary trichomes has evolved in only a few Drosophila species ([8,29]; D. Stern 2000–2013, unpublished data). Within the Drosophila virilis group, which diverged from D. melanogaster approximately 40 Myr ago, several species have evolved loss of quaternary trichomes similar to D. sechellia [29]. Genetic studies between species of the D. virilis species group showed that trichome differences appeared to segregate as a single Mendelian locus on the X chromosome (the location of the svb locus), with the hairy phenotype dominant to the naked phenotype. Furthermore, the pattern of svb expression in the D. virilis group species closely matched the final pattern of trichomes in each species [29]. In addition, previous studies had demonstrated that multiple genes that act upstream of svb display conserved expression patterns across the D. virilis species group [6]. In addition, we showed that the function of shavenbaby as a genetic switch for trichome development has been evolutionarily conserved, because knockdown of svb levels in D. virilis leads to loss of larval trichomes [30]. Combined, these results suggested that the differences between hairy and naked species in the D. virilis group resulted from regulatory variation at svb or at another X-linked gene that regulates svb [29].
If the observed differences in svb expression in the D. virilis group resulted from changes to the svb cis-regulatory region, and not from changes in a trans regulatory factor, then there should be differences in the regulatory activity of svb enhancers. To test this hypothesis, we needed first to identify the svb enhancers in species of the D. virilis group. Within this group, the hairy species D. virilis was the only member with a sequenced genome, a fact that led us to explore the sequence of the svb locus in the D. virilis genome. We found that the putative approximately 132 kb cis-regulatory region of shavenbaby in D. virilis showed low levels of conservation with the approximately 90 kb shavenbaby cis-regulatory region from D. melanogaster. However, we did find multiple regions of 30 bp or longer that were identical between the two species (‘anchors’). These ‘anchors’ were scattered across the entire cis-regulatory region and did not cluster in or near characterized enhancers. We also did not observe islands of conservation corresponding to the characterized D. melanogaster enhancers in a multiple-species alignment of the svb locus (figure 3). These observations raised the possibility that the positions of the svb enhancers might be evolutionarily flexible.
Given the lack of promising candidate regions, we decided that the safest course of action was to perform an unbiased, comprehensive reporter gene analysis of the entire 132 kb region upstream of the svb first exon in D. virilis [30]. This experiment revealed six regions that drove reporter expression in patterns similar to parts of the native svb mRNA embryonic expression pattern. To our surprise, the D. virilis and D. melanogaster svb enhancers were located in approximately the same genomic positions (‘anchors’ served as landmarks for comparisons) and positional homologues drove extremely similar expression patterns ([30]; figures 5 and 6). We found only two exceptions. First, the D. melanogaster A enhancer does not appear to have a homologue in D. virilis. Second, expression in primary and tertiary cells driven by D. melanogaster 7 has been lost in the positional homologue of D. virilis and, instead, we observed a similar pattern driven by enhancer 24 in D. virilis (figure 6). In conclusion, six enhancer pairs appeared to be orthologues between D. virilis and D. melanogaster. Reciprocal BLAST analyses (using enhancer sequences as queries) revealed weak, but significant, sequence similarity, confirming the hypothesis that these enhancers are indeed orthologous. Previous reports had shown that individual enhancers retain their relative positions in distantly related species [31–33]. The positional conservation of most enhancers in the cis-regulatory region of svb raises additional questions, as yet unanswered, about whether a particular spatial arrangement of enhancers is required for svb function.
Figure 5.
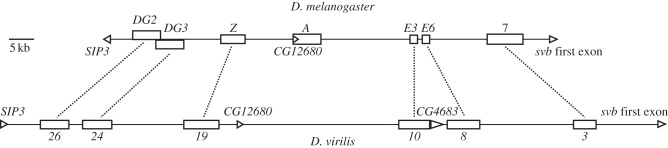
Conservation of shavenbaby cis-regulatory structure between D. melanogaster and D. virilis. Horizontal lines schematize the svb cis-regulatory region in D. melanogaster (above) and D. virilis (below). Empty boxes indicate the position of transcriptional enhancers. Dotted lines connect orthologous enhancers. Triangles denote coding regions.
Figure 6.

Parallel changes in orthologous enhancers underlie the appearance of convergent morphologies in D. sechellia and D. ezoana. The black regions within trichome domains (figure 1) schematize the expression patterns of shavenbaby enhancers in (a) D. melanogaster, (b) D. sechellia, (c) D. virilis and (d) D. ezoana. Orthologous enhancers are positioned in the same column. Dotted boxes highlight the loss of enhancer activity in D. sechellia and D. ezoana. NT, not tested.
Once we had identified the D. virilis enhancers, we tested whether changes to the svb cis-regulatory region in species from the D. virilis species group could explain the observed differences in svb expression patterns, and the resulting trichome patterns. We focused on two sister species, Drosophila ezoana, which does not produce any quaternary trichomes and D. littoralis, which produces abundant quaternary trichomes (figure 1). The convergent phenotypes of D. sechellia and D. ezoana (figure 1) are mirrored by parallel loss of svb expression in quaternary cells in both species [8,29]. In the D. sechellia lineage, enhancers DG2, Z, A, E6 and 7 lost their activity in quaternary cells, resulting in production of ‘naked’ cuticle (figure 6). In D. virilis, expression of svb in quaternary cells is determined mostly by enhancers 8 and 19 (the orthologues of D. melanogaster Z and E6; figure 6; [30]). We therefore tested the activity of enhancers 8 and 19 from D. ezoana and D. littoralis. As a control, we analysed the activity of enhancer 3, a regulatory element expressed in ventral cells in D. virilis (figure 6). The cloned enhancer fragments were tested with reporter assays in transgenic D. virilis embryos, thus providing a trans regulatory environment capable of driving expression of svb enhancers in quaternary cells. We observed that all three D. littoralis enhancers drove expression in the same spatial domains as the orthologous D. virilis enhancers. By contrast, D. ezoana enhancers 8 and 19 were completely inactive, whereas enhancer 3 drove expression in ventral rows ([30]; figure 6). The lack of activity of enhancers 8 and 19 is consistent with the loss of expression of svb in quaternary cells and with the cuticle pattern of D. ezoana (figure 1).
These observations suggest that convergence in cuticular morphology between D. sechellia and D. ezoana was caused by changes in orthologous enhancers of shavenbaby (figure 6). That is, parallel genetic changes in distantly related lineages generated similar phenotypes [30].
8. Concluding remarks
We are often asked what function the quaternary trichomes on first-instar larvae serve and why these trichomes have been lost in D. sechellia and in other species, such as D. ezoana. At the moment, the answers to both questions are unknown. Curiously, all species of the D. melanogaster species group, and many other drosophilids, produce naked cuticle in the quaternary domain in second and third instar larvae ([34]; D. Stern & N. Frankel 2000–2013, unpublished data), suggesting that the largely conserved pattern of quaternary trichomes is important to first-instar larvae. Whatever the ecological function of these trichomes, our work on svb has revealed that we can gain significant insights into genetic evolution even when we do not know the ecological setting in which particular phenotypic features have evolved.
We have discovered that multiple svb enhancers drive overlapping patterns of expression. This apparent redundancy ensures robust svb expression and trichome morphogenesis under stressful conditions. We used the genetic tools available in D. melanogaster to generate a precise deletion within the svb locus and exploited the production of trichomes as a quantitative readout of svb activity. This quantitative readout of svb activity, a unique feature of this system among the currently available systems for studying morphological evolution, also allowed us to discover that a seemingly simple morphological transition, the loss of quaternary trichomes in first-instar larvae, required a large number of small-effect substitutions. The observation that, during evolution, a single locus can accumulate many substitutions of small effect may harmonize seemingly divergent views of the genetic basis for evolution. Classical population genetics theory and many empirical studies of the genetic basis for phenotypic evolution within and between populations have consistently supported the view that evolution occurs through genetic changes of relatively small phenotypic effect [35,36]. By contrast, other studies of phenotypic variation between species have often revealed variation at single loci with a substantial phenotypic effect [37–39]. Our results and those of others [40–44] suggest that, at least sometimes, loci of large effect may have evolved through the accumulation of many mutations of much smaller effect. Most studies of the genetic causes of phenotypic variation have not reached the level of resolution provided by our studies of svb, so it is not yet clear whether the patterns we have observed are true more generally.
Finally, we uncovered that the convergent loss of dorsal trichomes in D. sechellia and D. ezoana was generated by parallel genetic changes in orthologous enhancers of svb. This case and others suggest that, often, similar genetic routes underlie the appearance of repeated phenotypes [18]. Evolutionarily relevant genetic changes can be clustered in genes that act in key positions in developmental networks. The position of svb within the developmental network defining trichome development and the observed changes in svb enhancers suggest, for the following reasons, that these genetic modifications were selected due to their limited pleiotropic effects [18]. First, the svb cis-regulatory region integrates signals from many patterning systems active in the early embryo. Modifications to genes active earlier in development could modify trichome patterns, but these changes would also be likely to modify other phenotypic features. Second, svb acts as a genetic switch, directly and indirectly regulating many genes required for trichome development. Thus, changes in svb expression influence an entire module of morphogenesis. Third, changes in the cis-regulatory regions that drive expression in specific anatomical domains limit changes in gene expression to just those domains, providing the greatest specificity of effect on the phenotype while, simultaneously, regulating an entire module of morphogenesis.
While our detailed studies have revealed much about the structure and evolution of the svb locus, many important facts remain unresolved. First, we do not yet know the identity of the transcription factors that regulate the activity of svb enhancers. Most of the sites implicated in our dissection of the E6 enhancer do not resemble known transcription factor binding sites. Second, we do not know whether, in the native context of the svb cis-regulatory region, enhancers act independently or whether they interact to modulate svb expression levels. Recent reports demonstrate that the multiple regulatory elements of a gene display physical interactions, and that these interactions are critical for proper gene expression [45,46]. The evolutionary conservation of svb cis-regulatory structure suggests that the position and spacing of individual enhancers may be important to foster physical interactions. Third, we do not understand why the embryonic enhancers are positioned so far apart in a large genomic region. The DNA flanking enhancers is approximately as well conserved as are the enhancers themselves and it is likely that these intervening regions encode functional elements. Previously, it has been almost impossible to ask specific questions about large genomic regions such as the svb cis-regulatory region. However, the recent development of technologies for manipulating large DNA fragments [47] and for easier transgene integration [48,49] provide new opportunities to address these questions about the large-scale structure of genes.
Occasionally, systems biology approaches for the study of cis-regulation are judged superior to single-gene studies. However, high-throughput studies involving hundreds or thousands of genes cannot provide the type of cis-regulatory information that can be obtained with the precise analysis of a single gene. In the svb case, the simple and quantitative phenotypic readout of svb function enabled our assays of genetic structure and phenotypic evolution. These advantages will continue to make the svb gene and the evolution of trichome patterning a valuable system for studying fundamental questions for years to come.
Funding statement
N.F. is supported by grants from Fundación Bunge y Born (FByB), Agencia Nacional de Promoción Científica y Tecnológica (PICT 0428-2011) and Consejo Nacional de Investigaciones Científicas y Técnicas (PIP 11220110100745, 2012–2014). N.F. is a career investigator of CONICET.
References
- 1.Averof M, Akam M. 1995. Hox genes and the diversification of insect and crustacean body plans. Nature 376, 420–423 (doi:10.1038/376420a0) [DOI] [PubMed] [Google Scholar]
- 2.Cohn MJ, Tickle C. 1999. Developmental basis of limblessness and axial patterning in snakes. Nature 399, 474–479 (doi:10.1038/20944) [DOI] [PubMed] [Google Scholar]
- 3.Warren RW, Nagy L, Selegue J, Gates J, Carroll S. 1994. Evolution of homeotic gene regulation and function in flies and butterflies. Nature 372, 458–461 (doi:10.1038/372458a0) [DOI] [PubMed] [Google Scholar]
- 4.Averof M, Patel NH. 1997. Crustacean appendage evolution associated with changes in Hox gene expression. Nature 388, 682–686 (doi:10.1038/41786) [DOI] [PubMed] [Google Scholar]
- 5.Stern DL. 1998. A role of Ultrabithorax in morphological differences between Drosophila species. Nature 396, 463–466 (doi:10.1038/24863) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickinson WJ, Tang Y, Schuske K, Akam M. 1993. Conservation of molecular prepatterns during the evolution of cuticle morphology in Drosophila larvae. Evolution 47, 1396–1406 (doi:10.2307/2410155) [DOI] [PubMed] [Google Scholar]
- 7.Bokor P, DiNardo S. 1996. The roles of hedgehog, wingless and lines in patterning the dorsal epidermis in Drosophila. Development 122, 1083–1092 [DOI] [PubMed] [Google Scholar]
- 8.Sucena E, Stern DL. 2000. Divergence of larval morphology between Drosophila sechellia and its sibling species caused by cis-regulatory evolution of ovo/shaven-baby. Proc. Natl Acad. Sci. USA 97, 4530–4534 (doi:10.1073/pnas.97.9.4530) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wieschaus E, Nusslein-Volhard C, Jurgens G. 1984. Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. Roux's Arch. Dev. Biol. 193, 296–307 (doi:10.1007/BF00848158) [DOI] [PubMed] [Google Scholar]
- 10.Mevel-Ninio M, Terracol R, Salles C, Vincent A, Payre F. 1995. Ovo, a Drosophila gene required for ovarian development, is specifically expressed in the germline and shares most of its coding sequences with shavenbaby, a gene involved in embryo patterning. Mech. Dev. 49, 83–95 (doi:10.1016/0925-4773(94)00305-7) [DOI] [PubMed] [Google Scholar]
- 11.Delon I, Chanut-Delalande H, Payre F. 2003. The ovo/shavenbaby transcription factor specifies actin remodelling during epidermal differentiation in Drosophila. Mech. Dev. 120, 747–758 (doi:10.1016/S0925-4773(03)00081-9) [DOI] [PubMed] [Google Scholar]
- 12.Payre F, Vincent A, Carreno S. 1999. Ovo/svb integrates wingless and DER pathways to control epidermis differentiation. Nature 400, 271–275 (doi:10.1038/22330) [DOI] [PubMed] [Google Scholar]
- 13.Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, Kobayashi S, Payre F, Kageyama Y. 2010. Small peptides switch the transcriptional activity of shavenbaby during Drosophila embryogenesis. Science 329, 336–339 (doi:10.1126/science.1188158) [DOI] [PubMed] [Google Scholar]
- 14.Pueyo JI, Couso JP. 2011. Tarsal-less peptides control notch signalling through the shavenbaby transcription factor. Dev. Biol. 355, 183–193 (doi:10.1016/j.ydbio.2011.03.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Payre F. 2004. Genetic control of epidermis differentiation in Drosophila. Int. J. Dev. Biol. 48, 207–215 (doi:10.1387/ijdb.15272387) [PubMed] [Google Scholar]
- 16.Chanut-Delalande H, Fernandes I, Roch F, Payre F, Plaza S. 2006. Shavenbaby couples patterning to epidermal cell shape control. PLoS Biol. 4, e290 (doi:10.1371/journal.pbio.0040290) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandes I, Chanut-Delalande H, Ferrer P, Latapie Y, Waltzer L, Affolter M, Payre F, Plaza S. 2010. Zona pellucida domain proteins remodel the apical compartment for localized cell shape changes. Dev. Cell 18, 64–76 (doi:10.1016/j.devcel.2009.11.009) [DOI] [PubMed] [Google Scholar]
- 18.Stern DL, Orgogozo V. 2009. Is genetic evolution predictable? Science 323, 746–751 (doi:10.1126/science.1158997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai X, Schonbaum C, Degenstein L, Bai W, Mahowald A, Fuchs E. 1998. The ovo gene required for cuticle formation and oogenesis in flies is involved in hair formation and spermatogenesis in mice. Genes Dev. 12, 3452–3463 (doi:10.1101/gad.12.21.3452) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delon I.2003. Etude de la morphogénèse au cours du développement et de l’évolution: caractérisation fonctionnelle du gène ovo/shavenbaby chez la drosophile. These de doctorat de la Université de Toulouse, France.
- 21.Frankel N, Erezyilmaz DF, McGregor AP, Wang S, Payre F, Stern DL. 2011. Morphological evolution caused by many subtle-effect substitutions in regulatory DNA. Nature 474, 598–603 (doi:10.1038/nature10200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frankel N, Davis GK, Vargas D, Wang S, Payre F, Stern DL. 2010. Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466, 490–493 (doi:10.1038/nature09158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parks AL, et al. 2004. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 36, 288–292 (doi:10.1038/ng1312) [DOI] [PubMed] [Google Scholar]
- 24.Waddington CH. 1942. Canalization of development and the inheritance of acquired characters. Nature 150, 563–565 (doi:10.1038/150563a0) [DOI] [PubMed] [Google Scholar]
- 25.Perry MW, Boettiger AN, Bothma JP, Levine M. 2010. Shadow enhancers foster robustness of Drosophila gastrulation. Curr. Biol. 20, 1562–1567 (doi:10.1016/j.cub.2010.07.043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frankel N. 2012. Multiple layers of complexity in cis-regulatory regions of developmental genes. Dev. Dyn. 241, 1857–1866 (doi:10.1002/dvdy.23871) [DOI] [PubMed] [Google Scholar]
- 27.McGregor AP, Orgogozo V, Delon I, Zanet J, Srinivasan DG, Payre F, Stern DL. 2007. Morphological evolution through multiple cis-regulatory mutations at a single gene. Nature 448, 587–590 (doi:10.1038/nature05988) [DOI] [PubMed] [Google Scholar]
- 28.Orr HA. 1998. Testing natural selection vs. genetic drift in phenotypic evolution using quantitative trait locus data. Genetics 149, 2099–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sucena E, Delon I, Jones I, Payre F, Stern DL. 2003. Regulatory evolution of shavenbaby/ovo underlies multiple cases of morphological parallelism. Nature 424, 935–938 (doi:10.1038/nature01768) [DOI] [PubMed] [Google Scholar]
- 30.Frankel N, Wang S, Stern DL. 2012. Conserved regulatory architecture underlies parallel genetic changes and convergent phenotypic evolution. Proc. Natl Acad. Sci. USA 109, 20 975–20 979 (doi:10.1073/pnas.1207715109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cande J, Goltsev Y, Levine MS. 2009. Conservation of enhancer location in divergent insects. Proc. Natl Acad. Sci. USA 106, 14 414–14 419 (doi:10.1073/pnas.0905754106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hare EE, Peterson BK, Iyer VN, Meier R, Eisen MB. 2008. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 4, e1000106 (doi:10.1371/journal.pgen.1000106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lang M, Hadzhiev Y, Siegel N, Amemiya CT, Parada C, Strahle U, Becker M-B, Müller F, Meyer A. 2010. Conservation of shh cis-regulatory architecture of the coelacanth is consistent with its ancestral phylogenetic position. EvoDevo 1, 11 (doi:10.1186/2041-9139-1-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuhn DTSM, Ventimiglia J, Sprey TE. 1992. Cuticle morphology changes with each larval molt in D. melanogaster. Drosophila Inf. Ser. 71, 218–222 [Google Scholar]
- 35.Rockman MV. 2012. The QTN program and the alleles that matter for evolution: all that's gold does not glitter. Evolution 66, 1–17 (doi:10.1111/j.1558-5646.2011.01486.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flint J, Mackay TF. 2009. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res. 19, 723–733 (doi:10.1101/gr.086660.108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graze RM, Barmina O, Tufts D, Naderi E, Harmon KL, Persianinova M, Nuzhdin SV. 2007. New candidate genes for sex-comb divergence between Drosophila mauritiana and Drosophila simulans. Genetics 176, 2561–2576 (doi:10.1534/genetics.106.067686) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bradshaw HD, Jr, Otto KG, Frewen BE, McKay JK, Schemske DW. 1998. Quantitative trait loci affecting differences in floral morphology between two species of monkeyflower (Mimulus). Genetics 149, 367–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reed RD, et al. 2011. Optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 333, 1137–1141 (doi:10.1126/science.1208227) [DOI] [PubMed] [Google Scholar]
- 40.Stam LF, Laurie CC. 1996. Molecular dissection of a major gene effect on a quantitative trait: the level of alcohol dehydrogenase expression in Drosophila melanogaster. Genetics 144, 1559–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bickel RD, Kopp A, Nuzhdin SV. 2011. Composite effects of polymorphisms near multiple regulatory elements create a major-effect QTL. PLoS Genet. 7, e1001275 (doi:10.1371/journal.pgen.1001275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loehlin DW, Werren JH. 2012. Evolution of shape by multiple regulatory changes to a growth gene. Science 335, 943–947 (doi:10.1126/science.1215193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Studer AJ, Doebley JF. 2011. Do large effect QTL fractionate? A case study at the maize domestication QTL teosinte branched1. Genetics 188, 673–681 (doi:10.1534/genetics.111.126508) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Linnen CR, Poh YP, Peterson BK, Barrett RD, Larson JG, Jensen JD, Hoekstra HE. 2013. Adaptive evolution of multiple traits through multiple mutations at a single gene. Science 339, 1312–1316 (doi:10.1126/science.1233213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marinic M, Aktas T, Ruf S, Spitz F. 2013. An integrated holo-enhancer unit defines tissue and gene specificity of the fgf8 regulatory landscape. Dev. Cell 24, 530–542 (doi:10.1016/j.devcel.2013.01.025) [DOI] [PubMed] [Google Scholar]
- 46.Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, Splinter E, de Laat W, Spitz F, Duboule D. 2011. A regulatory archipelago controls Hox genes transcription in digits. Cell 147, 1132–1145 (doi:10.1016/j.cell.2011.10.023) [DOI] [PubMed] [Google Scholar]
- 47.Hollenback SM, Lyman S, Cheng J. 2011. Recombineering-based procedure for creating BAC transgene constructs for animals and cell lines. Curr. Protoc. Mol. Biol. 95, 23.14.1–23.14.28 (doi:10.1002/0471142727.mb2314s95) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wesolowska N, Rong YS. 2013. Long-range targeted manipulation of the Drosophila genome by site-specific integration and recombinational resolution. Genetics 193, 411–419 (doi:10.1534/genetics.112.145631) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robert VJ, Katic I, Bessereau JL. 2009. Mos1 transposition as a tool to engineer the Caenorhabditis elegans genome by homologous recombination. Methods 49, 263–269 (doi:10.1016/j.ymeth.2009.02.013) [DOI] [PubMed] [Google Scholar]