

Abstract
The therapeutic potential of adult neural stem cells (aNSCs) has been shown in EAE, an animal model of MS, administered by either i.c.v. or i.v. injection. However, i.c.v. is an invasive approach, while the i.v. route of aNSCs is associated with a non-specific immune suppression in the periphery. Here we demonstrate that intranasal (i.n.) delivery of fluorescently labeled aNSCs resulted in their appearance in the olfactory bulb, cortex, hippocampus, striatum, brainstem, and spinal cord. These cells induce functional recovery from ongoing EAE similar to that achieved with i.v. injected aNSCs, with comparable anti-inflammatory and remeylination effects in CNS inflammatory foci. Importantly, unlike the peripheral immune suppression brought about by i.v. NSCs, intranasal delivery did not influence peripheral immune responses. We conclude that aNSCs can be reliably delivered to the CNS via the nasal route to induce functional recovery and confer immunomodulation and remyelination in EAE. Intranasal administration of NSCs provides a highly promising, noninvasive and CNS-specific alternative to current cell-based approaches in treating EAE.
Keywords: Neural stem cells, EAE, Intranasal
Introduction
Although the etiology of multiple sclerosis (MS) remains elusive, it is considered an autoimmune disease of the central nervous system (CNS), which is characterized by multifocal inflammation, demyelination, axonal loss and gliosis in the brain and spinal cord [1]. Inflammatory infiltration and demyelination of the CNS are the key pathological features of MS and its animal model, EAE [2,3]. Transplantation of adult neural stem cells is a promising approach for treating CNS diseases. Adult neural stem cells (aNSCs) are a specific type of multipotent stem cell in the ependymal cell layers of the adult subventricular zone (SVZ), and the cortical and limbic regions of the brain. Their capacity for migration into the CNS and neural differentiation makes these cells an attractive candidate for cell replacement therapy. Indeed, aNSCs have been shown to be effective, not only in EAE, but in animal models of other types of neurodegenerative diseases [4–7]. Although the exact mechanisms underlying symptomatic improvement due to aNSC therapy remain to be elucidated, studies have demonstrated the ability of aNSCs to differentiate into oligodendrocytes, to repair injured myelin tissue and to reduce inflammatory lesions of the brain and spinal cord in EAE mice [5]. In a recent open-label clinical trial, patients with secondary progressive MS, whose visual pathways were impaired, showed improvement following intravenous infusion of autologous bone marrow-derived mesenchymal stem cells (MSCs). This study showed that cell-based therapy can be safe in MS, with patients showing structural, functional, and physiological improvement in several visual endpoints after treatment [8].
aNSC administration from the periphery to the CNS is difficult due to limitations presented by the blood-brain barrier (BBB). Currently, the principal transplantation routes for cellular therapy are by intracranial or intravenous (i.v.) injection. While the intracranial route is frequently used in animal studies, less invasive approaches are preferred for clinical applications. Other problems with this route include the consequences of direct tissue trauma such as inflammation, cerebral edema, and reactive gliosis [9]. I.v. administration is a less invasive delivery method; however, it may lead to widespread systemic distribution and retention of NSCs in peripheral organs such as lungs, liver, and spleen [10,11]. The BBB normally prevents movement of aNSCs from micro-circulation to brain. Our previous work demonstrated that at week 2 post transfer (p.t.), only a few aNSCs were found in the brain parenchyma, while most of them remained in perivascular areas of the CNS and the peripheral organs [12]. Another recent study reported retention of NSCs in lung capillaries post injection, leading to inflammation and apoptosis in lung tissue [13].
Intranasal (i.n.) delivery of stem cells is a potential strategy to overcome the obstacles created by the BBB and is a promising option because of its non-invasiveness. Interestingly, fluorescently labeled rat MSC have been detected 1h after i.n. delivery in the olfactory bulb, hippocampus, thalamus, cortex, subarachnoid space and spinal cord of mice, and 24% of these cells survived at least 4.5 months [14,15]. Behavioral analyses of a rat model of Parkinson's disease showed significant improvement in forepaw motor function 40–110 days after i.n. delivery of 1×106 stem cells [15]. MSCs delivered i.n. 10 days after the induction of hypoxia–ischemia led to significant improvement in sensorimotor function and decreased brain lesion size 18 days after treatment [16].
In this study we tested the therapeutic effects of i.n. aNSCs in an ongoing chronic-progressive EAE mouse model. Our results demonstrate that i.n. delivery of fluorescently labeled aNSCs resulted in the appearance of these cells in the olfactory bulb, cortex, and spinal cord. I.n. delivery of aNSCs induced earlier functional recovery, and exhibited anti-inflammatory and remyelination effects in CNS inflamed foci equivalent to those achieved with i.v. injection. Importantly, the peripheral immunological response was not affected by i.n. delivery. These results illustrate the therapeutic potential of i.n. delivery of NSCs for CNS disorders.
Materials and Methods
EAE induction and aNSC treatment
Female C57BL/6 mice, 7–9 weeks of age, were injected s.c. with 200 μg MOG35–55 in CFA containing 5 mg/ml Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) at 2 sites on the back. All mice received 200 ng pertussis toxin (Campbell, CA) i.p. on days 0 and 2 p.i. Clinical score was checked daily by two researchers blindly according to a 0–5 scale as follows: 1, limp tail or waddling gait with tail tonicity; 2, waddling gait with limp tail (ataxia); 2.5, ataxia with partial limb paralysis; 3, full paralysis of one limb; 3.5, full paralysis of one limb with partial paralysis of second limb; 4, full paralysis of two limbs; 4.5, moribund; and 5, death. All animal protocols were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University following NIH guidelines.
GFP-aNSCs were prepared as described [17]. At days 14 and 21 p.i., GFP-aNSCs or vehicle (PBS) was delivered intranasally (i.v.) or intravenously (i.n.) as described previously [14]. Briefly, animals were divided into 3 groups (n=7 in each group); the first group received GFP-aNSCs via i.n. delivery; the second group received GFP-aNSCs via i.v. injection; the third group was treated with vehicle (PBS). For i.n. administration, 30 minutes before GFP-aNSCs or vehicle administration, two doses of 3 μl hyaluronidase (total 100 U; Sigma-Aldrich Chemical Co.) in PBS were applied to each nostril and spontaneously inhaled. Subsequently, a total of 1×106 GFP-aNSCs in 12 μl PBS or vehicle were administered as two doses of 3 μl applied to each nostril. For i.v., aNSCs were given by injection of single aNSCs (1.0×106 cells in 200 μl PBS/each mouse) i.v. via the tail vein. Untreated control mice received hyaluronidase nasally, followed by nasal PBS (12 μl) and i.v. PBS (200 μl) at the same time as the treated mice.
Immunohistochemistry
Animals were sacrificed at days 1, 7 and 21 p.t. and extensively perfused with PBS. Brains and spinal cords were cryoprotected with 30% sucrose in PBS and embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Zoeterwoude, The Netherlands). In particular, spinal cords were carefully excised from the brain stem to the lumbar region. The lumbar enlargement was identified and then transected at the exact midpoint of the lumbar enlargement to standardize a site along the longitudinal axis of the cord, ensuring that the same lumbar spinal cord regions were analyzed for all conditions. Transverse sections of brain and spinal cord were cut and immunohistochemistry was performed using various antibodies.
Anti-myelin basic protein (MBP) antibody staining slices were assessed for quantification of demyelination:0, none; 1, rare foci; 2, a few areas of demyelination; 3, large (confluent) areas of demyelination. Quantification of CNS damage was performed on 6 sections per mouse; 6–8 mice per group were analyzed.
Immunofluorescence controls were routinely performed with incubations in which primary antibodies were not included. Finally, slides were covered with mounting medium (Vector Laboratories) containing 1 μM DAPI. Results were visualized by fluorescent microscopy (Nikon Eclipse 800) or confocal microscopy (Zeiss LSM 510).
Proliferation assays
T-cell proliferation was assayed by 3H-thymidine incorporation as described previously [18]. Briefly, triplicate aliquots of 5×105 splenocytes in 96 well plates were stimulated with anti-CD3/CD28 or MOG35–55 peptide (10 μg/ml). After 60 hrs of incubation, cells were pulsed with 1 μCi 3H-thymidine/well for 18 hrs and then were harvested on fiberglass filters. Thymidine incorporation was determined using a Wallac 1450 MicroBeta TriLux scintillation counter (PerkinElmer, Turku, Finland).
Histopathology
At the end of experiments (21 days p.t.), H&E for inflammation was performed on 7-μm spinal cord sections. Slides were assessed in a blinded fashion for inflammation [19]: 0, none; 1, a few inflammatory cells; 2, organization of perivascular infiltrates; and 3, increasing severity of perivascular cuffing with extension into the adjacent tissue.
Statistical analysis
Clinical scores were analyzed by calculating the area under the curve for each mouse over the clinical period of the experiment. Differences between multiple groups were evaluated by the Kruskal-Wallis one-way analysis of variance (ANOVA). Experiments with two groups were tested for statistical significance using unpaired, two-tailed, Student's t tests. Differences were considered significant at a level of p<0.05.
Results
Effective suppression of EAE by i.n. administration of GFP-aNSCs
aNSCs were isolated and expanded from the SVZ of adult C57BL/6 mice. After 3–5 days of culture, various sizes of primary neurospheres containing 20–200 aNSCs were formed from single aNSCs. Free-floating primary neurospheres were collected, dissociated and cultured for the next generation. aNSCs at passages of 4–10 were used in all experiments. To trace them after transplantation into EAE mice, we infected these cells with the bicistronic lentiviral vector Lv.GFP, encoding GFP. At day 3 post-infection of NSCs, more than 80% infection efficiency for both vectors in aNSCs was observed (data not shown). GFP+ cells were then sorted by FACS to reach >99% purity and transferred into growth medium (Figure 1A).
Figure 1. Effective suppression of EAE by i.n. administration of GFP-aNSCs.

(A) A modified neurosphere is shown. GFP+ cells were sorted by FACS to reach >99% purity, then transferred into growth medium where they became neurospheres. Nuclei in A are stained with DAPI (blue). Magnification ×40. aNSCs transduced with Lv.GFP were dissociated into single cells and washed twice with PBS. 1.0×106 cells/mouse were injected once, at different stages of disease, EAE mice that received the same volume of PBS either i.n. or served as a sham injection control. Symbols represent mean values and SD of 6–7 mice from each group. (B) NSC-i.n. delivery or NSC-i.v. injection at day 14 p.i.; (C) NSC-i.n. delivery or i.v. injection at the peak of disease (day 21 p.i.). *p<0.05, **p<0.01, comparisons between sham-EAE group and other groups; #p<0.05, comparison between NSC-i.n. and NSC-i.v. injection.
To determine the efficacy of i.n. delivery of aNSCs in EAE, these cells were dissociated into single cell suspensions, and delivered i.n. at 1.0×106 cells in 20 μl PBS/per mouse either at disease onset (day 14 post-immunization, p.i.) or at peak of disease (day 21 p.i.). For comparison, a separate group of mice received the same number of NSCs i.v., and mice that received PBS i.n. served as untreated controls.
As shown in Figure 1B, untreated mice developed typical EAE, while both i.n. and i.v. delivery of GFP-aNSC treatment at onset of EAE resulted in significantly decreased disease severity. Significant suppression of the progression and severity of EAE was obtained by both i.n. and i.v. delivery of GFP-aNSC treatment compared with control, 21 days following aNSC application. Mice treated via the i.n. route (day 14 p.i.) displayed significantly lower clinical scores than those treated via i.v. injection at 4 days following aNSC application. The therapeutic effect of transplantation at day 21 p.i. (peak of EAE) was similar to that at day 14 p.i. (Figure 1C).
Engraftment of i.n. aNSCs in the CNS
Mice treated with aNSCs i.n. and i.v. starting from day 21 p.i. were extensively perfused. Brain and spinal cords were harvested at days 1, 7 and 21 p.t. and extensively studied for NSC distribution, and all other pathology/immunohistochemistry was focused at standard 500 μm2 specific fields within the ventral column of the lumbar spinal cord (L3) as described [12]. At day 1 p.t., a few i.n. delivered GFP+ cells were found in the olfactory bulb, cortex, hippocampus, striatum, and brainstem, but not the spinal cord (Figure 2A), similar to a previous observation [14]. At day 7 p.t., GFP-aNSCs that had been i.n. delivered, but not those delivered via i.v. injection (data not shown), were found in both brain (Figure 2B) and spinal cord (Figure 2C). At day 21 p.t., the majority of NSCs delivered i.n. or i.v. had migrated into inflammatory foci of the CNS parenchyma (including the brain and spinal cord), which were identified as tissues within white matter areas (e.g., ventral column of spinal cord) (Figures 2D and 2E). A large number of GFP-NSCs were detected in the demyelinated foci following i.n. or i.v. delivery (21–25 cells/mm2 in brains and 15–17 cells/mm2 in spinal cords (Figure 2F).
Figure 2. Localization and migration of transplanted GFP-aNSCs in the CNS.

Mice treated with aNSCs i.n. and i.v. were sacrificed at days 1, 7 and 21 p.t., and brains and spinal cords were harvested for immunohistology. At day 21 p.t., all groups were examined in the same region of the corpus callosum and spinal cord. Transplanted aNSCs (green) from i.n. delivery were primarily confined to brain perimengingeal sites at day 1 p.t. (A), but not spinal cord (data not shown), while at day 7 p.t. these cells were found in both brain (B) and spinal cord (C). At day 21 p.t., the majority of these cells from both i.n. (D) and i.v. (E) had migrated to the parenchyma of the corpus callosum (not shown) and spinal cord. Nuclei are stained with DAPI (blue). (F) Quantitative analysis of GFP-aNSCs that reached the parenchyma of EAE at days 1, 7 and 21 p.t. Symbols represent mean values and SD of 6–8 mice from each group. *p<0.05, comparison between 7 and 21 days p.t.; #p<0.05, comparison between i.n. delivery and i.v. injection.
Anti-inflammatory effect of i.n.-aNSCs in the CNS
Consistent with clinical observations, inflammatory infiltration in spinal cords was significantly suppressed by aNSCs i.n. and i.v. compared to untreated-EAE. At week 3 p.t., inflammatory infiltration was significantly attenuated following i.n. and i.v. injection, as shown by H & E staining (Figure 3A). Significantly lower numbers of CD4+ T cells (Figure 3B) and F4/80+ macrophages (Figure 3C) were detected in i.n. delivery and i.v. injection compared with untreated mice (p<0.05–0.01, Figure 3D).
Figure 3. Anti-inflammatory effect of aNSCs in the CNS.
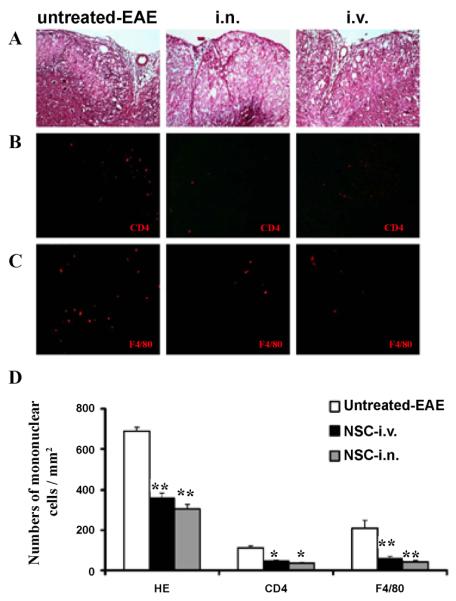
Mice described in Figure 1 were sacrificed 21 days p.t., and lumbar spinal cords were harvested for H&E staining and immunostaining. All groups were examined in the same region of the ventral column at L3. Reduced inflammatory infiltration (A) and numbers of CD4+ cells (B) and F4/80+ cells in (C) were found in spinal cord lesions of i.n. delivery and i.v. injection mice compared to untreated EAE mice. Magnification ×20 for A, B and C. (D) Quantitative analysis of H&E scores and numbers of CNS-infiltrating mononuclear cells. Symbols represent mean values and SD of 6–8 mice from each group. *p<0.05, **p<0.01, comparison between untreated-EAE group and other groups. There was no significant difference between the i.n. delivery and i.v. injection groups.
Lack of effect of i.n. aNSCs on peripheral immune responses
To study the autoantigen-induced immune cell proliferation responses of i.n. aNSC-treated mice compared with the other treatment groups, splenocytes were harvested 3 weeks p.t. and cultured with MOG35–55 peptide and anti-CD3/CD28 for 3 days. Spleen cell proliferation was significantly lower in i.v. aNSC-injected groups (starting from day 14 p.i.) as compared to i.n. and untreated control (p<0.05–0.01; Figure 4A). There was no significant difference between i.n. delivery and untreated control groups (p>0.1; Figure 4A). The same result was obtained in mice that received NSC starting from day 21 p.t. (Figure 4B). These results suggest a direct effect of i.n. delivered NSC on the CNS, without inhibiting proliferation of immune cells in the periphery.
Figure 4. Non-interference of immune responses outside the CNS in vivo with i.n.-aNSCs.
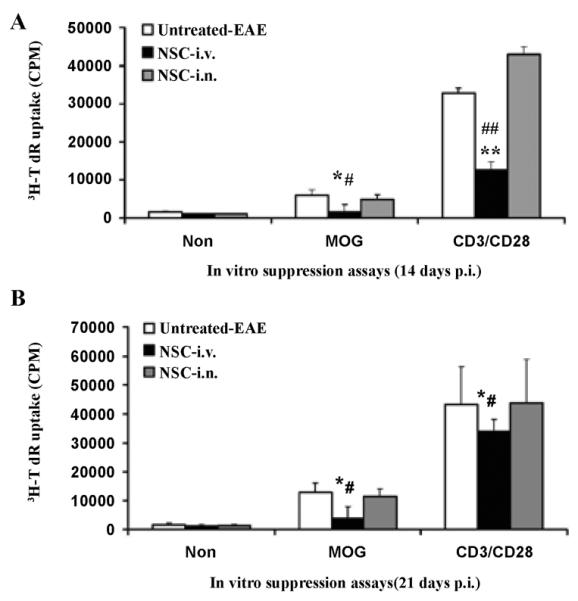
Spleen cells from mice were stimulated in vitro with 10 μg/ml MOG35–55 peptide and anti-CD3/CD28 for 3 d. Lymphocyte proliferation production of cells was measured by 3H-TdR incorporation (mean ± SD of triplicate). Symbols represent mean values and SD of 6–8 mice from each group. *p<0.05, **p<0.01, comparison between untreated-EAE group and other groups. #p<0.05, ##p<0.01, comparison between i.n. group and i.v. injection group. There was no significant difference between the i.n. delivery and untreated-EAE group.
I.n. aNSCs promote remyelination
To compare the regenerative potential of i.n. and i.v. aNSCs vs. the untreated group in EAE mice, the ventral column at L3 in the spinal cord (shown in Figure 5A) was examined in all groups at the end of the experiment (21 days p.t.). Multi-focal myelin loss in EAE mice was also detected, with markedly decreased MBP in the ventral column of spinal cord (Figure 5B). Spinal cords of i.n.-aNSC-treated and i.v. mice showed rare demyelination foci and a significantly lower score of demyelination than untreated EAE mice (Figure 5C).
Figure 5. Transplanted aNSCs promote remyelination.
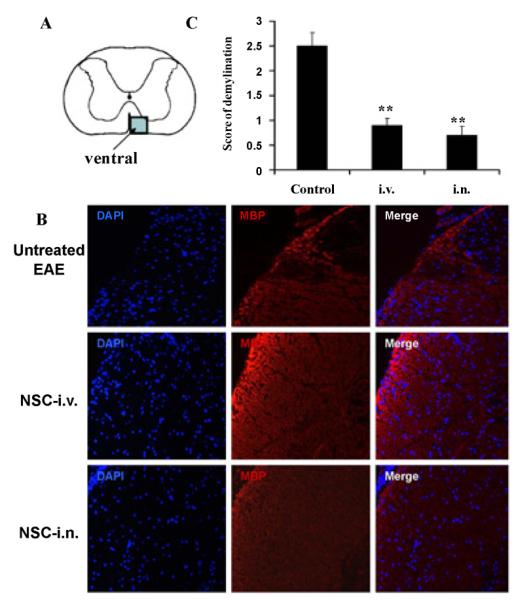
(A) shows the ventral column of the lumbar spinal cord (L3). (B) Myelin basic protein (MBP) immunostaining in the ventral column of spinal cord sections at day 21 p.t. for detection of demyelination. Magnification ×20. (C) Mean scores of myelination. Symbols represent mean values and SD of 6–8 mice from each group. *p<0.05, **p<0.01, comparison between untreated-EAE and other groups.
To characterize the phenotype of demyelinated lesions in mice that received i.n. or i.v. aNSCs, spinal cords were harvested at the end of the experiment (day 21 p.t.), and triple immunostaining was performed in spinal cord sections with cell type-specific antibodies. Co-localization of GFP and neural specific markers in the spinal cord revealed that some of the transplanted cells differentiated into NG2+ oligodendrocyte progenitors (Figure 6A) and GalC+ mature oligodendrocytes (Figure 6B). While oligodendrocyte-like cells could be observed in all groups, i.n. and i.v. aNSC-treated mice exhibited a significantly higher absolute number of oligodendrocytes (GalC+) than untreated groups (p<0.01; Figure 6C).
Figure 6. I.n.-GFP-aNSCs promote remyelination of demyelinated axons in spinal cord.
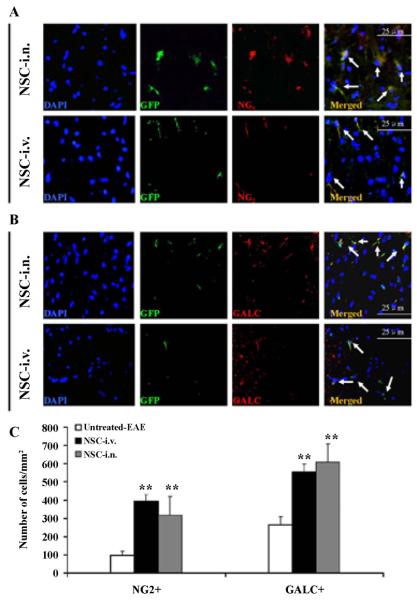
Spinal cords were harvested for immunohistology at day 21 p.t. All groups were examined in the same region: a specific site on the ventral column of the lumbar spinal cord (L3). Cells co-labeled with GFP (green), cell nucleus (blue) and oligodendrocyte precursor cells (NG2+; red) (A) and oligodendrocytes (GalC+; red) (B) derived from transplanted aNSCs (arrows). (C) Quantification of total NG2+GFP+ and GalC+GFP+ cell numbers. **p<0.01, comparison between untreated-EAE and other groups. Symbols represent mean values and SD of 6–8 mice from each group. Magnification ×40 for A–B.
Co-localization of GFP and neural specific markers in the spinal cord showed that some transplanted cells differentiated into β-III-tubulin neurons (Figure 7A) and GFAP+ astrocytes (Figure 7B). GFP+ cells retained undifferentiated SOX2+ morphological features (Figure 7C), consistent with our previously published experimental results [12].
Figure 7. I.n.-GFP-aNSCs selectively expand neuron and oligodendrocyte populations in vivo.
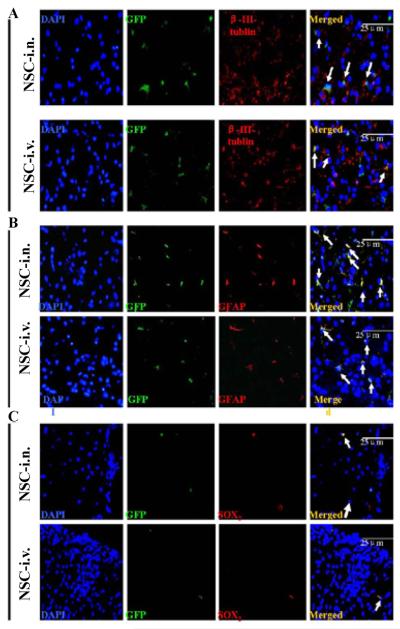
Mice treated with aNSCs at day 21 p.i. were sacrificed at day 21 p.t., and lumbar spinal cords were harvested for immunohistology. Cells co-labeled with GFP (green), cell nucleus (blue) and neural specific markers (red), including β-III-tublin (A) GFAP+ (B) were identified as differentiated cells derived from transplanted GFP-aNSCs (arrows). Some transplanted GFP-aNSCs remained SOX2+ (C) undifferentiated NSCs. Magnification ×40.
Discussion
This study demonstrates the therapeutic efficacy of i.n.-delivered aNSCs to the CNS, especially in the spinal cord, in an animal model of EAE. Administration of i.n. aNSCs improved functional outcome and attenuated the immunological and pathological features of EAE. These results indicate that intranasal delivery could be an efficient route for stem cell transplantation in neuroimmunological diseases, such as multiple sclerosis. We demonstrate that i.n. aNSCs not only effectively suppress CNS inflammation, but also promote remyelination. Of note, i.n. administered NSCs had no effect on systemic immunological response, except in the CNS. I.n. delivered NSCs had therapeutic effects comparable to those delivered i.v. as measured by cell replacement, neuroprotection and immunomodulation; however, i.n. aNSCs had the significant advantage of not interfering with systemic immunological functions.
Neither the precise pathways by which aNSCs travel, nor their mechanisms of action, have been fully elucidated. Three routes have been proposed for cell migration into the brain and spinal cord after nasal delivery: 1) the peripheral olfactory system connecting the nasal passages with the olfactory bulb and rostral brain regions (e.g. anterior olfactory nucleus and frontal cortex); 2) the peripheral trigeminal system connecting the nasal passages with brainstem and spinal cord regions; 3) entry into the CSF with movement along the surface of the cortex followed by entry into the CNS parenchyma [14,20]. Our study is the first to deliver NSCs nasally as a therapy for EAE, and to confirm that cells can migrate into the brain rapidly, reach distant CNS areas, including the spinal cord, and survive there until the animals are sacrificed (in our experiments, 3 weeks). In addition, nasal delivery of NSCs leads to their more rapid appearance in the CNS than with i.v. injection, another advantage in view of possible future application in human cell-based therapy. Taken together, these results indicate the potential of i.n. NSCs as a novel and efficient approach for NSC delivery in EAE/MS therapy.
NSCs have recently emerged as a potential effective therapy in the treatment of MS [4,13,21]. The capacity of NSCs for CNS migration and neural cell differentiation makes these cells attractive resources for cell-based therapy, not only in EAE, but also in other types of neurological diseases [22,23]. Therapeutic effects of transplanted NSCs mainly include cell replacement, neuroprotection, and immunomodulation. Systemic and i.c.v. transplantation of NSCs halted EAE progression and decreased severity of clinical signs [4,13,21]. These NSCs can differentiate into myelin-producing cells that engage in subsequent remyelination, resulting in a decrease in net myelin and axonal loss. Grafted aNSCs have been reported to survive in the CNS for up to 15 months, without any sign of deleterious outcomes such as adverse immune responses, inappropriate anatomical accumulation, or tumor formation [24]. However, a degree of caution is warranted in future studies in particular for the possibility of the latter complication [25,26].
Transplanted NSCs, when injected peripherally, transiently colonize systemic organs, e.g., lymph nodes and spleen, and can inhibit T-cell activation and proliferation [25,27–29], as well as dendritic cell function [30]. Due to their nonspecific properties, a systemic immunosuppressive effect may occur when NSCs are present in the periphery before migration into the CNS (approx. 18–20 days post i.v. injection) [12,21,31]. Nasally administrated NSCs, in contrast, migrate into the CNS directly, without systemic distribution, thus minimizing the potential peripheral immunosuppression caused by NSCs.
In MS and EAE, a few autoreactive T cells in an autoimmune infiltrate are thought to control a vast population of nonspecific cells; therefore, the suppression of autoreactive T cells could lead to a significant reduction in other inflammatory infiltrates [32]. Given that transplanted NSCs in the inflamed foci of the CNS interact more closely with infiltrating, pathogenic immune cells than with those in the periphery, suppression of inflammation in the target organ by NSCs is likely to be more effective than in the periphery. Consistent with this, it has also been found that, while systemic administration of IL-10 failed to suppress EAE, local delivery of this cytokine had a significant effect [33]. Our current study has shown that, while a comparable therapeutic effect was obtained in both i.v. and i.n. administration of NSCs, splenocytes from mice that received i.v. NSCs exhibited suppressed proliferation after culture of both MOG and anti-CD3/CD28 ex vivo, indicating a systemic immunosuppression, which was not observed in mice that received i.n. NSCs. These data demonstrate that, in contrast to i.v. NSCs, suppression of EAE by i.n. NSCs is CNS-specific and does not interfere with systemic immune responses.
While demyelination in EAE is thought to be induced by a specific, myelin-reactive immune response, spontaneous remyelination is often incomplete or lacking, resulting in substantial axonal and neuronal loss [34]. Axonal loss, in turn, makes remyelination unsuccessful due to the absence of remyelination signals from the axons [35]. In MS/EAE, remyelination takes place in a microenvironment containing elements that are intrinsically hostile to the oligodendrocyte lineage [35]. Reducing autoimmune responses at inflammatory foci in the CNS by transplanted aNSCs would thus convert such a microenvironment into one supportive of the remyelination process. These effects would result in a significantly increased number of oligodendrocytes than in untreated EAE mice, which would provide a basis for enhanced remyelination [36].
Functional benefits resulting from NSC transplantation correlate only marginally to the number of fully differentiated neural cells obtained from grafted cells [12,21]. This inability to complete the differentiation process and the tendency to maintain an undifferentiated phenotype within the host tissue suggest that transplanted NSCs partially exert their therapeutic effect by alternative mechanisms [37]. In our study,although our data showed that NSCs delivered by both routes differentiated into oligodendrocytes and neurons, these exogenous cells (GFP+GalC+) were only a small proportion of all the GalC+ cells, while endogenous myelinating cells (GFP−GalC+) are the major source of remyelinating cells when both routes are used. aNSCs thus promote neuron survival, axonal growth and remyelination of both transplanted (exogenous) and resident (endogenous) precursors. On the other hand, cell replacement cannot fully explain the significant improvement in clinical signs, and immunomodulation and neuroprotection in the CNS also have important effects on remyelination and neurological recovery in NSC therapy.
Success of cell-based therapy in neurological disorders depends on the therapeutic properties of the cell type, the method and safety of administration, the number of cells delivered to the site of disease and, finally, on avoiding excessive migration of the therapeutic cells into other organs and systems. In our study, i.n. delivery of aNSCs showed clinical recovery effects and attenuation of pathological features comparable to the i.v. injection route of aNSCs. However, i.n. delivery is a noninvasive approach with minimal or no effects on systemic immunological function outside of the CNS. Further efforts should be devoted to this targeted approach to human therapy.
Taken together, our data demonstrate that aNSCs applied via the nasal route have therapeutic effects that appear to be equivalent to those induced by i.v. aNSCs and lead to protection from CNS inflammation and promotion of remyelination and neural repair. Such an effective alternative route of administration could significantly improve the efficacy of NSC-based therapy in EAE/MS.
Acknowledgments
This study was supported by the NIH/NINDS (R01NS075260), the Gilbert Trust and the Groff Foundation. Drs. Wu, Han and Guan are supported by the National Natural Science Foundation of China (81070959) and the Key Project of National Natural Science Foundation of China (81230027). We thank Katherine Regan for editorial assistance.
Abbreviations
- aNSCs
Adult Neural Stem Cells
- MBP
Myelin Basic Protein
- MOG
Myelin Oligodendrocyte Glycoprotein
- p.i.
Post immunization
- p.t.
Post transplantation
- SVZ
Subventricular Zone
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of Interest
The authors have no conflicting financial interest.
References
- 1.Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS) Br J Pharmacol. 2011;164:1079–1106. doi: 10.1111/j.1476-5381.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pahan K. Neuroimmune pharmacological control of EAE. J Neuroimmune Pharmacol. 2010;5:165–167. doi: 10.1007/s11481-010-9219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- 4.Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688–694. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- 5.Einstein O, Grigoriadis N, Mizrachi-Kol R, Reinhartz E, Polyzoidou E, et al. Transplanted neural precursor cells reduce brain inflammation to attenuate chronic experimental autoimmune encephalomyelitis. Exp Neurol. 2006;198:275–284. doi: 10.1016/j.expneurol.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Bacigaluppi M, Pluchino S, Peruzzotti-Jametti L, Kilic E, Kilic U, et al. Delayed post-ischaemic neuroprotection following systemic neural stem cell transplantation involves multiple mechanisms. Brain. 2009;132:2239–2251. doi: 10.1093/brain/awp174. [DOI] [PubMed] [Google Scholar]
- 7.Glavaski-Joksimovic A, Virag T, Chang QA, West NC, Mangatu TA, et al. Reversal of dopaminergic degeneration in a parkinsonian rat following micrografting of human bone marrow-derived neural progenitors. Cell Transplant. 2009;18:801–814. doi: 10.3727/096368909X470801. [DOI] [PubMed] [Google Scholar]
- 8.Connick P, Kolappan M, Crawley C, Webber DJ, Patani R, et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurol. 2012;11:150–156. doi: 10.1016/S1474-4422(11)70305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coyne TM, Marcus AJ, Woodbury D, Black IB. Marrow stromal cells transplanted to the adult brain are rejected by an inflammatory response and transfer donor labels to host neurons and glia. Stem Cells. 2006;24:2483–2492. doi: 10.1634/stemcells.2006-0174. [DOI] [PubMed] [Google Scholar]
- 10.Kraitchman DL, Tatsumi M, Gilson WD, Ishimori T, Kedziorek D, et al. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 2005;112:1451–1461. doi: 10.1161/CIRCULATIONAHA.105.537480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauger O, Frost EE, van Heeswijk R, Deminière C, Xue R, et al. MR evaluation of the glomerular homing of magnetically labeled mesenchymal stem cells in a rat model of nephropathy. Radiology. 2006;238:200–210. doi: 10.1148/radiol.2381041668. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Jiang Z, Fitzgerald DC, Ma C, Yu S, et al. Adult neural stem cells expressing IL-10 confer potent immunomodulation and remyelination in experimental autoimmune encephalitis. J Clin Invest. 2009;119:3678–3691. doi: 10.1172/JCI37914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Reekmans KP, Praet J, De Vocht N, Tambuyzer BR, Bergwerf I, et al. Clinical potential of intravenous neural stem cell delivery for treatment of neuroinflammatory disease in mice? Cell Transplant. 2011;20:851–869. doi: 10.3727/096368910X543411. [DOI] [PubMed] [Google Scholar]
- 14.Danielyan L, Schäfer R, von Ameln-Mayerhofer A, Buadze M, Geisler J, et al. Intranasal delivery of cells to the brain. Eur J Cell Biol. 2009;88:315–324. doi: 10.1016/j.ejcb.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Danielyan L, Schäfer R, von Ameln-Mayerhofer A, Bernhard F, Verleysdonk S, et al. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson disease. Rejuvenation Res. 2011;14:3–16. doi: 10.1089/rej.2010.1130. [DOI] [PubMed] [Google Scholar]
- 16.van Velthoven CT, Kavelaars A, van Bel F, Heijnen CJ. Nasal administration of stem cells: a promising novel route to treat neonatal ischemic brain damage. Pediatr Res. 2010;68:419–422. doi: 10.1203/PDR.0b013e3181f1c289. [DOI] [PubMed] [Google Scholar]
- 17.Couillard-Despres S, Winner B, Karl C, Lindemann G, Schmid P, et al. Targeted transgene expression in neuronal precursors: watching young neurons in the old brain. Eur J Neurosci. 2006;24:1535–1545. doi: 10.1111/j.1460-9568.2006.05039.x. [DOI] [PubMed] [Google Scholar]
- 18.Xu H, Wawrousek EF, Redmond TM, Nickerson JM, Wiggert B, et al. Transgenic expression of an immunologically privileged retinal antigen extraocularly enhances self tolerance and abrogates susceptibility to autoimmune uveitis. Eur J Immunol. 2000;30:272–278. doi: 10.1002/1521-4141(200001)30:1<272::AID-IMMU272>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 19.Zhang GX, Gran B, Yu S, Li J, Siglienti I, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–2160. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- 20.Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev. 2012;64:614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Pluchino S, Zanotti L, Rossi B, Brambilla E, Ottoboni L, et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005;436:266–271. doi: 10.1038/nature03889. [DOI] [PubMed] [Google Scholar]
- 22.Lee JA, Kim BI, Jo CH, Choi CW, Kim EK, et al. Mesenchymal stem-cell transplantation for hypoxic-ischemic brain injury in neonatal rat model. Pediatr Res. 2010;67:42–46. doi: 10.1203/PDR.0b013e3181bf594b. [DOI] [PubMed] [Google Scholar]
- 23.Park D, Lee HJ, Joo SS, Bae DK, Yang G, et al. Human neural stem cells over-expressing choline acetyltransferase restore cognition in rat model of cognitive dysfunction. Exp Neurol. 2012;234:521–526. doi: 10.1016/j.expneurol.2011.12.040. [DOI] [PubMed] [Google Scholar]
- 24.Lepore AC, Neuhuber B, Connors TM, Han SS, Liu Y, et al. Long-term fate of neural precursor cells following transplantation into developing and adult CNS. Neuroscience. 2006;139:513–530. doi: 10.1016/j.neuroscience.2005.12.043. [DOI] [PubMed] [Google Scholar]
- 25.Einstein O, Fainstein N, Vaknin I, Mizrachi-Kol R, Reihartz E, et al. Neural precursors attenuate autoimmune encephalomyelitis by peripheral immunosuppression. Ann Neurol. 2007;61:209–218. doi: 10.1002/ana.21033. [DOI] [PubMed] [Google Scholar]
- 26.De Feo D, Merlini A, Laterza C, Martino G. Neural stem cell transplantation in central nervous system disorders: from cell replacement to neuroprotection. Curr Opin Neurol. 2012;25:322–333. doi: 10.1097/WCO.0b013e328352ec45. [DOI] [PubMed] [Google Scholar]
- 27.Ben-Hur T, Einstein O, Mizrachi-Kol R, Ben-Menachem O, Reinhartz E, et al. Transplanted multipotential neural precursor cells migrate into the inflamed white matter in response to experimental autoimmune encephalomyelitis. Glia. 2003;41:73–80. doi: 10.1002/glia.10159. [DOI] [PubMed] [Google Scholar]
- 28.Einstein O, Karussis D, Grigoriadis N, Mizrachi-Kol R, Reinhartz E, et al. Intraventricular transplantation of neural precursor cell spheres attenuates acute experimental allergic encephalomyelitis. Mol Cell Neurosci. 2003;24:1074–1082. doi: 10.1016/j.mcn.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 29.Ben-Hur T. Immunomodulation by neural stem cells. J Neurol Sci. 2008;265:102–104. doi: 10.1016/j.jns.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 30.Pluchino S, Zanotti L, Brambilla E, Rovere-Querini P, Capobianco A, et al. Immune regulatory neural stem/precursor cells protect from central nervous system autoimmunity by restraining dendritic cell function. PLoS One. 2009;4:e5959. doi: 10.1371/journal.pone.0005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J, Yan Y, Ciric B, Yu S, Guan Y, et al. Evaluation of bone marrow-and brain-derived neural stem cells in therapy of central nervous system autoimmunity. Am J Pathol. 2010;177:1989–2001. doi: 10.2353/ajpath.2010.091203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steinman L. A few autoreactive cells in an autoimmune infiltrate control a vast population of nonspecific cells: a tale of smart bombs and the infantry. Proc Natl Acad Sci U S A. 1996;93:2253–2256. doi: 10.1073/pnas.93.6.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Croxford JL, Feldmann M, Chernajovsky Y, Baker D. Different therapeutic outcomes in experimental allergic encephalomyelitis dependent upon the mode of delivery of IL-10: a comparison of the effects of protein, adenoviral or retroviral IL-10 delivery into the central nervous system. J Immunol. 2001;166:4124–4130. doi: 10.4049/jimmunol.166.6.4124. [DOI] [PubMed] [Google Scholar]
- 34.Ben-Hur T. Cell therapy for multiple sclerosis. Neurotherapeutics. 2011;8:625–642. doi: 10.1007/s13311-011-0073-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jadasz JJ, Aigner L, Rivera FJ, Küry P. The remyelination Philosopher's Stone: stem and progenitor cell therapies for multiple sclerosis. Cell Tissue Res. 2012;349:331–347. doi: 10.1007/s00441-012-1331-x. [DOI] [PubMed] [Google Scholar]
- 36.Yasuda A, Tsuji O, Shibata S, Nori S, Takano M, et al. Significance of remyelination by neural stem/progenitor cells transplanted into the injured spinal cord. Stem Cells. 2011;29:1983–1994. doi: 10.1002/stem.767. [DOI] [PubMed] [Google Scholar]
- 37.Bonnamain V, Neveu I, Naveilhan P. Neural stem/progenitor cells as a promising candidate for regenerative therapy of the central nervous system. Front Cell Neurosci. 2012;6:17. doi: 10.3389/fncel.2012.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]