

Abstract
A series of ruthenium catalysts bearing five-membered chelating NHC architectures that exhibit very high Z-selectivity in a variety of metathesis reactions have recently been reported. It was envisioned that catalysts possessing sixmembered chelates could similarly exhibit high Z-selectivity and address limitations of this methodology. We thus prepared a number of new catalysts and systematically investigated the impact of the NHC and anionic ligand on their stereoselectivity. In standard metathesis assays, only catalysts containing six-membered chelated NHC structures and η2-bound anionic ligands favored the Z-olefin products compared to traditional ruthenium catalysts. In addition, substitution with bulkier N-aryl groups led to improved Z-selectivity. The effect of ligand structure on stereoselectivity discovered in this study will be useful in the future design of highly active and Z-selective ruthenium catalysts.
INTRODUCTION
Olefin metathesis is a convenient and powerful method for the construction of carbon-carbon double bonds, and is widely used in a variety of fields including natural product synthesis,1 biochemistry,2 green chemistry3 and polymer chemistry.4 Since its discovery in the 1950’s, this methodology has developed at a fast pace due to the discovery of well-defined transition metal catalysts and advances in catalyst efficiency.5 While molybdenum and tungsten catalysts initially gained popularity due to their high activity,6 the discovery of highly active and functional group tolerant ruthenium catalysts has enabled the wide-spread use of this methodology in both academic laboratories and industry processes.7
Metathesis reactions generally proceed under thermodynamic control, meaning that most catalysts predominantly provide E-olefin products.8 Recently, however, catalysts capable of preferentially forming the Z-isomer have been discovered. 9, 10, 11 The first breakthrough in this field was reported by the Hoveyda and Schrock groups.9a The reported monoaryloxidepyrrolide (MAP) molybdenum and tungsten catalysts provided a very high proportion of cis(Z)-olefin products in the homo-coupling of terminal olefins, and more complicated metathesis reactions.9 This Z-selectivity was attributed to the difference in size of the two axial ligands in relevant metallacyclobutane intermediates. Based on this same concept, Jensen and coworkers reported a ruthenium catalyst bearing a single bulky thiolate ligand that also achieved high Z-selectivity in olefin homocoupling.10
We have reported a series of ruthenium-based catalysts with unique chelating N-heterocyclic carbene (NHC) architectures (Figure 1).11 Cross metathesis (CM) products formed using these catalysts had a significantly lower E/Z ratio compared to previous generations of ruthenium catalysts. NHC-chelated structures derived from ruthenium alkylidene complexes had been previously observed, however they were formed as decomposition products and were not metathesis active.12 Catalysts 1a and 2a-c were synthesized via an intramolecular carboxylate-driven C-H bond insertion and contained an intact alkylidene. Complexes containing a five-membered chelate (2a-c) exhibited very high Z-selectivity in a variety of metathesis reactions including the homocoupling of terminal olefins, CM, macrocyclic ring-closing metathesis (RCM) and ring-opening metathesis polymerization (ROMP). This methodology has started to be applied to the synthesis of insect pheromones and more complicated natural products.13 Recent density functional theory (DFT) calculations for catalysts 1a, 2a and 2b suggest that reactions involving these chelated catalysts proceed under kinetic control through ‘side-bound’ ruthenacyclobutane intermediates. 14 It was proposed that the chelating NHC positions the N-aryl group above the ruthenacyclobutane, increasing the steric penalty for ruthenacycle substituents that point toward the N-aryl group and thus leading to productive formation of Z-olefins.
Figure 1.

Ruthenium-based NHC-chelated catalysts for Z-selective olefin metathesis.
In order to design highly efficient Z-selective catalysts containing six-membered chelating architectures, a more thorough understanding of the influence of ligands on stereoselectivity and activity is of paramount importance. In this report, we have prepared and crystallographically characterized a number of new NHC-chelated and non-chelated catalysts, and subsequently tested their activity in a variety of metathesis reactions. In this way, we attempted to clarify the relationship between catalyst structure and stereoselectivity with this class of catalysts.
RESULTS and DISCUSSION
Catalyst Syntheses
Modifications to the non-chelating N-aryl group of chelated catalysts were achieved by straightforward ligand and catalyst synthesis (Figure 2). Complex 3, bearing an N-Mes, N’-DIPP substituted NHC ligand, was reacted with silver pivalate, affording the bulkier NHC-chelated catalyst 4 (Scheme 1).15 The salt metathesis and C-H activation was monitored by 1H NMR and revealed that a dipivalate complex was initially formed and slowly converted to catalyst 4 by intramolecular C-H activation of the benzylic position of the N-Mes group. It should be noted that C-H activation of the N-DIPP group was not observed over the course of the reaction and two possible explanations are envisioned: (1) the benzylic hydrogen on the N-DIPP group is highly hindered and would lead to an unstable tertiary carbon- metal bond, and (2) the methyl groups are not activated and the resulting 7-membered chelating architecture is thermodynamically less stable.16 The crystal structure of complex 4 confirmed formation of this 6-membered chelated structure (Figure 3) and showed essentially the same structural features as catalyst 1a, except for the obvious change from N-Mes to N-DIPP.11a
Figure 2.

Proposed modifications to catalyst 1a. Mes = 2,4,6-trimethylphenyl, DIPP = 2,6-diisopropylphenyl.
Scheme 1.

Synthesis of NHC-chelated catalyst 4
Figure 3.

X-ray crystal structure of catalyst 4 is shown. Displacement ellipsoids are drawn at 50% probability. For clarity, hydrogen atoms have been omitted. Selected bond length (Å) for 4: C1-Ru1 1.931, C12-Ru1 2.094, C25-Ru1 1.842, O1-Ru1 2.305, O2-Ru1 2.360, O3-Ru1 2.235;
In the same manner as 1a, chelated catalysts bearing bulkier carboxylate ligands were synthesized by the salt metathesis and C-H activation of catalyst 5a. Reaction of starting complex 5a with Ag(OCOCMe2Ph) afforded 1b directly in high yield (Scheme 2). The bulkier salts Ag(OCOCMePh2) and Ag(OCOCPh3) were also reacted with complex 5a to afford the corresponding NHC-chelated catalysts, however decomposition upon silica gel column chromatography prevented isolation of 1c and 1d. In order to synthesize the desired chelated species, a new synthetic route was devised (Scheme 2). Bistriflate complex 5b was prepared by treating 5a with excess AgOTf,17 and subsequently reacted with sodium carboxylates to provide catalysts 1c and 1d cleanly; these species could then be purified by a simple solvent wash. As such, 5b is a versatile precursor for forming NHC chelated catalysts under mild conditions.18 In each of these activation reactions, the dicarboxylate complexes are formed as intermediates, followed by intramolecular C-H activation to yield the NHC-chelated catalysts, with concomitant formation of the corresponding carboxylic acids.19 X-ray crystal structures of catalysts 1b and 1d are shown in Figure 4 and display the same structural features as complex 1a: a 6-membered chelating NHC ligand and an η2-bound carboxylate ligand with the two oxygen atoms lying in the equatorial plane.
Scheme 2.

Syntheses of NHC-chelated catalysts 1a-d
Figure 4.


X-ray crystal structures of catalysts (a) 1a,11a (b) 1b and (c) 1d are shown. Displacement ellipsoids are drawn at 50% probability. For clarity, hydrogen atoms have been omitted. Selected bond length (Å) for 1a: C1-Ru1 1.945, C12-Ru1 2.095, C22-Ru1 1.840, O1-Ru1 2.296, O2-Ru1 2.332, O3-Ru1 2.230; for 1b: C1-Ru1 1.939, C12-Ru1 2.094, C22-Ru1 1.843, O1-Ru1 2.325, O2-Ru1 2.332, O3-Ru1 2.218; for 1d: C1-Ru1 1.926, C12-Ru1 2.102, C22-Ru1 1.843, O1-Ru1 2.314, O2-Ru1 2.252, O3-Ru1 2.301.
We had previously reported that the pivalate ligand of complex 2a, bearing a five-membered chelate, could be replaced by a variety of anionic ligands in a facile manner,11c however, attempts to exchange the anionic ligand of catalyst 1a were generally unsuccessful.20 We were nonetheless able to synthesize an NHC-chelated catalyst substituted with a less bulky chloride ligand. Reaction of complex 1a with one equivalent of HCl immediately afforded catalyst 6 (Scheme 3). Interestingly, excess HCl caused unchelation of the NHC ligand and provided the dichloride complex 5a. Presumably, the excess HCl protonated the chelating methylene carbon and broke the ruthenium-carbon bond of 6 while keeping the alkylidene intact. As a result, addition of exactly one equivalent of HCl is required to selectively form complex 6.
Scheme 3.

Synthesis of NHC-chelated catalyst 6
In order to supplement our investigations of chelated catalysts, we investigated some non-chelated analogs containing a single bulky anionic ligand.21 Recently, Jensen and coworkers reported that a bulky thiolate ligated catalyst derived from catalyst 5a showed high cis-selectivity in simple homocoupling reactions (Figure 5).10 It was thus proposed that a non-chelated catalyst substituted with a single bulky carboxylate could potentially be Z-selective. Initial attempts to synthesize 7a from 5a by ligand exchange with one equivalent of AgOPiv were unsuccessful due to formation of a mixture of unreacted 5a and the bispivalate complex, which subsequently converted to chelated catalyst 1a. As such, a new synthetic route to this type of nonchelated species was required. Complex 5c was prepared by treating 5a with one equivalent of AgOTf.17 Complex 5c could then be reacted with sodium carboxylates to yield 7a (R = Me) and 7d (R = Ph) in excellent yields (Scheme 4). It should be noted that 7a and 7d did not undergo intramolecular C-H activation. Considering that C-H activation and subsequent formation of a chelated structure was observed when the bistriflate complex 5b was exposed to sodium carboxylates as shown in Scheme 1, it seems that substitution with two carboxylate ligands is required for C-H activation to occur. It is proposed that the congested environment around the ruthenium center in the dicarboxylate intermediate positions one of the carboxylate ligands close enough to interact with the benzylic position of the mesityl group to promote a C-H activation event. The X-ray crystal structure of 7d is shown in Figure 6. In contrast to the aforementioned chelated catalysts, the carboxylate ligand of 7d is coordinated to the ruthenium center in a monodentate fashion as a formal 16e- complex, similar to catalyst 5a.
Figure 5.

A reported ruthenium catalyst bearing a bulky thiolate ligand.
Scheme 4.

Syntheses of non-chelated catalysts 7a and 7d
Figure 6.

X-ray crystal structure of catalyst 7d is shown. Displacement ellipsoids are drawn at 50% probability. For clarity, hydrogen atoms have been omitted. Selected bond length (Å) for 7d: C1-Ru1 1.975, C22-Ru1 1.830, Cl1-Ru1 2.341, O1-Ru1 2.261, O2-Ru1 2.021.
Ligand Effects on Metathesis Reactivity
The standard CM reaction of allylbenzene (8) and cis-1,4-diacetoxy-2-butene (9) was carried with catalysts 1a-7d under using previously reported conditions.22 This reaction provided a good general method to directly compare the catalysts under investigation (Table 1).
Table 1.
Selected data for the CM of 8 and 9 a
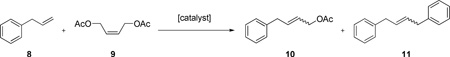 | ||||||||
|---|---|---|---|---|---|---|---|---|
| 10 |
11 |
|||||||
| entry | cat. | cat. load., mol %b | solvent | time, min | conv, %c | E /Z d | conv, %c | E /Z d |
| 1e | 1a | 2.5 | C6H6 | 2 | 50 | 1.4 | 2.8 | 0.75 |
| 30 | 58 | 1.5 | 3.1 | 1.4 | ||||
| 2 | 1b | 2.5 | C6H6 | 2 | 51 | 1.3 | 2.2 | 0.79 |
| 30 | 57 | 1.5 | 2.7 | 0.75 | ||||
| 3 | 1c | 2.5 | C6H6 | 2 | 54 | 1.6 | 2.0 | 1.4 |
| 30 | 63 | 1.9 | 3.1 | 1.2 | ||||
| 4 | 1d | 2.5 | C6H6 | 2 | 39 | 2.0 | (NA)f | (NA)f |
| 30 | 58 | 2.3 | 2.9 | 1.3 | ||||
| 5 | 3 | 2.5 | C6H6 | 2 | 70 | 4.1 | 5.4 | 3.4 |
| 30 | 84 | 9.1 | 8.3 | 6.5 | ||||
| 6 | 4 | 2.5 | C6H6 | 2 | 62 | 0.95 | 3.0 | 0.94 |
| 30 | 72 | 1.2 | 4.7 | 1.0 | ||||
| 7e | 5a | 2.5 | C6H6 | 2 | 70 | 11 | 7.6 | 6.0 |
| 30 | 66 | 11 | 10 | 6.9 | ||||
| 8 | 6 | 2.5 | C6H6 | 2 | 19 | 3.2 | (NA)f | (NA)f |
| 30 | 56 | 4.2 | 1.3 | 5.3 | ||||
| 9 | 7a | 2.5 | C6H6 | 2 | 31 | 3.1 | 2.1 | 2.7 |
| 30 | 75 | 6.8 | 7.0 | 6.5 | ||||
| 10 | 7d | 2.5 | C6H6 | 2 | 33 | 3.1 | 1.0 | 2.2 |
| 30 | 81 | 8.9 | 7.5 | 5.3 | ||||
All reactions were carried out using 0.005 mmol of catalyst, 0.20 mmol of 8, 0.40 mmol of 9 and 0.10 mmol of tridecane (internal standard for GC analysis) in 1.0 ml of C6H6 at 23°C.
Based on 8.
Conversion of 8 to the product determined by GC analysis.
Molar ratio of E isomer and Z isomer of the product determined by GC analysis.
Ref. 10a.
GC signal of the product was too small to quantify.
Catalyst 4, bearing the bulkier N-DIPP group, showed slightly higher conversion compared to catalyst 1a, but their trends were quite similar (Figure 7). After reaching a plateau at 60–70% conversion, the E/Z ratios remained relatively unchanged over time (Figure 7(b)). It is thought that this is due to the propensity of catalysts 1a and 4 to decompose under the reaction conditions, preventing metathesis events from isomerizing the Z-olefins to the E-isomer.23 Despite reaching similar conversions, differences in stereoselectivity were quite obvious between these two catalysts. Catalyst 4 provided a noticeably lower E/Z ratio of cross product 10 (49% E) compared to catalyst 1a (59% E) at ~ 60% conversion (Figure 7(b)). The higher Z-selectivity of 4 compared to 1a can be explained as follows: because the ruthenacyclobutane forms side-bound and is thus located below the N-aryl group, the bulkier N-DIPP group repels the substituent in the intermediate leading to E-olefin formation (R1 in Figure 8) more strongly than the smaller N-Mes group of 1a, contributing to the higher Z-selectivity (Figure 8).25
Figure 7.

Plots for (a) conversion vs time and (b) E/Z ratio vs conversion for CM of 8 and 9. a Conversion of 8 to 10 determined by GC analysis. b Molar ratio of E isomer and Z isomer of 10 determined by GC analysis.
Figure 8.

Plausible side-bound intermediates that lead to E-olefins for catalysts (a) 1a and (b) 4.
In order to further explore the effect of the bulkier N-aryl group on Z-selectivity, the metathesis homocoupling of substrate 8 (Table 2), and macrocyclic RCM reactions (Table 3) were attempted. As expected from the CM assay above, catalyst 4 exhibited higher Z-selectivity compared to 1a in both assays. In addition, catalyst 4 was superior to 1a in terms of selectivity for the desired homocoupled product 11 compared to the undesired isomerization product 12. In contrast to catalyst 1a, catalyst 4 reached a much higher ratio of 11/12 and maintained a high value even when the conversion reached a plateau at 120 minutes. Since the isomerization results from decomposed catalyst, the greater stability of 4 appears to arise from the bulky N-DIPP group of 4.27
Table 2.
Selected data for the metathesis homocoupling of 8 a
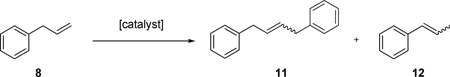 | |||||||
|---|---|---|---|---|---|---|---|
| 11 |
11/12e | ||||||
| entry | cat. | cat. load., mol %b | solvent | time, min | conv, %c | E/Z d | |
| 1 | 1a | 2.5 | C6H6 | 15 | 25 | 0.97 | 13 |
| 120 | 32 | 1.4 | 2.9 | ||||
| 2 | 4 | 2.5 | C6H6 | 45 | 24 | 0.97 | 26 |
| 120 | 29 | 1.1 | 19 | ||||
All reactions were carried out using 0.005 mmol of catalyst, 0.20 mmol of 8 and 0.10 mmol of tridecane (internal standard for GC analysis) in 1.0 ml of C6H6 at 23°C.
Based on 8.
Conversion of 8 to 11 determined by GC analysis.
Molar ratio of E isomer and Z isomer of 11 determined by GC analysis.
Determined by GC analysis.
Table 3.
Selected data for the macrocyclic RCM of 16 a
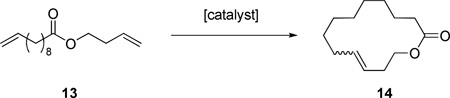 | ||||||
|---|---|---|---|---|---|---|
| 14 |
||||||
| entry | cat. | cat. load., mol %b | solvent | time, min | conv, %c | E/Zd |
| 1 | 1a | 5.0 | C6H | 30 | 17 | 1.1 |
| 120 | 24 | 1.1 | ||||
| 2 | 4 | 5.0 | C6H6 | 30 | 12 | 0.77 |
| 120 | 19 | 0.83 | ||||
All reactions were carried out using 0.003 mmol of catalyst, 0.060 mmol of 13 and 0.10 mmol of tridecane (internal standard for GC analysis) in 20 ml of C6H6 at 50°C.
Based on 13.
Conversion of 13 to 14 determined by GC analysis.
Molar ratio of E isomer and Z isomer of 14 determined by GC analysis.
The effect of the anionic ligand was evaluated by comparing the activity of catalysts 1a-d bearing increasingly bulkier carboxylate ligands in the standard CM reaction (Figure 9); the steric effects of these anionic ligands can be visualized from the crystal structures of the corresponding catalysts in Figure 4. As shown in Figure 9(a), all of the presented catalysts showed similar trends in conversion, reaching a plateau at around 60% within 30–90 minutes, however the stereoselectivity varied dramatically depending on the identity of the carboxylate ligand (Figure 9(b)). Proceeding from catalyst 1a to 1d, as the carboxylate ligand becomes bulkier, the E/Z ratio increases. Taking into account the side-bound intermediates proposed for 1a and 2a,14 plausible metallacyclobutane intermediates are presented (Figure 10). It is envisioned that the bulkier carboxylate ligand (i.e. Ph3CCOO (1d)) repels the metallacyclobutane substituents (R1 and R2 in Figure 10), destabilizing the ruthenacyclobutane intermediate that leads to productive formation of Z-olefins (Figure 10(b)) when compared to the less bulky pivalate ligand (i.e. tBuCOO (1a), Figure 10(a)). In the future design of Z-selective catalysts containing six-membered chelates, it is envisioned that substitution with less bulky ligands capable of binding in an η2-fashion will lead to improved catalysts.
Figure 9.

Plots for (a) conversion vs time and (b) E/Z ratio vs conversion for CM of 8 and 9. a Conversion of 8 to 10 determined by GC analysis. b Molar ratio of E isomer and Z isomer of 10 determined by GC analysis.
Figure 10.

Plausible side-bound intermediates for (a) 1a and (b) 1d.
Next, we briefly investigated the effects of substituting a chelated catalyst with a monodentate chloride ligand. Despite containing a chelating architecture that normally imparts improved Z-selectivity, catalyst 6 provided a high E/Z ratio of cross product 10 in the standard CM assay (Table 1). The E/Z ratio was significantly higher than chelated catalyst 1a, and more closely resembled that of non-chelated ruthenium metathesis catalysts like 5a. One possible explanation for this is that CM reactions catalyzed by 6 could proceed not via a side-bound intermediate (Figure 11(a)) as proposed for Z-selective catalysts 1a and 2a-b, but via a bottom-bound ruthenacyclobutane intermediate (Figure 11(b)) as proposed for non-chelated catalysts.28 In the bottom-bound intermediate, the ruthenacyclobutane substituents (R1 and R2 in Figure 11) would be located farther away from the NHC ligand, meaning that there would be less steric influence on the substituents, seemingly providing the thermodynamically preferred E-isomer. Given the lack of experimental and theoretic evidence, this hypothesis is conjecture at this point.
Figure 11.

Possible side-bound intermediate (a) and bottom-bound intermediate (b) for 6.
Previously, Jensen and coworkers reported that a non-chelated catalyst bearing a single bulky thiolate ligand (Figure 5) catalyzed highly Z-selective olefin metathesis and proceeded through a bottom-bound ruthenacyclobutane intermediate.10b Catalysts 7a and 7d bearing a single bulky carboxylate ligand were therefore prepared and tested in the standard CM assay. The trends in stereoselectivity for the non-chelated catalysts 7a and 7d were very similar to that of catalyst 5a, favoring formation of the E-isomer (Figure 12(b)). Clearly, the presence of a single carboxylate ligand does not lead to any observable Z-selectivity. It is envisioned that CM reactions catalyzed by 7a and 7d proceed via bottom-bound ruthenacyclobutane intermediates (Figure 13(b)), however the carboxylate ligand does not seem bulky enough to force the ruthenacyclobutane substituents (R1 and R2 in Figure 13) in the same direction to productively form Z-olefin products. Even if these catalysts proceeded via side-bound intermediates (Figure 13(a)), because of free rotation around the Ru-C(NHC) bond which is allowed in non-chelated structures, the N-Mes group of the NHC ligand cannot effectively repel the ruthenacyclobutane substituents. Either way, the thermodynamically favored E-isomer dominates in reactions catalyzed by 7a and 7d.
Figure 12.

Plots for (a) conversion vs time and (b) E/Z ratio vs conversion for CM of 8 and 9. a Conversion of 8 to 10 determined by GC analysis. b Molar ratio of E isomer and Z isomer of 10 determined by GC analysis.
Figure 13.

Possible (a) side-bound intermediate and (b) bottom-bound intermediate for 7a and 7d.
CONCLUSION
In summary, we have synthesized a series of catalysts containing six-membered chelating architectures and their non-chelated analogs, and determined their performance in various olefin metathesis assays. From these studies, new information has been gained about how changes to the NHC and anionic ligand affect a catalyst’s Z-selectivity. In designing a highly efficient Z-selective catalyst, understanding the roles of each ligand on a catalyst’s reactivity and selectivity is very useful. It was shown that catalysts require I) an NHC-chelated structure and II) an η2-bound carboxylate ligand to maintain high levels of Z-selectivity. When the bidentate pivalate ligand on catalyst 1a was replaced with a monodentate chloride (6), there was a marked decrease in Z-selectivity. Additionally, as demonstrated with catalyst 4, a bulky N-aryl group enhances Z-selectivity due to increased steric repulsion with ruthenacyclobutane substituents. As was previously shown, these NHC-chelated structures lock rotation of the NHC ligand and force the N-aryl group above the ruthenacyclobutane, allowing it to better influence it’s substituents and favoring formation of a cis-disubstituted ruthenacycle in order to productively form Z-olefins. In considering the difference in selectivity between catalysts 1a-1d bearing carboxylate ligands of different sizes, a less bulky η2-ligand seems to promote higher Z-selectivity, presumably because it enables more effective repulsion from the N-aryl group. In order to design highly Z-selective catalysts with a six-membered chelated architecture, catalyst activity and selectivity must be improved to compliment the family of five-membered chelated catalysts.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. D. Benitez, Dr. B. K. Keitz and Dr. P. Teo for helpful discussions and suggestions for this work. Materia, Inc. is thanked for the generous donation of catalysts. Dr. M. W. Day and Mr. L. M. Henling are acknowledged for X-ray crystallography analysis. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF CRIF:MU award to the California Institute of Technology, CHE-0639094. This work was financially supported by National Institutes of Health (NIH 5R01GM031332-27) and Mitsui Chemicals, Inc.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental details, NMR spectra, and crystallographic data are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Cossy J, Arseniyadis S, Meyer C. Metathesis in Natural Product Synthesis: Strategies, Substrates, and Catalysts. 1st ed. Weinheim, Germany: Wiley-VCH; 2010. [Google Scholar]
- 2.Binder JB, Raines RT. Curr. Opin. Chem. Biol. 2008;12:767. doi: 10.1016/j.cbpa.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrodi Y, Ung T, Vargas A, Mkrtumyan G, Lee CW, Champagne TM, Pederson RL, Hong SH. CLEAN - Soil, Air, Water. 2008;36:669. [Google Scholar]
- 4.(a) Leitgeb A, Wappel J, Slugovc C. Polymer. 2010;51:2927. [Google Scholar]; (b) Sutthasupa S, Shiotsuki M, Sanda F. Polymer Journal. 2010;42:905. [Google Scholar]; (c) Liu X, Basu A. J. Organomet. Chem. 2006;691:5148. [Google Scholar]
- 5.Fürstner A. Angew. Chem. Int. Ed. 2000;39:3012. [PubMed] [Google Scholar]
- 6.Schrock RR, Hoveyda AH. Angew. Chem. Int. Ed. 2003;42:4592. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]
- 7.(a) Trnka TM, Grubbs RH. Acc. Chem. Res. 2001;34:18. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]; (b) Samojlowicz C, Bieniek M, Grela K. Chem. Rev. 2009;109:3708. doi: 10.1021/cr800524f. [DOI] [PubMed] [Google Scholar]; (c) Vougioukalakis G, Grubbs RH. Chem. Rev. 2010;110:1746. doi: 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]
- 8.Grubbs RH, editor. Handbook of Metathesis. Vols. 1–3. Weinheim: Wiley-VCH; 2003. [Google Scholar]
- 9.(a) Flook MM, Jiang AJ, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2009;131:7962. doi: 10.1021/ja902738u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jiang AJ, Zhao Y, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:16630. doi: 10.1021/ja908098t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Marinescu SC, Schrock RR, Müller P, Takase MK, Hoveyda AH. Organometallics. 2011;30:1780. doi: 10.1021/om200150c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Meek SJ, O’Brien RV, Llaveria J, Schrock RR, Hoveyda AH. Nature. 2011;471:461. doi: 10.1038/nature09957. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Flook MM, Ng VWL, Schrock RR. J. Am. Chem. Soc. 2011;132:1784. doi: 10.1021/ja110949f. [DOI] [PubMed] [Google Scholar]; (f) Yu M, Wang C, Kyle AF, Jukubec P, Dixon DJ, Schrock RR, Hoveyda AH. Nature. 2011;479:88. doi: 10.1038/nature10563. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Townsent EM, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2012;134:11334. doi: 10.1021/ja303220j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wang C, Haeffner F, Schrock RR, Hoveyda AH. Angew. Chem. Int. Ed. 2013;52:1939. doi: 10.1002/anie.201209180. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wang C, Yu M, Kyle AF, Jakubec P, Dixon DJ, Schrock RR, Hoveyda AH. Chem. Eur. J. 2013;19:2726. doi: 10.1002/chem.201204045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Occhipinti G, Hansen FR, Törnroos KW, Jensen VR. J. Am. Chem. Soc. 2013;135:3331. doi: 10.1021/ja311505v. [DOI] [PubMed] [Google Scholar]; (b) Jensen VR, Occhipinti G, Hansen FR. Novel Olefin Metathesis Catalysts Int. Patent Appl. WO 2012032131. 2012 [Google Scholar]
- 11.(a) Endo K, Grubbs RH. J. Am. Chem. Soc. 2011;133:8525. doi: 10.1021/ja202818v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Keitz BK, Endo K, Herbert MB, Grubbs RH. J. Am. Chem. Soc. 2011;133:9686. doi: 10.1021/ja203488e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Keitz BK, Endo K, Patel PR, Herbert MB, Grubbs RH. J. Am. Chem. Soc. 2012;134:693. doi: 10.1021/ja210225e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rosebrugh LE, Herbert MB, Marx VM, Keitz BK, Grubbs RH. J. Am. Chem. Soc. 2013;135:1276. doi: 10.1021/ja311916m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Trnka TM, Morgan JP, Sanford MS, Wilhelm TE, Scholl M, Choi TL, Ding S, Day MW, Grubbs RH. J. Am. Chem. Soc. 2003;125:2546. doi: 10.1021/ja021146w. [DOI] [PubMed] [Google Scholar]; (b) Leitao EM, Dubberley SR, Piers WE, Wu Q, McDonald R. Chem. Eur. J. 2008;14:11565. doi: 10.1002/chem.200801584. [DOI] [PubMed] [Google Scholar]
- 13.(a) Herbert MB, Marx VM, Pederson RL, Grubbs RH. Angew. Chem. Int. Ed. 2013;52:310. doi: 10.1002/anie.201206079. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Marx VM, Herbert MB, Keitz BK, Grubbs RH. J. Am. Chem. Soc. 2013;135:94. doi: 10.1021/ja311241q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Liu P, Xu X, Dong X, Keitz BK, Herbert MB, Grubbs RH, Houk KN. J. Am. Chem. Soc. 2012;134:1464. doi: 10.1021/ja2108728. [DOI] [PubMed] [Google Scholar]; (b) Dang Y, Wang ZX, Wang X. Organometallics. 2012;31:7222. [Google Scholar]; (c) Dang Y, Wang ZX, Wang X. Organometallics. 2012;31:8654. [Google Scholar]; (d) Miyazaki H, Herbert MB, Liu P, Dong X, Xu X, Keitz BK, Ung T, Mkrtumyan G, Houk KN, Grubbs RH. J. Am. Chem. Soc. 2013;135:5848. doi: 10.1021/ja4010267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.We recently reported that an intramolecular C-H bond activation for adamantyl analogue 2c could be achieved only when sodium pivalate was used. See ref. 11d.
- 16.When [H2I (DIPP)2]RuCl2[=CH-o-(OiPr)C6H4] was reacted with silver pivalate, a complex bearing two pivalte ligands was formed but no intramolecular C-H bond activation was observed even upon heating.
- 17.5b and 5c were synthesized by modification of reported procedure: Krause JO, Nuyken O, Wurst K, Buchmeiser MR. Chem. Eur. J. 2004;10:777. doi: 10.1002/chem.200305031.
- 18.A mild method to effect the salt metathesis and C-H activation directly using NaOPiv was recently reported (ref 11d). However, a reaction of 5a with NaOAc only led to catalyst decomposition. This result may suggest that bulky carboxylate is necessary for formation and/or stabilization of the NHC-chelating catalyst.
- 19.All the acidic protons of the carboxylic acids were detected around δ = 10.5–11.5 ppm in 1H NMR spectra.
- 20.Attempts to replace the pivalate ligand on catalyst 1a with a nitrate ligand were unsuccessful.
- 21.Teo P, Grubbs RH. Organometallics. 2010;29:6045. [Google Scholar]
- 22.Ritter T, Hejl A, Wenzel AG, Funk TW, Grubbs RH. Organometallics. 2006;25:5740. [Google Scholar]
- 23.Reported decomposition pathways for 2a, in which the alkylidene moiety inserts into the Ru-C bond, followed by hydride elimination, were suggested from isolated decomposed product and supported by computational results (See Ref. 24). It is proposed that reaction intermediates of catalyst with six-membered chelates, including methylidene complexes, tend to suffer from analogous decomposition pathways. Although none of decomposed product was isolated, dramatic color change of the reaction solution from green to yellow were observed for 1a and 4, respectively. In RCM of diethyldiallyl malonate and ROMP of 1,5-cyclooctadiene by catalysts 1a and 4, conversion also reached a plateau within 15–60 minutes. See detail in SI.
- 24.Herbert MB, Lan Y, Keitz BK, Liu P, Endo K, Day MW, Houk KN, Grubbs RH. J. Am. Chem. Soc. 2012;134:7861. doi: 10.1021/ja301108m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.We have also reported higher Z-selectivity for 2c substituted with an N-DIPP group compared to 2b substituted with an N-Mes group. See ref. 11d.
- 26.(a) Ivin KJ, Mol JC. Olefin Metathesis and Metathesis Polymerization. San Diego, CA: Academic Press; 1997. [Google Scholar]; (b) Pederson RL, Fellows IM, Ung TA, Ishihara H, Hajela S. Adv. Synth. Catal. 2002;344:728. [Google Scholar]; (c) Lehman SE, Schwendeman JE, O’Donnell PM, Wagener KB. Inorg. Chim. Acta. 2003;345:190. [Google Scholar]; (d) Schmidt B. Eur. J. Org. Chem. 2004:1865. and references therein. [Google Scholar]
- 27.We have also reported that 2c gave higher proportion of the metathesis product than 2b in the homocoupling of 8. See ref 11d.
- 28.(a) Adlhart C, Chen P. J. Am. Chem. Soc. 2004;126:3496. doi: 10.1021/ja0305757. [DOI] [PubMed] [Google Scholar]; (b) Cavallo L, Correa A. J. Am. Chem. Soc. 2006;128:13352. doi: 10.1021/ja064924j. [DOI] [PubMed] [Google Scholar]; (c) Benitez D, Tkatchouk E, Goddard WA., III Chem. Commun. 2008:6194. doi: 10.1039/b815665d. [DOI] [PubMed] [Google Scholar]
- 29.DFT calculation for 1a, 2a and 2b shows that side-bound pathways are energetically more favored than the bottom-bound pathways. See ref. 14.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.