

Abstract
Activation of the serotonin (5-hydroxytryptamine, 5-HT) 5HT2C G protein-coupled receptor (GPCR) is proposed as novel pharmacotherapy for obesity and neuropsychiatric disorders. In contrast, activation of the 5-HT2A and 5-HT2B GPCRs is associated with untoward hallucinogenic and cardiopulmonary effects, respectively. There is no crystal structure available to guide design of 5-HT2C receptor-specific ligands. For this reason, a homology model of the 5-HT2C receptor was built based on the crystal structure of the human β2 adrenoceptor GPCR to delineate molecular determinants of ligand–receptor interactions for drug design purposes. Computational and experimental studies were carried out to validate the model. Binding of N(CH3)2-PAT [(1R, 3S)-(−)-trans-1-phenyl-3-N,N-dimethylamino-1,2,3,4-tetrahydronaphthalene], a novel 5-HT2C agonist/5-HT2A/2B inverse agonist, and its secondary [NH(CH3)-PAT] and primary (NH2-PAT) amine analogs were studied at the 5-HT2C wild type (WT) and D3.32A, S3.36A, and Y7.43A 5-HT2C point-mutated receptors. Reference ligands included the tertiary amines lisuride and mesulergine and the primary amine 5-HT. Modeling results indicated that 5-HT2C residues D3.32, S3.36, and Y7.43 play a role in ligand binding. Experimental ligand binding results with WT and point-mutated receptors confirmed the impact of D3.32, S3.36, and Y7.43 on ligand affinity.
Keywords: drug design, GPCR homology modeling, serotonin 5-HT2C receptor
Introduction
Ligands that activate the serotonin 5-hydroxy-tryptamine (5-HT2C) G protein-coupled receptor (GPCR) may be therapeutic for obesity, psychoses, psychostimulant addiction, and other neuropsychiatric disorders [1, 2]. Clinically, there is essentially no tolerance for activation of 5-HT2A and 5-HT2B receptors that can lead to untoward hallucinogenic and cardiopulmonary effects, respectively. The 5-HT2 GPCR family shares approximately 80% sequence homology [3] and have a common primary signaling pathway, complicating 5-HT2C receptor-specific agonist development.
GPCRs putatively share a three-dimensional (3D) structure consisting of a bundle of seven transmembrane (TM) alpha helices, connected by alternating intracellular and extracellular loops, with the N-terminus in the extracellular domain and C-terminus in the intracellular domain. To date, GPCR crystal structures have been reported for bovine rhodopsin (bRho) [4–8], opsin, [9, 10], turkey β1 adrenoceptor (β1AR) [11], human β2 AR (β2AR) in an inactive state [12–15], human A2A adenosine receptor (AA2AR) [16], a nanobody-stabilized active state of the β2AR [17], and the structure of an irreversible agonist–β2AR complex [18]. The 3D structures of the serotonin GPCR families (5-HT1, 5-HT2, 5-HT4, 5-HT5, 5-HT6, and 5-HT7) remain elusive, as is the case for most other GPCRs. A recent review summarizes the GPCRs structure known to date [19].
All ligands for aminergic neurotransmitter GPCRs contain a basic amine moiety. The endogenous neurotransmitters have a primary amine moiety, but, ligands with secondary or tertiary amine groups also are known, including, N(CH3)2-PAT [(1R, 3S)-(−)-trans-1-phenyl-3-N,N-dimethylamino-1,2,3,4-tetrahydronaphthalene], a novel 5-HT2C agonist/5-HT2A/2B inverse agonist [20]. Mutagenesis studies indicate that the fully conserved aspartate residue D3.32 of aminergic neurotransmitter GPCRs interacts with the positively charged amine moiety of endogenous agonists and other ligands at physiological pH; this ionic interaction is required for ligand binding [21–23]. In 5-HT2 receptors, there is a conserved serine residue S3.36, approximately one helical turn from D3.32. Mutagenesis experiments and molecular modeling studies (based on the bovine rhodopsin structure) of the 5-HT2A receptor [24, 25] suggest that the 5-HT primary amine moiety forms a hydrogen bond to S3.36 and D3.32. Mutation of the 5-HT2A S3.36 residue to alanine (S3.36A) decreases affinity of primary amine more than secondary amine ligands, whereas, affinity of tertiary amines is barely affected. For the 5-HT2C receptor, the effects of the D3.32A, S3.36A, and Y7.43A single point mutations on ligand binding have been reported recently (Canal et al., submitted for publication; Refs. [26–28]). Such experimental ligand binding and mutagenesis studies have been used to validate the accuracy of GPCR homology models, including, for the 5-HT2C receptor. In this study, we describe the building of a 5-HT2C model based on homology to the human β2AR and analyze its structure using molecular dynamics (MD) simulations and ligand docking techniques. Results are presented that compare ligand docking outcomes to experimental binding results using several tertiary, secondary, and primary amine ligands at wild type (WT) and D3.32A, S3.36A, and Y7.43A point-mutated 5-HT2C receptors.
Methods
MODEL BUILDING
A homology model of the human serotonin 5-HT2C receptor was built based on the crystal structure of the β2AR/T4-lysozyme chimera (Protein Data bank entry 2rh1) [14]. The 5-HT2C native sequence was aligned to the β2AR sequence using ClustalW multiple sequence alignment [29, 30]. Point mutations were performed as needed and the gaps were analyzed, followed by the appropriate sequence additions and deletions to match the 5-HT2C receptor amino acid sequence. The TM domains were built using the Biopolymer module of Sybyl 8.1 (Tripos International, St. Louis, MO). The inverse agonist carazolol, present in the crystal structure of the human hβ2AR (2rh1), was deleted and the resulting 7-TM bundle was optimized using Tripos force field [31]. Regions outside the 7-TM bundle such as the 5-HT2C N-terminus, C-terminus, extracellular and intracellular loop residues were built using loop database PRODAT in Sybyl 8.1. The disulfide bond formed between the cysteine amino acids C3.25 (at the end of TM domain 3) and C6.31 (in extracellular loop 3) in the β2AR structure was conserved in the 5-HT2C model. The crude model was minimized using the Powell method implemented in Sybyl with Tripos force field [31] and AMBER charges [32]. The resulting model was inserted into a rectangular box containing a pre-equilibrated 1-palmitoyl-2-oleyl-sn-glycero phosphatidyl choline (POPC) bilayer [33] that consisted of 200 lipid molecules, forming a rectangular patch, and, 5,483 water molecules covering the head groups on each side of the bilayer. The system containing the WT 5-HT2C receptor model within the simulated membrane contained 37,775 atoms. The system was relaxed using the Tripos force field to a gradient 0.05 kcal/Å mol, prior to MD simulation in the POPC membrane. MD simulation conditions were time run 5000 ps, time step 1 fs, with snapshots collected every 5 fs. Other parameters were the constant number of particles (N), volume (V) and temperature (T) NVT canonical ensemble, 300 K temperature, Boltzmann initial velocities, and nonbonded cutoff set at 8 Å. Constraints for alpha carbons in the TM domains were used. Subsequently, the constraints were removed for a 1000 ps MD simulation run. The final model was obtained from the median structure after clustering analysis of the frames from the last 10 ps of the MD simulation and optimized using the Tripos force field to a convergence of 0.05 kcal/ Å mol. The 3D molecular models of the D3.32A, S3.36A, and Y7.43A 5-HT2C mutated receptors were obtained by point mutation of the WT 5-HT2C receptor followed by relaxation and MD procedures as described above.
LIGAND DOCKING
Ligands used in this study were the tertiary amines lisuride, mesulergine, (1R, 3S)-(−)-trans-1-phenyl-3-N,N-dimethylamino-1,2,3,4-tetrahydronaphthalene (N(CH3)2-PAT) [20] its secondary amine analog NHCH3-PAT, and its primary amine analog NH2-PAT, as well as, the primary amine endogenous agonist 5-HT (Fig. 1). The ligand structures were built as monocations (protonated amines) using HyperChem 8.0 (Hyper-Chem (TM) Professional 8.0, Hypercube, Gainesville, FL) and structures were optimized using PM3 model Hamiltonian to a gradient of 0.01 kcal/Å mol.
FIGURE 1.

Ligands used in 5-HT2C docking studies.
Ligands were prepositioned in the binding pocket by performing rigid docking with the PatchDock server [34]. The low-energy–high-score solutions were analyzed to select the initial configuration, ensuring the essential interaction between the carboxylate oxygen of receptor residue D3.32 and the ligand protonated amine moiety [21, 22]. The initial ligand–receptor complex configuration was used for flexible ligand docking with Flexidock in Sybyl 8.1. Flexidock uses an algorithm to probe the conformational space defining possible interactions between the ligand and its putative binding site. The binding site was defined by assigning residue D3.32 as a definitive binding site interaction point, and including residues within a 7 Å radius. Structure preparation was carried out prior to docking studies assigning AMBER [32] charges for the protein and Gesteiger-Marsili [35] charges for the ligand. Rotatable bonds in the ligand and the side chains of residues defining the receptor putative active site were screened for optimal positioning of the ligand and side chains in the conformational space; remaining residues were frozen during docking. Default FlexiDock parameters were set at 80,000 generation. The best docking solution, according to the highest FlexiDock score, was minimized using the Tripos force field to a gradient 0.05 kcal/Å mol, prior to MD simulation. The selected high-score pose of the docked ligand was subjected to a MD simulation run for 500 ps, with other parameters the same as above. The final structure of the ligand docked into the receptor was obtained from the average of last 10 ps of the dynamics simulation.
SATURATION AND COMPETITION BINDING STUDIES
Saturation and competition binding studies were carried out as previously described (Canal et al., submitted for publication). The WT, D3.32A, S3.36A, and Y7.43A 5-HT2C receptors were radiolabeled with [3H]-mesulergine (Perkin-Elmer), having specific activity of 92 Ci/mmol. Test ligands were: 5-HT hydrochloride (Alfa Aesar), lisuride hydrogen maleate (gift from Dr. Elaine Sanders-Bush (Vanderbilt University, Nashville, TN). Synthesis of N(CH3)2-, NH(CH3)-, and NH2-PAT was performed according to previous laboratory methods [20, 36] to obtain the hydrochloride salt of the trans-stereoisomers [ligand docking studies considered only the active (1R, 3S) enantiomer]; ligand structures are given in Figure 1. The cDNA encoding the human unedited WT 5-HT2C receptor was obtained from University of Missouri-Rolla (UMR) cDNA Resource Center (Rolla, MO). The D3.32A, S3.36A, and Y7.43A point-mutated 5-HT2C receptors were generated by polymerase chain reaction (PCR) using standard procedures (Canal et al., submitted for publication). For saturation binding, [3H]-mesulergine was used to obtain KD and Bmax values. Competitive displacement experiments were used to determine IC50 values. Ligand affinity is expressed as an approximation of Ki values by conversion of the IC50 data using the equation Ki = IC50/1 + L/KD where L is the concentration of radioligand having affinity KD [37]. Comparisons of Ki and KD values between WT, S3.36A, and Y7.43A 5-HT2C receptors were performed using one-way analysis of variance (ANOVA)s with Tukey’s multiple comparison post hoc tests.
Results and Discussion
HOMOLOGY MODELING
The sequence of the WT 5-HT2C receptor was aligned to the sequence of the template structure β2A receptor using ClustalW. Sequence alignment showing the TM domains is shown in Table I. Conserved residues are indicated in bold, and reference residues are labeled according to the standard Ballesteros nomenclature for GPCRs [23]. This nomenclature allows for comparison of GPCRs having different residue sequences. The most conserved residue in a given TM domain is labeled Xη.50, where X is the one-letter amino acid code and η is the TM helix (TMH) number; the other amino acids in each TMH are numbered according to this reference residue. Despite the low overall sequence identity between the β2A and 5-HT2C receptors (~30%), the majority of highly conserved residues among the 5-HT2 and β2A GPCRs are in the TM helices, thus allowing for accurate alignment.
TABLE I.
Alignment of 5-HT2C and β2ADR receptor sequences using ClustalW.

|
Conserved residues are indicated in bold. Reference residues are labeled according to Ballesteros nomenclature [23].
Star(*) indicated the boundaries of TM domains.
Conserved residue sequences among 5-HT2 receptors and the β2A receptor in the TM domains were used to verify the alignment, and included: the GNXLVI motif in TM I, including the reference residue N1.50, the TNYF, SLAXAD motifs in TM II, including the reference residue D2.50, the DVL motif in TM III, including the essential residue D3.32, and the TASI and DRY motifs, the KA motif and W4.50 in TM IV, the FXXPLXIM motif in TM V, including P5.50, the WXPFFIXNI motif in TM VI, including residues W6.48, P6.50, and the WIGY and NPLXY motifs in TM VII, including residue P7.50.
The alignment in Table I was used to generate the 3D model of the WT 5-HT2C receptor. The initial WT 5-HT2C model, generated as described in the Model Building section, was optimized in vacuum and subsequently equilibrated using MD simulation in the POPC membrane. The resulting TM bundle structure of the WT 5-HT2C receptor is shown in Figure 2, generated with PyMOL 1.3 [38]. TMH are spectrum color-coded, from blue for TM I, to red for TM VII. In Figure 2, panel A shows the TM bundle oriented with the extracellular domain on top and the intracellular domain at the bottom. Panel B is the view from the extracellular domain down into the receptor cavity. The overall structure of the D3.32A, S3.36A, and Y7.43A point-mutated 5-HT2C receptors (not shown) was similar to the WT 5-HT2C receptor.
FIGURE 2.
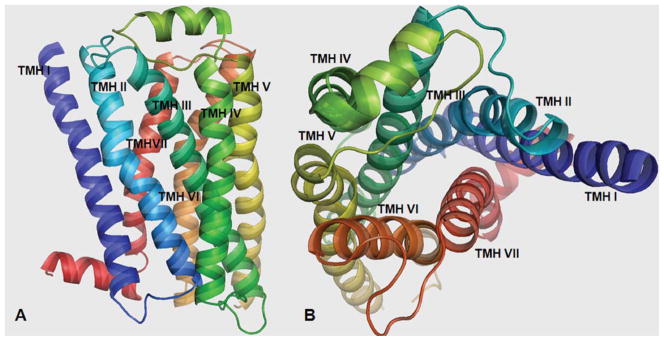
Three-dimensional 5-HT2C model structure: Panel A: TM domains with the extracellular domain on top and the intracellular domain at the bottom. Panel B: View from the extracellular domain showing the binding site cavity. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
At the end of the process, the 5-HT2C model was analyzed using PDBsum in PROCHECK 3.6.2 [39, 40]. Figure 3 shows the Ramachandran plot for the 5-HT2C model; PROCHECK statistics are reported in Table II. Analysis of the Ramachandran plot results show that 96.9% of residues are in the allowed regions, 89.1% are in the most favored regions (Psi angle values vary from −180° to 0°; Phi angle values vary from 0° to 180°; bottom-left quadrant, Fig. 3), and 7.8% are in additional allowed regions; supplementary 2.7% are in generously allowed regions, confirming the structure to a reasonable model.
FIGURE 3.
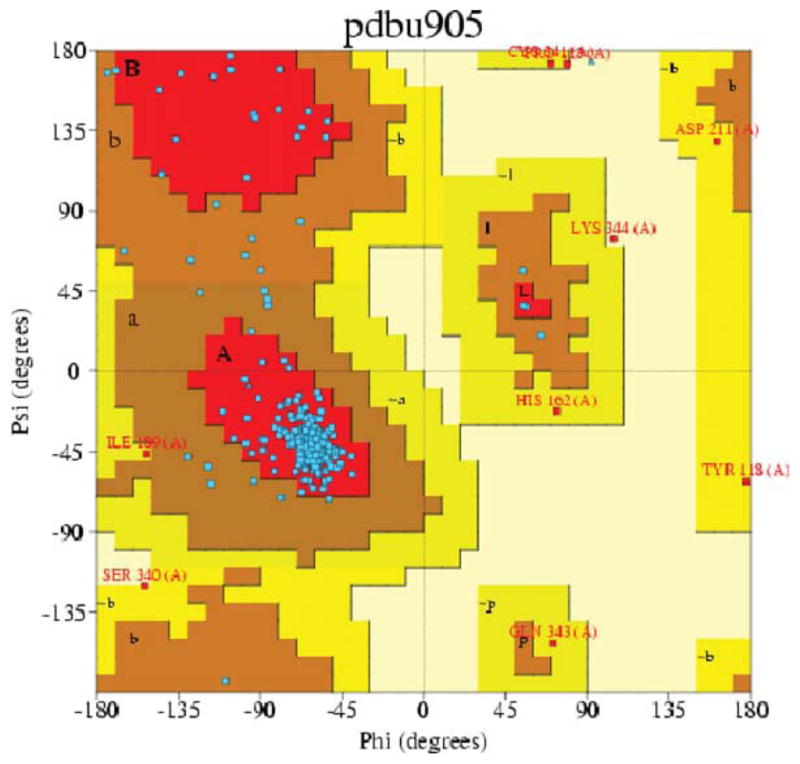
Ramachandran Plot of the 5-HT2C homology model generated with PROCHECK. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
TABLE II.
Procheck statistics of TM residues in the 5-HT2C homology model.
| Number of residues | % tage | |
|---|---|---|
| Most favored regions | 228 | 89.1 |
| Additional allowed regions | 20 | 7.8 |
| Generously allowed regions | 7 | 2.7 |
A description of the residues contained in each TM domain is given in Table III. The TM spanning bundle is composed of seven helices (TMH I–TMH VII) of variable length (23–34 residues), with the longest being TMH III that orients diagonally with respect to the whole TM bundle (Fig. 2). TMH VII is the shortest (23 residues) and continues to helix VIII (12 residues) that orients nearly perpendicular to the TM spanning bundle.
TABLE III.
5-HT2C transmembrane domain residues.
| Domain | Residues in TM | Reference residue |
|---|---|---|
| TMH I | P49-M80, 31 residues | N71 (N1.50) |
| TMH II | A87-Y116, 29 residues | D99 (D2.50) |
| TMH III | P123-R157, 34 residues | R152 (R3.50) |
| TMH IV | R168-V191, 23 residues | W179 (W4.50) |
| TMH V | P212-Q244, 32 residues | P226 (P5.50) |
| TMH VI | N305-L336, 31 residues | P326 (P6.50) |
| TMH VII | E347-L370, 23 residues | P365 (P7.50) |
| TMH VIII | K373-C385, 12 residues |
The overall structure of the 7-TM bundle is similar to the previously reported rhodopsin-based 5-HT2C homology model [20]; however, some differences are found mainly arising from the template receptor structure used. For example, there was a more pronounced kink in TM V near P5.50 in the Rho-based model compared to the β2AR-based 5-HT2C model, a stronger helix kink in TM VI near P6.50 in the β2AR-based 5-HT2C model compared to the Rho-based model, and a small helix in the extracellular domain loop connecting TM IV and TM V for the βAR-based 5-HT2C model that is not present in the Rho-based model.
LIGAND BINDING
No specific binding of [3H]-mesulergine to the D3.32A point-mutated 5-HT2C receptor was detected (data not shown), consistent with results previously reported (Canal et al., submitted for publication). Values for ligand binding (Ki) at WT, S3.36A, and Y7.43A 5-HT2C receptors are shown in Table IV. The S3.36A mutation caused significant reduction in affinity (higher Ki values) for the primary amine ligands 5-HT and NH2PAT, and, modestly decreased affinity of the secondary and tertiary amines NHCH3-PAT and N(CH3)2-PAT, but, did not affect affinity of the tertiary amine lisuride. The Y7.43A mutation caused a large reduction in affinity for 5-HT and a moderate reduction for the secondary and tertiary amines, NHCH3-PAT and N(CH3)2-PAT. In contrast, no effect was observed for the primary amine NH2-PAT, and a moderate increase in affinity was found for the tertiary amine lisuride (Canal et al., submitted for publication). These experimental mutagenesis results provide important information regarding ligand–receptor molecular interactions. Thus, ligand affinity is impacted when, for example, hydrogen and/or electrostatic binding interactions are lost due to residue mutation to alanine. The experimental results were used to validate molecular interactions of the ligands in the 5-HT2C binding pocket proposed from molecular modeling and ligand docking experiments.
TABLE IV.
Ki values of ligands tested at WT, S3.36A, and Y7.43A 5-HT2C receptors.
| Ligand | WT |
Ki (nM)
|
Fold change relative to WT
|
||
|---|---|---|---|---|---|
| S3.36A | Y7.43A | S3.36A | Y7.43A | ||
| N(CH3)2-PAT | 80 | 183 | 238 | 2.3 | 3.0 |
| NHCH3-PAT | 183 | 564 | 505 | 3.1 | 2.8 |
| NH2-PAT | 1,800 | 8,992 | 1,736 | 5.0 | 1.0 |
| 5-HT | 7 | 89 | 112 | 12.4 | 15.6 |
| Lisuride | 10 | 9 | 6 | 0.9 | 0.6 |
LIGAND DOCKING
Residues observed in the putative binding pocket of the 5-HT2C receptor model [Fig. 4(A)] include, W3.28, I3.29, L3.31, D3.32, V3.33, S3.36, T3.37, S5.43, F5.47, M6.47, W6.48, C6.49, F6.51, F6.52, N6.55, N7.36, V7.39, W7.40, G7.42, and Y7.43. With no ligand bound, the model showed that the para-hydroxy group of the Y7.43 residue was in close proximity (3.35 Å) to the carboxylate group of D3.32 [Fig. 4(A)]; this close interaction (3.38 Å) was also observed in the template β2AR structure bound to the inverse agonist carazolol [12–14]. This result suggests that Y7.43 plays a role in stabilizing the negative charge of the D3.32 carboxylate; however, this result may vary depending on the ligand structure and conformation/activation state of the receptor. On binding of 5-HT [Fig. 4(B)], the distance between the Y7.43 para-hydroxy group and D3.32 carboxylate was shortened to 2.50 Å. The 5-HT protonated amine group formed a hydrogen bond to the D3.32 carboxylate group (1.85 Å) and also to S3.36 (2.20 Å), as well as, to Y7.43 over a longer distance (3.12 Å). The 5-hydroxy group of 5-HT was near N6.55 (4.28 Å) and D5.35 (5.31 Å), likely forming electrostatic interactions, but too far for hydrogen bonding. An alternative 5-HT pose (not shown) was found in which the protonated amino moiety of 5-HT is able to form hydrogen bond interactions with the side chain of D3.32 (1.85 Å), S3.36 (2.08 Å), and Y7.43 (3.10 Å), while the 5-hydroxy group was in close proximity to Y7.43 para-hydroxy group (2.46 Å). In the S3.36A and Y.743A 5-HT2C receptor models (not shown), hydrogen bond interactions between 5-HT and residues S3.36 and Y7.43 were not present, consistent with the lower affinity of 5-HT at the S3.36A and Y7.43A point-mutated 5-HT2C receptors compared to the WT receptor (Table IV).
FIGURE 4.
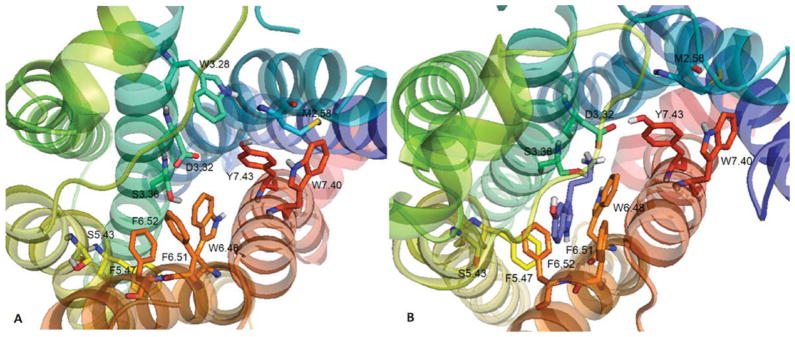
(A) Unbound WT 5-HT2C receptor and (B) serotonin (5-HT) docked to WT 5-HT2C receptor. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The effect of the 5-HT2C S3.36A point mutation on binding affinity of primary, secondary, and tertiary amine ligands studied here was similar to that observed for the 5-HT2A S3.36A receptor [24, 25]. Protonated primary amines (5-HT, NH2-PAT) having three hydrogen atoms available for interactions with D3.32 and S3.36 were more sensitive to mutation of these residues as compared to secondary (NHCH3-PAT) and tertiary amine [N(CH3)2-PAT, lisuride] ligands (Table IV).
For the Y7.43A mutation, factors other than or in addition to hydrogen bonding apparently come into play, depending on the ligand. For the primary amine 5-HT, the Y7.43A mutation produced an even greater loss of affinity than the S3.36A mutation. For the PAT analogs, however, the tertiary and secondary amines were more affected than the primary amine by the Y7.43A mutation. As results for the S3.36A mutation demonstrate, if hydrogen bonding was the primary interaction between Y7.43 and the PAT analogs, the Y7.43A mutation should negatively impact affinity of the primary amine more than the secondary and tertiary amines. Thus, the experimental binding results for the Y7.43A point-mutated receptor suggest the possibility of other interactions. For example, in the case of N(CH3)2-PAT, the ligand docked at 5-HT2C receptor such that aromatic interactions with the Y7.43 side chain were possible. In contrast, the primary amine NH2-PAT docked in a position that did favor π–π interactions with the Y7.43 side chain. Given that the affinity of N(CH3)2-PAT is greater than that of NHCH3-PAT and NH2-PAT at the WT 5-HT2C receptor (Table IV), it is likely that the PAT N-methyl groups not only increase the basicity of the amine nitrogen but also form van der Waals and perhaps other interactions that lead to a tight fit in the binding pocket. Unfortunately, MD simulations using the Y7.43F point-mutated 5-HT2C receptor indicated loss of electrostatic interactions between F7.43 and the D3.32 side chain, which allowed for greatly increased rotation of the F7.43 side chain that confounded interpretation.
Figures 5, 6, and 7 show N(CH3)2-PAT, NHCH3-PAT, and NH2-PAT, respectively, docked at the WT 5-HT2C binding pocket. For N(CH3)2-PAT, NHCH3-PAT, and NH2-PAT (respectively): the protonated amino groups form hydrogen bonds with D3.32 (1.84, 1.97, 1.93 Å) with S3.36 (3.27, 2.84, 2.62 Å) and Y7.43 (2.96, 2.78, 3.70 Å), albeit, over a longer distance. Docking poses show similar configurations of N(CH3)2-PAT and NHCH3-PAT, where the aromatic ring in the PAT bicycle structure is close to the aromatic ring of Y7.43 side chain. Moreover, for N(CH3)2-PAT, the bicycle aromatic ring is parallel to the Y7.43 aromatic ring, suggesting π–π stacking interactions. However, for NH2-PAT, the bicycle aromatic ring is not positioned for π–π stacking interactions with Y7.43. Loss of π–π stacking interactions for N(CH3)2-PAT and NHCH3-PAT apparently accounts for the reduced affinity of these analogs at the Y7.43A point-mutated receptor, whereas, no change in affinity was observed for NH2-PAT (Table IV).
FIGURE 5.

N(CH3)2-PAT docked in WT 5-HT2C receptor. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
FIGURE 6.

NHCH3-PAT docked in WT 5-HT2C receptor. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
FIGURE 7.

NH2-PAT docked in WT 5-HT2C receptor. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 8 shows lisuride docked at the 5-HT2C binding pocket. The lisuride configuration in the binding pocket allows for formation of a hydrogen bond to D3.32 (1.56 Å), but not with S3.36 (7.21 Å), or to Y7.43 (3.95 Å). Consistent with these modeling results, affinity of lisuride at the S3.36A point-mutated receptor was not different than the WT receptor (Table IV). Also in the molecular model, a hydrogen at the amide side chain of N6.55 is in close proximity to the carbonyl oxygen of lisuride (1.72 Å), likely, providing an additional stabilizing interaction that may explain the moderate increase in affinity of lisuride at the point-mutated compared to WT 5-HT2C receptor (Table IV) (see supporting information).
FIGURE 8.
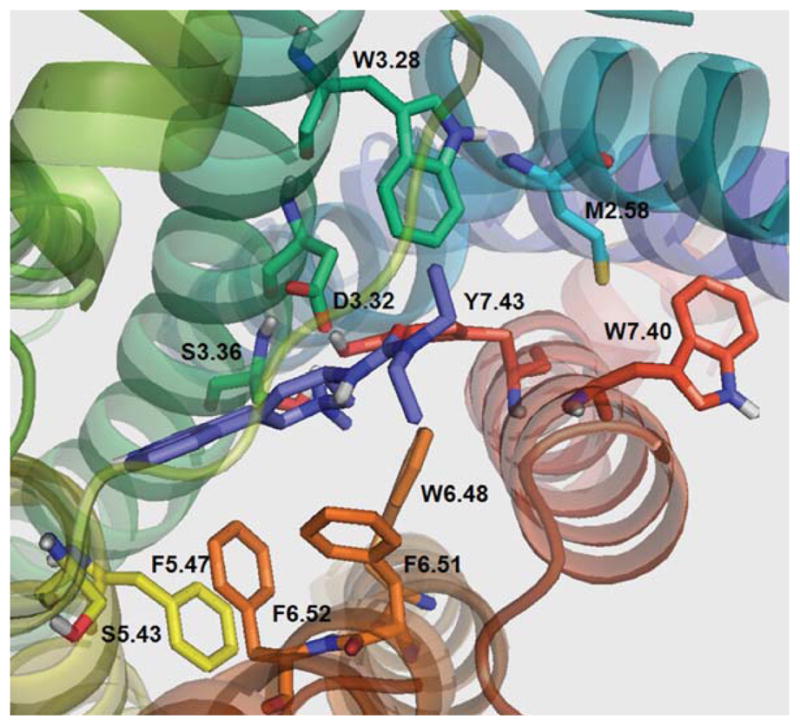
Lisuride docked in WT 5-HT2C receptor. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Conclusions
The 5-HT2C GPCR homology model built from the human β2AR crystal structure was tested using computational techniques and ligand docking results were validated by comparison to results obtained from point-mutated receptor binding experiments. The model structure was stable after unrestrained MD simulations in lipid bilayer membrane. PROCHECK statistics show that the majority of residue side chains are in allowed regions. The overall structure of the 7-TM bundle was similar to the previously reported rhodopsin-based 5-HT2C homology model [20]; however, some differences were apparent due to the different template structure used. The human 5-HT2C homology model herein explains the experimental binding results of the non-selective 5-HT2 agonist 5-HT and the selective 5-HT2C agonist N(CH3)2-PAT [20]. The carboxylate of 5-HT2C residue D3.32 was found to be in close proximity to the para-hydroxy phenyl group of Y7.43, which suggests that a hydrogen bond between these amino acids is involved in binding pocket stabilization. Additionally, it appears that ligand binding modulates the distance between the 5-HT2C D3.32 and Y7.43 side chains and that this distance varies, depending on the ligand chemical structure.
While results here document the usefulness of combining mutagenesis and molecular modeling/ computational chemistry approaches to predict GPCR ligand affinity, extending such results to predict function is fraught with complexity. A critical assumption of current GPCR signaling theory is that there exists a heterogeneous population of active and inactive receptor conformations. However, the molecular determinants governing ligand binding that lead to stabilization of agonist versus inverse agonist GPCR conformation states are poorly understood. Moreover, it is now well-established that the same ligand may stabilize both agonist and inverse agonist conformations of the same GPCR, and, that the same GPCR may activate multiple signaling pathways [41–43]. Accordingly, current GPCR drug discovery efforts may reliably be guided by computationally predicted ligand affinity; however, functional outcome(s) largely must still be experimentally determined.
Acknowledgments
Contract grant sponsor: National Institutes of Health.
Contract grant numbers: DA023928, DA030989, and MH081193.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Jensen NH, Cremers TI, Sotty F. Sci World J. 2010;10:1870. doi: 10.1100/tsw.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bubar MJ, Cunningham KA. Prog Brain Res. 2008;172:319. doi: 10.1016/S0079-6123(08)00916-3. [DOI] [PubMed] [Google Scholar]
- 3.Kroeze WK, Kristiansen K, Roth BL. Curr Top Med Chem. 2002;2:507. doi: 10.2174/1568026023393796. [DOI] [PubMed] [Google Scholar]
- 4.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Trong IL, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:793. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Edwards PC, Burghammer M, Villa C. J Mol Biol. 2004;343:1409. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 6.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J Mol Biol. 2004;342:571. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 7.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Proc Natl Acad Sci USA. 2002;99:5982. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. Biochemsitry. 2001;40:7761. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Nature. 2008;454:183. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 10.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauβ N, Choe HW, Hofmann KP, Ernst OP. Nature. 2008;455:497. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 11.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AGW, Tate CG, Schertler GFX. Nature. 2008;454:486. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Ns WI, Kobilka BK, Stevens RC. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. Structure. 2008;16:897. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Nature. 2007;450:383. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 15.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. Science. 2007;318:1266. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 16.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. Science. 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rassmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, DeVree BT, Rosembaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Nature. 2011;469:175. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosembaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, DeVree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK. Nature. 2011;469:236. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Congreve M, Langmead CJ, Mason JS, Marshall FH. J Med Chem. 2011;54:4283. doi: 10.1021/jm200371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Booth RG, Fang L, Huang Y, Wilczynski A, Sivendran S. Eur J Pharmacol. 2009;615:1. doi: 10.1016/j.ejphar.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kristiansen K, Dahl SG. Eur J Pharmacol. 1996;306:195. doi: 10.1016/0014-2999(96)00180-x. [DOI] [PubMed] [Google Scholar]
- 22.Kristiansen K, Kroeze WK, Willins DL, Gelber EI, Savage JE, Glennon RA, Roth BL. J Pharmacol Exp Ther. 2000;293:735. [PubMed] [Google Scholar]
- 23.(a) Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. J Biol Chem. 2001;276:29171. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]; (b) Ballesteros JA, Shi L, Javitch JA. Mol Pharmacol. 2001;60:1. [PubMed] [Google Scholar]
- 24.Almaula N, Ebersole BJ, Ballesteros JA, Weinstein H, Sealfon SC. Mol Pharmacol. 1996;50:34. [PubMed] [Google Scholar]
- 25.Ebersole BJ, Visiers I, Weinstein H, Sealfon SC. Mol Pharmacol. 2003;63:36. doi: 10.1124/mol.63.1.36. [DOI] [PubMed] [Google Scholar]
- 26.Muntasir HA, Takahashi J, Rashid M, Ahmed M, Komiyama T, Hossain J, Nashimoto M, Nagamoto T. Biol Pharm Bull. 2006;29:1645. doi: 10.1248/bpb.29.1645. [DOI] [PubMed] [Google Scholar]
- 27.Herrick-Davis K, Grinde E, Harrigan TJ, Mazurkiewicz J. Biol Chem. 2005;280:40144. doi: 10.1074/jbc.M507396200. [DOI] [PubMed] [Google Scholar]
- 28.Kristiansen K. Pharmacol Ther. 2004;103:21. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 31.Clark M, Cramer RD, III, van Opdenhosch N. J Comp Chem. 1989;10:982. [Google Scholar]
- 32.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:5179. [Google Scholar]
- 33.Heller H, Schaefer M, Schulten K. J Phys Chem. 1993;97:8343. [Google Scholar]
- 34.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Nucl Acids Res. 2005;33:W363. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasteiger J, Marsili M. Tetrahedron. 1980;36:3219. [Google Scholar]
- 36.Bucholtz EC, Brown RL, Tropsha A, Booth RG, Wyrick SD. J Med Chem. 1999;42:3041. doi: 10.1021/jm980428x. [DOI] [PubMed] [Google Scholar]
- 37.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 38.The PyMOL Molecular Graphics System, Version 1.3. Schrödinger, LLC; [Google Scholar]
- 39.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Cryst. 1993;26:283. [Google Scholar]
- 40.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Proteins. 1992;12:345. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 41.Warren L. Delano, the PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC. (please see http://www.pymol.org/citing, http://pymol.sourceforge.net/fag.html#CITE)
- 42.Moniri NH, Covington-Strachan D, Booth RG. J Pharmacol Exp Ther. 2004;311:274. doi: 10.1124/jpet.104.070086. [DOI] [PubMed] [Google Scholar]
- 43.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. J Pharmacol Exp Ther. 2007;320:1. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]