

Abstract
Overexpression of the tyrosine kinase receptor, ErbB2/HER2/Neu, occurs in 25–30% of invasive breast cancer (BC) with poor patient prognosis. Due to confounding factors, inconsistencies still remain regarding the protective effects of n-3 polyunsaturated fatty acids (PUFAs) on BC. We therefore evaluated whether fat-1 transgenic mice, endogenously synthesizing n-3 PUFAs from n-6 PUFAs, were protected against BC development, and we then aimed to study in vivo a mechanism potentially involved in such protection. E0771 BC cells were implanted into fat-1 and wild-type (WT) mice. After tumorigenesis examination, we analyzed the expression of proteins involved in the HER2 signaling pathway and lipidomic analyses were performed in tumor tissues and plasma. Our results showed that tumors totally disappeared by day 15 in fat-1 mice but continued to grow in WT mice. This prevention can be related in part to significant repression of the HER2/β-catenin signaling pathway and formation of significant levels of n-3 PUFA-derived bioactive mediators (particularly 15-hydroxyeicosapentaenoic acid, 17-hydroxydocosahexaenoic acid, and prostaglandin E3) in the tumors of fat-1 mice compared with WT mice. All together these data demonstrate an anti-BC effect of n-3 PUFAs through, at least in part, HER2 signaling pathway downregulation, and highlight the importance of gene-diet interactions in BC.
Keywords: polyunsaturated fatty acid-derived mediators, xenograft prevention, n-3 tissue enrichment
Breast cancer (BC) remains one of the most threatening mortality factors throughout the world despite significant advancements in early detection and therapy. With a current mortality rate of 40%, over one million women worldwide will fall victim to BC. Four closely related transmembrane tyrosine kinase receptors (HER1, -2, -3, and -4) have been implicated in the pathogenesis of cancer including BC. Binding of small peptide ligand molecules to HER receptors triggers homo- or heterodimerization and autophosphorylation, which results in enhanced cell proliferation, migration, and invasion (1, 2) via the PI3K/AKT/β-catenin downstream signaling pathway (3). The HER2/HER3 heterodimer is considered to be the most active HER dimer and is crucial for signaling in tumors containing amplification of HER2 (4, 5). HER2 has no defined ligand but possesses an active tyrosine kinase domain (6) while, in contrast, HER3 has several ligands, including the neuregulins 1–4, but lacks intrinsic tyrosine kinase activity. HER2 overexpression occurs in 25–30% of invasive BCs and is associated with a more aggressive phenotype and a poor patient prognosis with intrinsic resistance to endocrine and conventional chemotherapy (7, 8). HER3 is often expressed together with HER2 in this disease (9). While both receptors are considered promising targets for therapy, the overemphasis on HER2 has shadowed the important role of HER3 in resistance to HER2-targeted therapies (10, 11). In recent years, an increased understanding of the role of HER3 has fueled the development of HER3-targeting agents (12). As tumors overexpressing HER2 are generally resistant to therapeutic agents, nutrition intervention may be a promising therapeutic strategy in preventing and treating this aggressive subtype of cancer. This can be accomplished by ablating HER2/HER3 expression and/or interfering with the interaction of HER2/HER3 heterodimers.
Epidemiological and preclinical studies suggest a protective effect of fish oil in the prevention of BC (13). In addition, in vitro and in vivo evidence demonstrates that n-3 fatty acids or their metabolites are able to reduce cellular proliferation and increase apoptosis in BC models (14). Moreover, a very recent report showed that mice expressing MMTV-neu(ndl)-YD5 (mouse mammary tumor virus), an aggressive HER2-positive BC model, and fat-1 (synthesizing n-3 PUFAs from n-6 PUFAs) can mitigate tumor development (15). Nevertheless, when the lifelong tumor development has been investigated in this mammary tumorigenesis model over-expressing HER2, mechanisms underlying such an anti-cancer role of n-3 PUFAs have not been elucidated. Then, the relevance of HER2 pathway involvement remains to be explored, as inhibitory dietary effects of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), the two main n-3 PUFAs found in fish oils, have been reported on HER family members (16, 17). Whether HER3 expression is associated with the n-3 PUFA-mediated antitumor effect in BC remains largely unknown. Despite research providing evidence that dietary or exogenously derived fatty acids may play an important role in the etiology, evolution, and/or progression of BC, many inconsistencies and discrepancies preclude definitive conclusions (18). For example, Holmes et al. (19) found an increased risk of BC associated with higher dietary marine n-3 PUFAs in a cohort study with 88,795 women. Such conflicting results reflect many confounding dietary elements. Indeed, marine oil, generally used in nutritional studies, contains not only EPA and DHA but also other fatty acids, and is particularly rich in vitamin D. Thus, it has been shown that oleic acid (18:1n-9) activates phosphatidylinositol 3-kinase, promotes proliferation, and reduces apoptosis of MDA-MB-231 BC cells (20); and Chatterjee et al. (21) showed, in a multitargeted approach, that the combination of vitamin D with MaxEPA (a fish oil supplement) was twice as effective as the individual treatments in reducing tumor incidence and multiplicity. Consequently, it is still very difficult to understand the specific roles of n-3 PUFAs on BC prevention regarding many variables arising from the diets.
In the present study, we evaluated the role of high n-3 PUFA content in the pathogenesis of BC by inducing xenografts in the transgenic fat-1 mouse model. These mice carry the fat-1 gene from the roundworm Caenorhabditis elegans, encoding an n-3 PUFA desaturase, absent in mammals, that catalyzes conversion of n-6 PUFAs into n-3 PUFAs (22). Therefore, these mice have endogenously elevated n-3 PUFA tissue content and exhibit a lower n-6/n-3 PUFA ratio compared with their wild-type (WT) littermates when maintained on a high n-6 PUFA diet. This contrasts feeding procedures using fish oil supplementation, which may bring confounding factors attributed to differences in the dietary composition. Hence, the fat-1 transgenic mouse model is a useful in vivo system for giving new insights of the role of the n-6/n-3 fatty acid ratio in BC tumorigenesis.
We examined the impact of enhanced n-3 PUFA production toward the development of BC and the regulation of the HER2/HER3/β-catenin/c-Myc signaling pathway. Thus, we implanted these cells in the fat-1 transgenic and WT mice in order to evaluate their tumorigenicities. Our data indicate that modulation of BC development by n-3 PUFAs might be mediated in part through HER2 signaling pathway downregulation.
MATERIALS AND METHODS
Materials
RPMI 1640, fetal bovine serum (FBS), glutamine, and antibiotics were purchased from PAA Laboratories. The antibodies raised against phospho (p)-HER2 (Tyr1248), HER2, p-HER3 (Tyr1289), HER3, p-Akt (Ser473), p-GSK-3β (Ser9), E-cadherin, and β-catenin were purchased from Cell Signaling Technology (Beverly, MA). c-Myc antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The horseradish peroxidase-linked secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). Heregulin β-1 was purchased from Sigma-Aldrich. Prostaglandin (PG)E2, PGE3, and 17-hydroxydocosahexaenoic acid (HDHA) were obtained from Bertin Pharma (France).
Animals and diet
Transgenic fat-1 mice were generated as described previously (22) and backcrossed onto a C57BL/6J background. The presence of the fat-1 gene in each mouse was confirmed both by genotyping and tail tissue fatty acid analysis profile. Transgenic and WT animals were maintained on a 10% safflower oil diet (SAFE, Augy, France) ad libitum and kept under pathogen-free conditions in standard cages in temperature- and humidity-controlled conditions with a 12 h light/dark cycle. We used 10- to 12-week-old female fat-1 transgenic mice and nontransgenic littermate controls for this experiment. The diet contained (per 100 g diet) 4.5 g sucrose, 18.6 g casein, 8.6 g cellulose, 50 g wheat starch, 0.3 g DL-methionine, 7 g mineral mix, 1 g vitamin mix, and 10 g safflower oil. Safflower oil is high in linoleic acid (18:2n-6) with very little n-3 fatty acid (less than 0.1% of the total fat supplied). Under the 10% safflower oil regimen, all the transgenic animals presented a total n-6/n-3 PUFA ratio greater than (but close to) one in their tail tissue (n-6/n-3 = 1.6 ± 0.2 in fat-1 mice vs. 39.5 ± 3.2 in WT control animals; n = 10 per group). All procedures were carried out according to institutional guidelines for the use and care of laboratory animals and approved by the Ethical Committee of the University of Burgundy (#A1408).
Cancer cell lines
E0771 medullary breast adenocarcinoma cells were obtained from Dr. Enrico Mihich at Roswell Park Cancer Institute, New York, NY. E0771 cells were originally isolated from a spontaneous cancer in C57BL/6 mice. This C57BL/6 adenocarcinoma-derived BC E0771 cell line is, to our knowledge, the only cell model that can be grown to form breast tumors in immunocompetent fat-1 transgenic mice. The human BC cell lines SK-BR-3 and BT-474 were kindly provided by Dr. Sarab Lizard (Centre Georges François Leclerc, Dijon, France). E0771 cells were cultured in RPMI 1640 supplemented with 5% FBS with iron, 2 mmol/l l-glutamine, and 100 units/ml penicillin/streptomycin. SK-BR-3 and BT-474 cells were maintained in RPMI 1640 supplemented with 10% FBS, 2 mmol/l l-glutamine, and penicillin/streptomycin. All these cells were incubated in a humidified atmosphere of 5% CO2-95% air at 37°C. In DHA-treated cells, the experiments were conducted in medium containing 0.5% FBS.
Cell injections and tumor measurement
Cultured E0771 cells were collected with trypsin digestion (0.05% trypsin-EDTA, 3 min), washed twice with RPMI 1640 medium, and counted. Each female mouse was injected subcutaneously in the lower abdomen in or near the number 4 mammary fat pad with 5 × 105 viable mycoplasma-free cells diluted in 200 μl of RPMI 1640 medium. The day of the injection of E0771 cells was designated day 0. Tumor volume was measured with a caliper every 2 or 3 days, and calculated according to the following formula: tumor volume = length × width2 × 0.5.
Lipidomic analysis
The fatty acid composition in tails (to perform phenotyping), tumors, and plasma was determined by gas chromatography as described previously (23).
Lipid mediators were analyzed using liquid chromatography coupled to electrospray ionization tandem mass spectrometry (LC/ESI-MS/MS) following the methodology developed by Masoodi et al. (24, 25). In brief, each tumor sample was homogenized in ice-cold methanol (4 ml of 15% v/v solution). Internal standards PGB2-d4 and 12-HETE-d8 (40 ng per sample each) (Cayman Chemicals, Ann Arbor, MI) were added to each sample and the homogenate was centrifuged for 5 min at 5,000 rpm at 4°C. The clear supernatant was acidified to pH 3.0 using 0.1M HCL and further purified by solid phase extraction cartridge (C18-E; Phenomenex, Macclesfield, UK) using methyl formate to elute the lipid mediators. Chromatographic analysis of prostanoids was performed on a C18 Luna column (5 μm, 150 × 2.0 mm, Phenomenex) while all hydroxy fatty acids were analyzed using a C18 Kinetex column (2.6 μm, 100 × 2.1 mm, Phenomenex). The analysis was performed using a Waters Alliance 2695 pump coupled to a triple quadrupole mass spectrometer (Quatro Ultima; Waters, Elstree, Hertfordshire, UK). The following multiple reaction monitoring transitions were used: PGE2 m/z 351 > 271, PGD2 m/z 351 > 271, PGE3 m/z 349 > 269, thromboxane (TX)B3 m/z 367 > 167, TXB2 m/z 369 > 169, PGB2-d4 m/z 337 > 179, 5-hydroxyeicosapentaenoic acid (HEPE) m/z 317 > 115, 18-HEPE m/z 317 > 133, 15-HEPE m/z 317 > 175, 12-HEPE m/z 317 > 179, 10- HDHA m/z 343 > 153, 14-HDHA m/z 343 > 161, 13-HDHA m/z 343 > 193, 17-HDHA m/z 343 > 201, and 12-HETE-d8 m/z 327 > 184. Protein content was estimated by the Bio-Rad protein assay using BSA as reference standard (Bio-Rad Laboratories Ltd., Hemel Hempstead, UK). Results are expressed as pg/mg protein.
Cell viability
The numbers of viable cells exposed to DHA were evaluated by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] cell proliferation assay, according to the manufacturer's protocol (Sigma-Aldrich). Briefly, BT-474, SK-BR-3, and E0771 cells were seeded and cultured in 96-well flat bottom plates at a density of 3 × 103 per 100 μl in medium containing 10% FBS, allowed to attach overnight, and then treated with DHA complexes to BSA in a 4:1 (DHA/BSA) molar ratio at 20, 40, 60, 80, and 100 μM or vehicle for 72 h in medium containing 0.5% FBS. MTT reagent (20 μl) was added to each well (final concentration 0.5 mg/ml) and the plate incubated at 37°C. After 4 h, supernatant was carefully removed and 100 μl of dimethyl sulfoxide was added to each well and the plate incubated for 2 h. The absorbance was read at 450 nm on a microplate reader (Bio-Rad, France). Data are represented as mean percent vehicle-treated cell proliferation ± SE of triplicate experiments with internal triplicates.
Western blot analysis
Cells were harvested in Triton protein lysis buffer (20 mM Tris, 150 mM NaCl, 200 mM EDTA, 200 mM EGTA, and 1% Triton X-100) containing protease and phosphatase inhibitor cocktail (Sigma-Aldrich, Saint Quentin Fallavier, France). Proteins (50 μg) were separated by 10% SDS-PAGE and electroblotted to Protran nitrocellulose membranes (Whatman, Dassel, Germany). After blocking nonspecific binding sites with 5% BSA in Tris-buffered saline (TBS) (0.1% Tween-20 in TBS), blots were probed overnight at 4°C with primary antibody against p-HER2 (Tyr1248), HER2, p-HER3 (Tyr1289), HER3, p-Akt (Ser473), p-GSK-3β (Ser9), E-cadherin, β-catenin (Cell Signaling, Ozyme), c-Myc (Santa Cruz Biotechnology), and β-actin (Sigma-Aldrich) at a concentration of 1/2,000, washed in Tween-TBS (T-TBS), and incubated for 1 h at room temperature with horseradish peroxidase-conjugated goat anti-rabbit IgG for all the antibodies, except β-actin was incubated with goat anti mouse IgG (Jackson ImmunoResearch Laboratories). Detection was performed using the enhanced chemiluminescence (ECL) Western blotting analysis procedure (ECL Plus; Amersham, Freiburg, Germany).
In order to validate the potential link between the observed differences in PUFA-derived mediators and the differential tumorigenicities of E0771 cells in the mice, we studied the effects of PGE2, PGE3, and 17-HDHA on the protein expression of HER2, HER3, and c-Myc in these cells in culture. After treating E0771 cells with 1 μM PGE2, 1 μM PGE3, 1 μM 17-HDHA, or DMSO as control, cells were harvested and Western blotting was performed to detect HER2, HER3, and c-Myc expression. The level of β-actin expression was used as the internal control for equal loading.
p-HER3 immunolabeling
Tumors were dehydrated and included in paraffin. Paraffin blocks were sectioned (5 μm thick section, two different levels per block) and slices of tumors were deposited onto Superfrost Plus slides.
p-HER3 (two slides per block at two different levels) immunohistofluorescence was performed using an automated Leica Bond-Max. Briefly, after dewax, antigen retrieval with EDTA (pH 9) buffer, and inhibition of endogenous peroxidases with H2O2 (3%), slides were saturated in BSA (3%) in PBS for 20 min, then incubated with an avidin and biotin blockage kit (Vector Labs, SP2001) for 15 min twice. Then sections were incubated with primary antibody (anti-p-HER3; Cell Signaling, 4791, 1:100) for 1 h. The sections were then washed and incubated with secondary antibody linked to biotin (Southern Biotech, 6440-08, 1:500) for 45 min. After three washes, sections were incubated in streptavidin peroxidase (Invitrogen, T20934, 1:100) and revealed in tyramide-AlexaFluor 568 (Invitrogen, T20934, 1:100). Sections were counterstained with DAPI (Sigma, D9542, 2 μg/ml) for 10 min and rinsed. Negative controls (primary antibody omission) were included.
After processing, the sections were imaged by the Cell Observer station (Zeiss). This station is composed of an inverted motorized microscope, a mercury lamp for the fluorescence, a CCD camera (Zeiss HRm), a computer, and the AxioVision software which controls the station. Images were acquired using 10× objective; each image represents about 0.6 mm2. About three images were made by level (six images by tumor). Image analysis was carried out using Visilog software. For the p-HER3 surface, images were threshold, binarized, and surface measurement of the binarized images was done. These surfaces were divided by the number of nuclei present in each image. This number was found by determining the mean area of a nucleus.
Statistical analysis
Results were expressed as the arithmetical mean and SE (mean ± SE) for each group. Statistical significance in the tumor growth curves, tumor and plasma major fatty acid composition, total n-6, total n-3, n-6/n-3 ratio, and different lipid mediators in tumor samples between WT and fat-1 transgenic mice was determined using a Student's t-test (*P < 0.05; **P < 0.01). The statistical study of the tumor p-HER3 immunohistofluorescence quantification was performed using the Mann-Whitney U test by Tanagra software (**P < 0.01).
RESULTS
n-3 PUFAs inhibit the growth of BC xenografts in fat-1 transgenic mice
To test the hypothesis that a balanced ratio of n-6/n-3 fatty acids is able to decrease the risk of BC, we implanted E0771 mouse BC cells into the fat-1 and WT mice and examined the tumorigenicity of inoculated tumor cells. As shown in Fig. 1, there was a dramatic difference in the tumor volume between fat-1 transgenic (n = 10) and WT mice (n = 6). Over an observation period of 25 days, all mice initially developed a palpable tumor by day 7 but, importantly, all the tumors in fat-1 mice never grew more and all palpable tumors disappeared at day 18. By contrast, all the tumors in WT mice continued to grow up until host sacrifice. These findings clearly show that expression of fat-1 inhibits the growth of BC cells in vivo and results in mammary tumor regression.
Fig. 1.

Tumorigenicity of E0771 BC cells in fat-1 transgenic and WT mice. Cells (5 × 105 diluted in 200 μl of serum-free RPMI 1640 medium) were injected subcutaneously into each of 10 transgenic and 6 WT mice (10 weeks old, female). A: Representative photographs showing tumor formation at two different time points after cell implantation. B: Growth rates of melanomas in WT and fat-1 transgenic mice. Tumor growth was monitored at the indicated time points with a caliper and tumor volume was calculated on the basis of the following formula: tumor volume = (length × width2) × 0.5. The points represent mean tumor volume ± SE obtained from 6 WT mice or from 10 fat-1 transgenic mice. These observations have been done on four independent experiments.
Inhibition of HER2 signaling pathway by n-3 PUFAs in fat-1 transgenic mice
In order to define the regulation of HER2 and HER3 expression by a decreased ratio in n-6/n-3 fatty acids in fat-1 mice, tumor tissues were analyzed for the expression of HER2 and HER3 by Western blotting. Results presented in Fig. 2A demonstrate that HER2 and HER3 expressions were markedly downregulated in the tumor tissues of fat-1 mice 10 days after cell injection. Moreover, p-HER3 immunohistofluorescence was significantly (P < 0.01) decreased in the fat-1 tumor tissues compared with the WT mice (Fig. 2B). β-Catenin not only plays a crucial role in morphogenesis and human cancer as a transcriptional regulator in canonical and noncanonical Wnt signaling pathways, it also takes part in cell-cell adhesion with the adhesion molecule E-cadherin, which is a potent invasion/tumor suppressor in BC (26). To investigate whether the decrease of HER2 and HER3 would affect cell-cell adhesion and β-catenin signaling, Western blotting was used to check the expression of E-cadherin and β-catenin. As shown in Fig. 2A, E-cadherin was markedly upregulated in the tumors of fat-1 mice compared with those in WT mice. We observed a change in the β-catenin expression pattern. Indeed, two protein bands were obtained for β-catenin expression in the fat-1 tumor tissues. We also checked the protein expression of the known transcriptional target gene c-Myc of β-catenin. As shown in Fig. 2A, c-Myc expression was hugely inhibited in tumors of fat-1 mice, suggesting that E-cadherin might be involved in modulating β-catenin signaling.
Fig. 2.

Downregulation of HER2/HER3/β-catenin/c-Myc signaling pathway in fat-1 transgenic mouse tumors exhibiting enhanced n-3 PUFA tissue level. A: Western blotting of HER2, HER3, β-catenin, E-cadherin, c-Myc, NF-κB, cleaved PARP, and β-actin in BC xenograft tumors from three WT (lanes 1–3) and three fat-1 transgenic (lanes 4–6) mice. B: Representative immunohistofluorescence for p-HER3 in the tumors of the WT (top) and fat-1 transgenic (middle) mice (n = 5 and 4, respectively). p-HER3 quantification of WT and fat-1 mice is presented as number of p-HER3 positive cell index. Results are presented as mean ± SE and differences were analyzed using the Mann-Whitney U test by Tanagra software (**P < 0.01).
Nuclear factor κB (NF-κB), a pro-inflammatory and pro-survival transcriptional factor, is known to be highly involved in the initiation and progression of BC (27). As shown in Fig. 2A, fat-1 mouse tumor tissues exhibit a marked decrease of NF-κB protein expression compared with the WT mice. A low n-6/n-3 ratio in transgenic animals not only decreases expression of NF-κB but also induces apoptosis accompanied by an increased expression of cleaved poly (ADP-ribose) polymerase (PARP) (Fig. 2A).
Tumor n-3 fatty acid enrichment and formation of PUFA-derived mediators
As shown in Fig. 3A, fatty acid composition of tumor total lipids revealed higher levels of EPA (20:5n-3) and DPA (22:5n-3) in fat-1 transgenic mice compared with WT animals, whereas arachidonic acid (AA, 20:4n-6) was decreased by 70%. Interestingly, the n-6/n-3 PUFA ratio (Fig. 3B) was significantly reduced in tumors from fat-1 mice (8.92 ± 2.63) compared with WT animals (30.51 ± 6.99) despite the animals being fed the same diet. These results indicate expression of fat-1 enriches the transgenic animals in n-3 PUFAs at the expense of n-6 PUFAs, giving a lower n-6/n-3 ratio.
Fig. 3.

Tumor n-3 fatty acid enrichment and formation of n-6 and n-3 PUFA-derived mediators. A: Tumor major fatty acid composition, total n-6, and total n-3. B: The n-6/n3 ratio is indicated for WT and fat-1 transgenic mice as white and black bars respectively [results are presented as mean ± SE; *P < 0.05, **P < 0.01 (Student's t-test); n = 8 per group]. The n-6/n-3 ratio is given by (18:2n-6 + 20:4n-6 + 22:4n-6 + 22:5n-6)/(18:3n-3 + 20:5n-3 + 22:5n-3 + 22:6n-3). C: Quantification (ng/mg of tumor) of different n-6- and n-3-derived lipid mediators in tumors of WT (n = 4) and fat-1 transgenic mice (n = 4). *P < 0.05; **P < 0.01 (Student's t-test).
We then assessed n-6 and n-3 PUFA-derived metabolites from tumors [specifically n-6 AA (20:4n-6), n-3 EPA (20:5n-3), and DHA (22:6n-3) metabolites] by LC/ESI-MS/MS analysis to determine whether differences in tumor growth between WT and fat-1 mice were associated with these pathways. As shown in Fig. 3C, the EPA- (5-, 18-, 15-, and 12-HEPE) and DHA- (10-, 13-, 14-, and 17-HDHA) derived metabolites were identified at physiologically active levels within tumors from fat-1 mice. These metabolites were not found in tumors from WT mice. In addition to HEPE and HDHA lipid metabolites, significant amounts of n-3-derived PGE3 and TXB3 were formed in tumors of fat-1 mice. There was no significant difference in the level of PGE2 between the two genotypes, but the levels of PGD2 and TXB2 were significantly lower in the fat-1 transgenic mice compared with WT mice (−72 and −70%, respectively).
Plasma n-3 fatty acid enrichment and total lipid level
Analysis of total lipid extracts from plasma showed distinctly different profiles between fat-1 and WT mice (Fig. 4A). There are significantly higher levels of n-3 PUFAs [20:5n-3 (EPA) and 22:6n-3 (DHA)] and a much lower concentration of n-6 PUFAs (AA, 20:4n-6) in the plasma from fat-1 transgenic mice compared with WT animals. As such, the n-6/n-3 PUFA ratio was significantly reduced in the transgenic mice (WT, 20.4:1; fat-1, 6.8:1). These data confirm that plasma was enriched in n-3 PUFAs at the expense of n-6 PUFAs, giving a lower n-6/n-3 ratio, when plasma total lipid level was not statistically changed in both groups (Fig. 4C).
Fig. 4.

Plasma n-3 fatty acid enrichment and total lipid level. A: Plasma major fatty acid composition, total n-6, total n-3, and n–6/n–3 ratio are indicated for WT and fat-1 transgenic mice as white and gray bars, respectively (mean ± SE). ND, not detected. B: Plasma fatty acid ratios in WT and fat-1 mice. C: Plasma total lipid level in WT and fat-1 animals. Results are presented as a mean ± SE. *P < 0.05; **P < 0.01 (Student's t-test); n = 5 per group. The n-6/n-3 ratio is given by (18:2n-6 + 20:4n-6 +22:4n-6 + 22:5n-6)/(18:3n-3 + 20:5n-3 + 22:5n–3 + 22:6n-3).
DHA inhibits in vitro proliferation of BC cells
Given the significant growth inhibition of E0771 cells in fat-1 transgenic mice, we next assessed whether n-3 PUFAs can affect BC cell proliferation in vitro. Mouse E0771 cells and two human HER2-amplified BC cells, SK-BR-3 and BT-474, were examined for their response to DHA treatment. As shown in Fig. 5, DHA induced a concentration-dependent reduction of cell viability in all BC cell lines at 72 h. DHA treatment exhibited more robust growth inhibition in E0771 cells than in SK-BR-3 and BT-474 cells, exhibiting 50% of mortality with only 20 μM of DHA (IC50) and inducing over 98% reduction of viable E0771 cells with DHA (40 μM) treatment.
Fig. 5.

Effects of DHA on viability of BC cells. The effect on cell viability of DHA in BT-474, SK-BR-3, and E0771 cells is assessed and quantified by MTT assay. Cells were treated with various concentrations of DHA. BT-474, SK-BR-3, and E0771 cells were seeded and cultured for 24 h in 96-well plates at a density of 3 × 103 cells per well and cultured in medium supplemented with 10% FBS. After this period, the cells were washed twice with PBS and the medium was replaced with fresh medium with 0.5% FBS containing DHA at increasing concentrations (20–100 μM) for a further 72 h. The number of viable cells exposed to DHA was evaluated by a colorimetric MTT assay. Data represent the mean of eight values and results and are expressed as viability in comparison with controls (100%).
DHA inhibits HER2/HER3 expression and subsequent signaling
In order to assess the mechanism by which n-3 PUFAs might induce growth inhibition of BC cells, Western blotting was used to examine protein expressions in the HER2/HER3/β-catenin signaling pathway in DHA-treated cells in vitro. As shown in Fig. 6, DHA treatment decreases HER2 and HER3 protein levels in E0771, SK-BR-3, and BT-474 cell lines; this effect was observed in a time- and dose-dependent manner (observed 24, 48, and 72 h after treatment). Importantly, treatment of DHA in SK-BR-3 and BT-474 cells for 48 h slightly downregulated HER2 and HER3, but dramatically inhibited p-HER2 and p-HER3 induced by heregulin stimulation. Similarly, the p-HER3 protein level was downregulated in E0771 cells treated with DHA for 24 h, while HER3 expression was not. These results suggest that DHA treatment not only impacts on HER2 and HER3 expression, but also on the formation of HER2/HER3 heterodimers. Given the critical role of β-catenin signaling in the mammary tumorigenesis, the potential effect of n-3 PUFAs on the β-catenin signaling was examined. As shown in Fig. 6, treatment with DHA reduced the β-catenin protein level in a time- and dose-dependent manner in cultured cells. Cytoplasmic β-catenin is controlled by a glycogen synthase-3β (GSK-3β) containing destruction complex, in which GSK-3β is phosphorylated and inactivated leading to cytoplasmic accumulation of β-catenin. To determine whether DHA treatment-induced degradation of β-catenin could be through inhibition of the phosphorylation of GSK-3β and its upstream kinase Akt, p-GSK-3β and p-Akt protein expressions were examined. We observed that DHA treatment downregulated GSK-3β and Akt phosphorylation in cultured cells. Moreover, this downregulation was more obvious in E0771 cells than in SK-BR3 and BT-474 cells. To further validate whether DHA treatment-mediated degradation of β-catenin will repress the expression of target gene c-Myc, we examined the c-Myc protein level in cultured cells treated with DHA. Our results show that c-Myc expression was markedly downregulated in three BC cell lines treated with DHA for 48 h. We also examined the potential effect of DHA treatment on E-cadherin expression in cultured cells. Our results regarding E-cadherin protein expression indicate that E-cadherin is downregulated when E0771, BT-474, and SK-BR-3 cell lines are treated with DHA for at least 48 h. Together, our results provide evidence for the antitumor mechanism of n-3 PUFAs through inhibiting the HER2/HER3/β-catenin signaling pathway in BC cells.
Fig. 6.

DHA inhibits HER2/HER3 expression and subsequent signaling pathway in SK-BR-3, BT-474, and E0771 cell lines. SK-BR-3 (A) and BT-474 (B) cells were treated with DHA at 40 and 80 μM in medium supplemented with 0.5% FBS for 24, 48, and 72 h. E0771 (C) cells were treated with DHA at 40 and 80 μM in medium supplemented with 0.5% FBS for 24 and 48 h. Heregulin β-1 (50 ng/ml) was added 1 h before harvest and lysates were immunoblotted as indicated for HER2, p-HER2, HER3, p-HER3, E-cadherin, β-catenin, p-Akt, p-GSK-3β, c-Myc, and β-actin protein expression.
In vitro validation of the relation of PUFA-derived mediators to HER2, HER3, and c-Myc expression
In order to validate the potential link between the observed differences in PUFA-derived mediators and the differential tumorigenicities of E0771 cells in the mice, we studied the effects of PGE2, PGE3, and 17-HDHA on the protein expression of HER2, HER3, and c-Myc in these cells in culture. As shown in Fig. 7, when HER2 protein expression was only downregulated by 17-HDHA, the HER3 protein level was highly decreased by both DHA mediators PGE3 and 17-HDHA. Moreover, the exposure of E0771 cells to 1 μM PGE2, PGE3, and 17-HDHA for 24 h dramatically decreased c-Myc expression. These data are consistent with our in vivo results suggesting that the higher levels in DHA-derived metabolites (particularly 17-HDHA) and n-3-derived PGE3 in fat-1 tumors may, at least in part, mediate the antitumor effect observed in fat-1 transgenic mice.
Fig. 7.
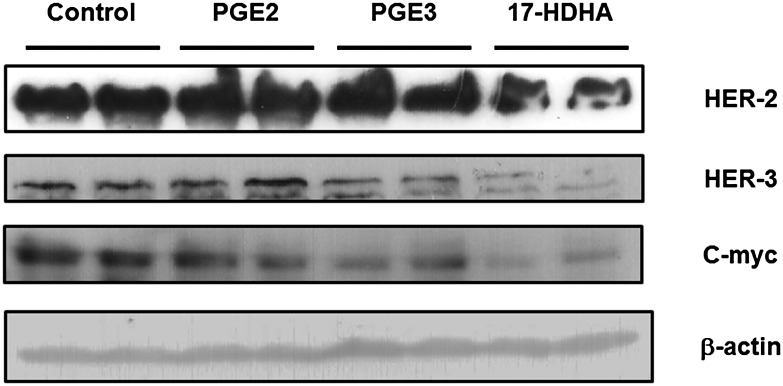
Effects of PGE2, PGE3, and 17-HDHA on HER2, HER3, and c-Myc expression in E0771 cells. After treatment of E0771 cells with PGE2 (1 μM), PGE3 (1 μM), 17-HDHA (1 μM), or DMSO as control, cells were harvested, and Western blotting was performed to detect HER2, HER3, c-Myc, and β-actin expression.
DISCUSSION
The role of PUFAs in BC development, progression, and prevention is not very well understood. PUFAs can mediate cancer development and progression through multiple mechanisms. For example, it has been shown that n-3 PUFAs, but not n-6 PUFAs, induced cell death in a mouse model of prostate cancer (28). Among important regulators of growth, survival, and apoptosis, n-3 PUFAs have been shown to induce growth inhibition of MDA-MB-231 cells by AKT phosphorylation and reduce DNA-binding activity of NF-κB (29). Similarly, treatment of BT-474 and SK-BR-3 BC cells with n-3 PUFAs suppresses the expression of HER2 oncoprotein via regulation of HER2/neu gene transcription (30).
Our study clearly demonstrates that the increase of endogenously synthesized n-3 PUFAs prevents BC development. Furthermore, the prevention of tumor growth is correlated with the formation of antitumor derivatives of n-3 PUFAs and with a downregulation of the HER2 signaling pathway.
A very recent study has shown that lifelong n-3 PUFA exposure was able to mitigate tumor development in an aggressive HER2-positive BC model, providing evidence that n-3 PUFAs can inhibit mammary tumorigenesis (15). Our results clearly demonstrate that more than decreasing the growth rate of the tumor, endogenous production of n-3 PUFAs in fat-1 mice induced mammary tumor regression (Fig. 1), whereas the xenografts in WT mice continued to grow. Evidence in the literature suggests that n-3 PUFAs reduce the risk of BC, however these studies show that n-3 PUFA feeding was unable to mimic the phenotype observed in fat-1 mice (31, 32). Expression of the fat-1 gene is a much more effective approach to modify fatty acid composition. By ubiquitously converting n-6 PUFAs to n-3 PUFAs, it significantly increases the absolute level of n-3 PUFAs as well decreases the level of n-6 PUFAs leading to a decreased ratio of n-6/n-3 PUFAs in mouse tissues, which cannot be achieved by conventional dietary intervention. Then, plasma and tumor tissue exhibited a reduced ratio of n-6 to n-3 PUFAs (Fig. 3A, B and Fig. 4A, B), compared with WT animals, in which the n-3 pathway is naturally compromised as n-3 and n-6 fatty acids share and compete for the same desaturase and elongase enzymes in their biosynthesis, which may also effect the levels of EPA- and DHA-derived metabolites.
The interesting data observed by MacLennan et al. (15) on a mammary tumorigenesis model overexpressing HER2, suggested the involvement of n-3 PUFAs on HER2 pathway regulation. Then, mechanisms underlying such involvement needed to be explored: one of the notable results of the present study was the downregulation of HER2 oncoprotein and the HER2/HER3/β-catenin signaling pathway in tumor tissues from fat-1 mice compared with WT mice. HER2/neu, one of the most commonly analyzed oncogenes in BC studies, is a frequent target of mammary oncogenesis (33). This orphan tyrosine kinase receptor regulates biological functions as diverse as cellular proliferation, transformation, differentiation, motility, and apoptosis (34). Recent studies have shown that fish oil and α-linolenic (18:3n-3) acid also downregulate HER2 expression (30, 32). Similarly, BC cells injected into nude mice fed fish, flaxseed, or canola oil (rich in n-3 PUFAs) formed smaller tumors with lower cell proliferation (35) and lower HER2 expression. HER2 heterodimerizes with HER3 to form an oncogenic unit where HER3 activates the PI3K/Akt pathway (5). Indeed, the inactivation of HER2 in BC cell lines (using trastuzumab) leads to decreased HER3 tyrosine phosphorylation and PI3K signaling (5, 36). In agreement with these data, we found here that HER3 protein expression was dramatically decreased in tumor tissues of the fat-1 transgenic mice (Fig. 2). This latter finding is noteworthy, as high expression of HER3 has been shown to predict early escape from HER-targeted therapies, such as the use of the anti-HER2 monoclonal antibody trastuzumab (37), and is consistent with a recent report showing that genetic ablation of HER3, or its knockdown by EZN-3920, decreased PI3K signaling and tumor growth in the mouse mammary tumor virus, MMTV-PyVmT, model of BC (38). Regarding the impact of n-3 PUFAs on HER2 and HER3 signaling, our studies were then extended to cultured cell lines exhibiting moderate expression of HER2 (E0771) and overexpressing HER2 (BT-474 and SK-BR-3), in which we investigated whether DHA could modulate cell proliferation and the HER2 signaling pathway. Interestingly, DHA treatment hugely inhibits proliferation/viability of the three cell lines (Fig. 5); and in addition to this inhibition, we also observed a downregulation of HER2 and HER3 and most importantly a DHA-dramatic decrease of p-HER2 and p-HER3 in the cell lines stimulated by the HER3-specific ligand heregulin (Fig. 6). This last result suggests that the formation of HER2/HER3 heterodimer might be hindered by DHA treatment. Additionally, two protein bands are seen for HER2 in both SK-BR-3 and BT-474 cells (Fig. 6) when only one band is observed in E0771. Recently, cancer-associated splice variants in several genes have been shown to be involved in tumorigenesis. A splice variant of HER2 (p68-HER2) has been shown to be involved in BC (39). Indeed, this spliced product of HER2 specifically prevents HER2/HER3 dimer phosphorylation by disrupting dimers formed with HER2, whereas it inhibits heregulin-dependent growth in BC cells.
In addition to the tumor regression observed in the transgenic animals, tumors of the fat-1 mice exhibit a higher level of cleaved PARP and a lower level of NF-κB protein expression compared with the WT mice. Moreover, treatment of the three BC cell lines with DHA for 72 h resulted in a concentration- and time-dependent inhibition of cell growth (Fig. 5). In regard to both in vivo and in vitro results, the n-3 PUFA-mediated effects involved apoptosis. One of the mechanisms of action attributed to the apoptosis augment in DHA/EPA-treated MDA-MB-231 cells was impairment of Akt phosphorylation and NF-κB activity (29). In fact, Akt directly promotes cell survival by phosphorylating and inactivating components of the apoptotic machinery. Akt also can activate transcription factors such as NF-κB, critical in tumorigenesis (40). In this sense, our in vitro results confirm our in vivo results, as we also observe a decrease of p-Akt in our DHA-treated cell lines (Fig. 6). In line with this, a recent in vivo study showed an increased apoptotic index of MCF-7 cells injected into flaxseed oil-fed nude mice (35), suggesting that it was probably due to the downregulation of tyrosine kinase receptors such as HER2, and the subsequent downregulation of Akt.
In addition, our results reveal a difference in the β-catenin expression pattern in tumors of fat-1 transgenic mice compared with the WT mice, in which E-cadherin is significantly upregulated. Moreover, we found reduced protein expression of β-catenin, p-GSK3 β, and c-Myc (a pro-oncogene of breast tumors) in all DHA-treated cell lines. β-catenin plays a crucial role in morphogenesis and human cancer through its dual function in cell-cell interactions, and as a transcriptional regulator in numerous signaling pathways (26). Besides the regulation of β-catenin signaling by Wnt, a number of adhesion molecules and other signaling pathways are involved in the control of β-catenin signaling (41). E-cadherin is a potent invasion/tumor suppressor. β-catenin can form a complex with E-cadherin through binding to the cytoplasmic tail of E-cadherin, which sequesters it at the plasma membrane and hinders its nucleus translocation (42). We hypothesize that the upregulation of E-cadherin observed in the tumors of fat-1 transgenic mice could play at least two potential roles: 1) it may inhibit tumor cell growth because E-cadherin has been established as both a tumor suppressor and an invasive suppressor in BCs (43); and 2) the change in the β-catenin expression profile in the fat-1 tumor tissues and the huge decrease in expression of its target gene c-Myc, suggest that increased expression of E-cadherin reduces the availability of cytoplasmic β-catenin by holding it in the plasma membrane, and thereby blocking its signaling to the nucleus. This would prevent, in the nucleus, the binding of β-catenin to the transcription factor T-Cell Factor/Lymphoid Enhancer Factor (LEF/TCF) that induces transcription of important downstream target genes implicated in cell proliferation, differentiation, and apoptosis such as c-Myc (44, 45). Results from Bonvini et al. (46), showing that inactivation of HER2 increases binding of β-catenin to E-cadherin leading to a decrease in β-catenin-mediated gene transcription, strengthens our results regarding E-cadherin upregulation in tumors of fat-1 mice and its potential roles played in these tissues. With regard to what we observe in the tumors of the transgenic mice, we failed to observe the upregulation of E-cadherin in cells treated with DHA (Fig. 6). This might be due to the fact that DHA-treated cell lines exhibit shrinkage and membrane rupture (data not shown) leading to decreased junctions of adhesion, which is responsible for the decrease of E-cadherin protein expression. Indeed, it has been shown that E-cadherin expression is triggered upon cell contacts being established and E-cadherin interaction will increase the E-cadherin level (47).
Another significant finding of the current study is large differences in the levels of EPA metabolites (notably 15-, 12-, and 18-HEPE) and DHA-derived mediators (particularly 17-HDHA) in the tumors of fat-1 mice compared with controls (Fig. 3C). 15-HEPE and 17-HDHA have already been linked to antitumorigenic properties (48, 49) via 15-lipoxygenase activity. These observations can be related to the marked difference in the n-6/n-3 PUFA ratio between the fat-1 and WT mice (Fig. 3A, B). As 17-HDHA is the precursor of the neuroprotectin D1, which has been reported to promote cell apoptosis (50), it is possible that the high levels of 17-HDHA in the tumors of the fat-1 mice could be an indicator of increased formation of the instable intermediate peroxy-metabolite, 17-HpDHA, which was shown to be directly cytotoxic to fast-growing tumor cells (48), contributing to the antitumor effect beyond the mechanisms described in the above paragraph. Moreover, the tumor PUFA-derived metabolite analysis reveals an increased level of PGE3, derived from the n-3 fatty acid EPA, in the fat-1 mice compared with the WT mice, when the level of PGE2 is unchanged (Fig. 3C). When PGE2 has been shown to promote cancer development (51, 52), PGE3 has been found to have anticancer effects (53). Our results suggest that PGE3 and 17-HDHA might be anticancer metabolites, and these generated metabolites from EPA and DHA respectively may underlie the antitumor effect observed in the fat-1 transgenic mice. However, the concentration of PGE3 in the tumors of the fat-1 animals did not reach that of PGE2 suggesting that there is a role for AA-derived lipid mediators that cannot be totally replaced by EPA-derived lipid metabolites, EPA competing with AA as substrate for metabolite production. Interestingly, our in vitro experiments showed that addition of 17-HDHA downregulated HER2 and HER3 protein expression. Moreover, c-Myc expression was dramatically decreased by PGE2, PGE3, and 17-HDHA exposure.
These results suggest that PGE3 and 17-HDHA are anticancer mediators, and generation of PGE3 and 17-HDHA from n-3 PUFAs may underlie the antitumor effect observed in fat-1 transgenic mice. Thus, our data demonstrate a tumorigenesis effect of n-3 fatty acids, at least in part through activation of HER2/HER3/c-Myc signaling pathway mediated by n-3 PUFA-derived mediators.
Taken together, our results provide the first evidence that expression of the fat-1 gene, leading to tissue enrichment of n-3 PUFAs, prevents mammary tumor development. This prevention might occur by downregulating the HER2/HER3/Akt/β-catenin signaling pathway and promoting synthesis of antitumor n-3 PUFA-derived lipid mediators in the tumors of fat-1 mice versus WT mice. These results provide encouraging preclinical evidence and molecular mechanisms by which n-3 PUFAs may regulate the malignant behavior of BC cells. In combination with conventional treatments, supplementing the diet with n-3 PUFAs may be a nontoxic means to synergistically improve cancer treatment outcomes for BC in which HER2/neu is overexpressed and may slow or prevent recurrence of cancer. Moreover, our study shows that used alone, an n-3 supplement may be a useful dietary alternative therapy for patients who are not candidates for standard toxic cancer therapies.
Acknowledgments
J.B. thanks Joseph Gresti (UMR Physiopathologie des Dyslipidémies, Dijon, France) for his experience in chromatography and his help in lipid analysis, and Amandine Bataille, Amandine Chlémaire, and André Bouchot for their great expertise in histology. He also greatly thanks Laurence Decocq and Raymond Berges for taking care of the animals. A.N. and K.A.M. thank Andrew Healey (Analytical Centre, University of Bradford) for excellent technical assistance.
Footnotes
Abbreviations:
- AA
- arachidonic acid
- BC
- breast cancer
- DHA
- docosahexaenoic acid
- EPA
- eicosapentaenoic acid
- HDHA
- hydroxydocosahexaenoic acid
- HEPE
- hydroxyeicosapentaenoic acid, HETE, hydroxyeicosatetraenoic acid
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NF-κB
- nuclear factor κB
- PARP
- poly (ADP-ribose) polymerase
- PG
- prostaglandin
- TCF/LEF
- T-Cell Factor/Lymphoid Enhancer Factor
- TX
- thromboxane
- WT
- wild-type
This work was supported by a French Government grant managed by the French National Research Agency (ANR) under the program “Investissements d'Avenir” with reference ANR-11-LABX-0021-01-LipSTIC Labex. J.B. acknowledges support from the Région Bourgogne. This work was also partially supported by a grant from La Ligue contre le cancer and the Groupe Lipides et Nutrition (GLN). No potential conflicts of interest relevant to this article have been reported.
REFERENCES
- 1.Sliwkowski M. X., Schaefer G., Akita R. W., Lofgren J. A., Fitzpatrick V. D., Nuijens A., Fendly B. M., Cerione R. A., Vandlen R. L., Carraway K. L., 3rd 1994. Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J. Biol. Chem. 269: 14661–14665 [PubMed] [Google Scholar]
- 2.Gschwind A., Fischer O. M., Ullrich A. 2004. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer. 4: 361–370 [DOI] [PubMed] [Google Scholar]
- 3.Sithanandam G., Fornwald L. W., Fields J., Anderson L. M. 2005. Inactivation of ErbB3 by siRNA promotes apoptosis and attenuates growth and invasiveness of human lung adenocarcinoma cell line A549. Oncogene. 24: 1847–1859 [DOI] [PubMed] [Google Scholar]
- 4.Pinkas-Kramarski R., Soussan L., Waterman H., Levkowitz G., Alroy I., Klapper L., Lavi S., Seger R., Ratzkin B. J., Sela M., et al. 1996. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 15: 2452–2467 [PMC free article] [PubMed] [Google Scholar]
- 5.Holbro T., Beerli R. R., Maurer F., Koziczak M., Barbas C.F., 3rd, Hynes N. E. 2003. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA. 100: 8933–8938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burgess A. W., Cho H. S., Eigenbrot C., Ferguson K. M., Garrett T. P., Leahy D. J., Lemmon M. A., Sliwkowski M. X., Ward C. W., Yokoyama S. 2003. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell. 12: 541–552 [DOI] [PubMed] [Google Scholar]
- 7.Yu D., Hung M. C. 2000. Role of erbB2 in breast cancer chemosensitivity. Bioessays. 22: 673–680 [DOI] [PubMed] [Google Scholar]
- 8.Sørlie T., Perou C. M., Tibshirani R., Aas T., Geisler S., Johnsen H., Hastie T., Eisen M. B., van de Rijn M., Jeffrey S. S., et al. 2001. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA. 98: 10869–10874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Travis A., Pinder S. E., Robertson J. F., Bell J. A., Wencyk P., Gullick W. J., Nicholson R. I., Poller D. N., Blamey R. W., Elston C. W., et al. 1996. C-erbB-3 in human breast carcinoma: expression and relation to prognosis and established prognostic indicators. Br. J. Cancer. 74: 229–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gianni L., Pienkowski T., Im Y. H., Roman L., Tseng L. M., Liu M. C., Lluch A., Staroslawska E., de la Haba-Rodriguez J., Im S. A., et al. 2012. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 13: 25–32 [DOI] [PubMed] [Google Scholar]
- 11.Schoeberl B., Faber A. C., Li D., Liang M. C., Crosby K., Onsum M., Burenkova O., Pace E., Walton Z., Nie L., et al. 2010. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 70: 2485–2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell M. R., Amin D., Moasser M. M. 2010. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin. Cancer Res. 16: 1373–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacLean C. H., Newberry S. J., Mojica W. A., Khanna P., Issa A. M., Suttorp M. J., Lim Y. W., Traina S. B., Hilton L., Garland R., et al. 2006. Effects of omega-3 fatty acids on cancer risk: a systematic review. JAMA. 295: 403–415 [DOI] [PubMed] [Google Scholar]
- 14.Sun H., Hu Y., Gu Z., Owens R. T., Chen Y. Q., Edwards I. J. 2011. Omega-3 fatty acids induce apoptosis in human breast cancer cells and mouse mammary tissue through syndecan-1 inhibition of the MEK-Erk pathway. Carcinogenesis. 32: 1518–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacLennan M. B., Clarke S. E., Perez K., Wood G. A., Muller W. J., Kang J. X., Ma D. W. 2013. Mammary tumor development is directly inhibited by lifelong n-3 polyunsaturated fatty acids. J. Nutr. Biochem. 24: 388–395 [DOI] [PubMed] [Google Scholar]
- 16.Schley P. D., Brindley D. N., Field C. J. 2007. (n-3) PUFA alter raft lipid composition and decrease epidermal growth factor receptor levels in lipid rafts of human breast cancer cells. J. Nutr. 137: 548–553 [DOI] [PubMed] [Google Scholar]
- 17.Menendez J. A., Ropero S., Lupu R., Colomer R. 2004. Dietary fatty acids regulate the activation status of Her-2/neu (c-erbB-2) oncogene in breast cancer cells. Ann. Oncol. 15: 1719–1721 [DOI] [PubMed] [Google Scholar]
- 18.Terry P. D., Rohan T. E., Wolk A. 2003. Intakes of fish and marine fatty acids and the risks of cancers of the breast and prostate and of other hormone-related cancers: a review of the epidemiologic evidence. Am. J. Clin. Nutr. 77: 532–543 [DOI] [PubMed] [Google Scholar]
- 19.Holmes M. D., Hunter D. J., Colditz G. A., Stampfer M. J., Hankinson S. E., Speizer F. E., Rosner B., Willett W. C. 1999. Association of dietary intake of fat and fatty acids with risk of breast cancer. JAMA. 281: 914–920 [DOI] [PubMed] [Google Scholar]
- 20.Hardy S., Langelier Y., Prentki M. 2000. Oleate activates phosphatidylinositol 3-kinase and promotes proliferation and reduces apoptosis of MDA-MB-231 breast cancer cells, whereas palmitate has opposite effects. Cancer Res. 60: 6353–6358 [PubMed] [Google Scholar]
- 21.Chatterjee M., Janarthan M., Manivannan R., Rana A., Chatterjee M. 2010. Combinatorial effect of fish oil (Maxepa) and 1alpha,25-dihydroxyvitamin D(3) in the chemoprevention of DMBA-induced mammary carcinogenesis in rats. Chem. Biol. Interact. 188: 102–110 [DOI] [PubMed] [Google Scholar]
- 22.Kang J. X., Wang J., Wu L., Kang Z. B. 2004. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature. 427: 504. [DOI] [PubMed] [Google Scholar]
- 23.Bellenger J., Bellenger S., Clément L., Mandard S., Diot C., Poisson J. P., Narce M. 2004. A new hypotensive polyunsaturated fatty acid dietary combination regulates oleic acid accumulation by suppression of stearoyl CoA desaturase 1 gene expression in the SHR model of genetic hypertension. FASEB J. 18: 773–775 [DOI] [PubMed] [Google Scholar]
- 24.Masoodi M., Nicolaou A. 2006. Lipidomic analysis of twenty-seven prostanoids and isoprostanes by liquid chromatography/electrospray tandem mass spectrometry. Rapid Commun. Mass Spectrom. 20: 3023–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masoodi M., Mir A. A., Petasis N. A., Serhan C. N., Nicolaou A. 2008. Simultaneous lipidomic analysis of three families of bioactive lipid mediators leukotrienes, resolvins, protectins and related hydroxy-fatty acids by liquid chromatography/electrospray ionisation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 22: 75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson W. J., Nusse R. 2004. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 303: 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao Y., Karin M. J. 2003. NF-kappaB in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia. 8: 215–223 [DOI] [PubMed] [Google Scholar]
- 28.Berquin I. M., Min Y., Wu R., Wu J., Perry D., Cline J. M., Thomas M. J., Thornburg T., Kulik G., Smith A., et al. 2007. Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. J. Clin. Invest. 117: 1866–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schley P. D., Jijon H. B., Robinson L. E., Field C. J. 2005. Mechanisms of omega-3 fatty acid-induced growth inhibition in MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 92: 187–195 [DOI] [PubMed] [Google Scholar]
- 30.Menéndez J. A., Vázquez-Martín A., Ropero S., Colomer R., Lupu R. 2006. HER2 (erbB-2)-targeted effects of the omega-3 polyunsaturated fatty acid, alpha-linolenic acid (ALA; 18:3n-3), in breast cancer cells: the “fat features” of the “Mediterranean diet” as an “anti-HER2 cocktail”. Clin. Transl. Oncol. 8: 812–820 [DOI] [PubMed] [Google Scholar]
- 31.Rose D. P., Connolly J. M., Rayburn J., Coleman M. 1995. Influence of diets containing eicosapentaenoic or docosahexaenoic acid on growth and metastasis of breast cancer cells in nude mice. J. Natl. Cancer Inst. 87: 587–592 [DOI] [PubMed] [Google Scholar]
- 32.Yee L. D., Young D. C., Rosol T. J., Vanbuskirk A. M., Clinton S. K. 2005. Dietary (n-3) polyunsaturated fatty acids inhibit HER-2/neu-induced breast cancer in mice independently of the PPARgamma ligand rosiglitazone. J. Nutr. 135: 983–988 [DOI] [PubMed] [Google Scholar]
- 33.Allred D. C., Clark G. M., Molina R., Tandon A. K., Schnitt S. J., Gilchrist K. W., Osborne C. K., Tormey D. C., McGuire W. L. 1992. Overexpression of HER-2/neu and its relationship with other prognostic factors change during the progression of in situ to invasive breast cancer. Hum. Pathol. 23: 974–979 [DOI] [PubMed] [Google Scholar]
- 34.Daly R. J. 1999. Take your partners, please–signal diversification by the erbB family of receptor tyrosine kinases. Growth Factors. 16: 255–263 [DOI] [PubMed] [Google Scholar]
- 35.Truan J. S., Chen J. M., Thompson L. U. 2010. Flaxseed oil reduces the growth of human breast tumors (MCF-7) at high levels of circulating estrogen. Mol. Nutr. Food Res. 54: 1414–1421 [DOI] [PubMed] [Google Scholar]
- 36.Motoyama A. B., Hynes N. E., Lane H. A. 2002. The efficacy of ErbB receptor-targeted anticancer therapeutics is influenced by the availability of epidermal growth factor-related peptides. Cancer Res. 62: 3151–3158 [PubMed] [Google Scholar]
- 37.Smith B. L., Chin D., Maltzman W., Crosby K., Hortobagyi G. N., Bacus S. S. 2004. The efficacy of Herceptin therapies is influenced by the expression of other erbB receptors, their ligands and the activation of downstream signalling proteins. Br. J. Cancer. 91: 1190–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cook R. S., Garrett J. T., Sánchez V., Stanford J. C., Young C., Chakrabarty A., Rinehart C., Zhang Y., Wu Y., Greenberger L., et al. 2011. ErbB3 ablation impairs PI3K/Akt-dependent mammary tumorigenesis. Cancer Res. 71: 3941–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koletsa T., Kostopoulos I., Charalambous E., Christoforidou B., Nenopoulou E., Kotoula V. 2008. A splice variant of HER2 corresponding to Herstatin is expressed in the noncancerous breast and in breast carcinomas. Neoplasia. 10: 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicholson K. M., Anderson N. G. 2002. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 14: 381–395 [DOI] [PubMed] [Google Scholar]
- 41.Polakis P. 2002. Casein kinase 1: a Wnt'er of disconnect. Curr. Biol. 12: R499–R501 [DOI] [PubMed] [Google Scholar]
- 42.Orsulic S., Huber O., Aberle H., Arnold S., Kemler R. 1999. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell Sci. 112: 1237–1245 [DOI] [PubMed] [Google Scholar]
- 43.Berx G., Cleton-Jansen A. M., Nollet F., de Leeuw W. J., van de Vijver M., Cornelisse C., van Roy F. 1995. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 14: 6107–6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clevers H. 2006. Wnt/beta-catenin signaling in development and disease. Cell. 127: 469–480 [DOI] [PubMed] [Google Scholar]
- 45.Gordon M. D., Nusse R. 2006. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 281: 22429–22433 [DOI] [PubMed] [Google Scholar]
- 46.Bonvini P., An W. G., Rosolen A., Nguyen P., Trepel J., Garcia de Herreros A., Dunach M., Neckers L. M. 2001. Geldanamycin abrogates ErbB2 association with proteasome-resistant beta-catenin in melanoma cells, increases beta-catenin-E-cadherin association, and decreases beta-catenin-sensitive transcription. Cancer Res. 61: 1671–1677 [PubMed] [Google Scholar]
- 47.Conacci-Sorrell M., Simcha I., Ben-Yedidia T., Blechman J., Savagner P., Ben-Ze'ev A. 2003. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J. Cell Biol. 163: 847–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gleissman H., Yang R., Martinod K., Lindskog M., Serhan C. N., Johnsen J. I., Kogner P. 2010. Docosahexaenoic acid metabolome in neural tumors: identification of cytotoxic intermediates. FASEB J. 24: 906–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weylandt K. H., Krause L. F., Gomolka B., Chiu C. Y., Bilal S., Nadolny A., Waechter S. F., Fischer A., Rothe M., Kang J. X. 2011. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-α. Carcinogenesis. 32: 897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ariel A., Li P. L., Wang W., Tang W. X., Fredman G., Hong S., Gotlinger K. H., Serhan C. N. 2005. The docosatriene protectin D1 is produced by TH2 skewing and promotes human T cell apoptosis via lipid raft clustering. J. Biol. Chem. 280: 43079–43086 [DOI] [PubMed] [Google Scholar]
- 51.Rose D. P., Connolly J. M. 2000. Regulation of tumor angiogenesis by dietary fatty acids and eicosanoids. Nutr. Cancer. 37: 119–127 [DOI] [PubMed] [Google Scholar]
- 52.Wu T. 2005. Cyclooxygenase-2 and prostaglandin signaling in cholangiocarcinoma. Biochim. Biophys. Acta. 1755: 135–150 [DOI] [PubMed] [Google Scholar]
- 53.Xia S., Lu Y., Wang J., He C., Hong S., Serhan C. N., Kang J. X. 2006. Melanoma growth is reduced in fat-1 transgenic mice: impact of omega-6/omega-3 essential fatty acids. Proc. Natl. Acad. Sci. USA. 103: 12499–12504 [DOI] [PMC free article] [PubMed] [Google Scholar]