

Abstract
We report the synthesis of four series of 3,5-disubstituted-phenyl ligands targeting the metabotropic glutamate receptor subtype 5: (2-methylthiazol-4-yl)ethynyl (1a–j,), (6-methylpyridin-2-yl)ethynyl (2a–j), (5-methylpyridin-2-yl)ethynyl (3a–j,), and (pyridin-2-yl)ethynyl (4a–j,). The compounds were evaluated for antagonism of glutamate-mediated mobilization of internal calcium in an mGluR5 in vitro assay. All compounds were found to be full antagonists and exhibited low nanomolar to subnanomolar activity.
Keywords: Glutamate, mGluR5, MPEP, Antagonist
It is well established that glutamate is the main excitatory neurotransmitter in the central nervous system (CNS) and acts through two classes of receptors. Fast excitatory transmission at glutamate synapses is mediated by the ligand-gated ionotropic receptors (iGluRs), consisting of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), kainate (KA), and N-methyl-d-aspartate (NMDA) receptors. iGluRs are permeable to both potassium and sodium, and in the case of NMDA receptors and some AMPA receptors, calcium ions.1,2 In contrast, metabotropic glutamate receptors (mGluRs) mediate slower responses through G-proteins, coupled to various transduction cascades.1 Group I mGluRs (mGluR1 and 5) are positively associated with phospholipase C via Gq/11 proteins, leading to phosphoinositide hydrolysis and formation of two intracellular second messengers: inositol triphosphate (IP3), which induces intracellular Ca2+ release, and diacylglycerol (DAG), which can activate protein kinase C. The mGluR subtypes belonging to Group II (mGluR2 and 3) and group III (mGluR4, 6, 7 and 8) are negatively coupled to adenylate cyclase via Gi/o proteins and will therefore decrease the intracellular concentration of cAMP.3,4 In addition, glutamate can, through the mGluRs, modulate excitatory (AMPA and NMDA) and inhibitory (GABA) signaling pathways as well as other ion channel receptors.5–7 Modulation of mGluR5 through the action of specific antagonists has potential for treatment of various CNS related diseases. In rodent models of Parkinson’s disease (PD), the selective mGluR5 antagonist 2-methyl-6-(phenylethynyl)-pyridine8 (MPEP, Fig. 1) has been shown to elicit a neuro-protective effect and attenuates both dyskinesia (L-DOPA-induced) and akinesia (6-OHDA induced).9–11
Figure 1.
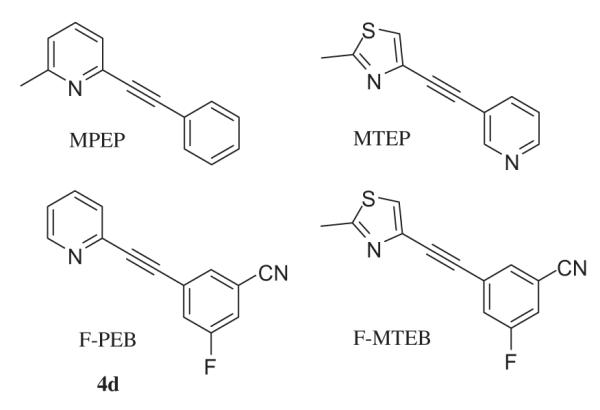
Selected mGluR5 antagonists described in the literature.
The finding that antidepressant treatments change the mGluR5 receptor expression in rat hippocampus12 prompted investigations to relate mGluR5 and affective disorders (anxiety and depression). Both MPEP and 3-[(2-methyl-1,3-thiazol-4yl)-ethynyl]-pyridine (MTEP, Fig. 1), a more selective and potent mGluR5 antagonist, show antidepressant-like activity in mouse and rat forced swim test, with an interesting synergistic effect with the tricyclic antidepressant imiprimine.13,14
Both peripheral and central mGluR5 receptors are implicated in pain transmission15 and MPEP was recently shown to dose-dependently attenuate pain in a rat post-operative model of pain (MPEP ED50 = 15 mg/kg ip; morphine ED50 = 1.3 mg/kg sc).16
The absence of reinforcing and locomotor stimulant effects of cocaine in mGluR5 null mutant mice was pivotal to link mGluR5 and addiction.17 Since then, MPEP has been shown to attenuate cocaine self-administration, cocaine-induced reinstatement of drug seeking, and the discriminative stimulus effects of cocaine at doses that did not markedly impair motor function or operant behavior.18 In the same manner, MPEP seems to decrease the reinforcing effects of ethanol in self-administrated mice19 and rats20, but divergent results have been obtained in models of nicotine and morphine addiction.21–23
These reports highlight the need for potent mGluR5 antagonists as therapeutic drugs and as radiotracers for clinical imaging studies. Among the radiotracers available to image mGluR5 receptors in vivo, the most studied is [18F]F-PEB ([18F]4d), which has shown excellent results in non human primates;24,25 however, this tracer suffers from the relatively short half-life of 18F (110 min), limiting its use to centers near a cyclotron facility. Other radiohalogens, such as 123I (T1/2 13.2 h), 76Br (T1/2 16.2 h) and 77Br (T1/2 55.9 h), have longer physical half lives, but because of limitations in availability and matching of radiation properties to imaging equipment, radioiodine is favored over radiobromine.26 In our efforts to develop new iodinated radiotracers that can be used with the 123I, we explored the structure–activity relationship between four groups of pyridyl and thiazolyl-ethynyl-3,5-disubstituted-phenyl derivatives and their mGluR5 antagonist properties. Ex vivo saturation experiments in rat reveal a density of mGluR5 receptors ranging from 21 nM in the hypothalamus to 83 nM in caudate/putamen27 (fmol/mg tissue = nM, assuming a specific gravity of 1 for the tissue and homogeneous distribution in the region of interest). It is generally accepted that a ratio Bmax/Kd >>1 is required to obtain a good imaging agent, which leads in the case of mGluR5 to compounds with an affinity of at least 2–5 nM. Low non specific binding is also a criterion for good radiotracer and is often associated with low lipophilicity; as a general rule, a value of Log P<3 usually leads to low non specific binding (higher values are acceptable for tracers with high Bmax/Kd ratio). Log P values <1 are usually too polar to penetrate the blood–brain-barrier and therefore render the tracer useless for brain imaging. Similarly, compounds that are substrates of the brain p-glycoprotein efflux pump (P-gp) will not achieve sufficient brain levels for imaging.
The same synthetic methodology was used in the preparation of the four different structural series and is exemplified in Scheme 1. The pyridylethynyltrimethylsilyl derivatives 6–8 were prepared by Sonogashira coupling reaction between the desired bromopyridine and trimethylsilylacetylene using bis(triphenylphosphine)palladium(II) chloride as catalyst and triethylamine as both base and solvent. The thiazole analog 5 was prepared from chloroacetyl chloride as previously reported.28 The 3,5-disubstituted phenyl bromide or iodide derivatives 9a–j were either commercially available (9i and j) or were synthesized using two different approaches (Scheme 2). The first one involves the introduction of a cyano group by dehydration of the corresponding amide with thionyl chloride, in which case the starting materials were the commercially available benzoic acids (9a,d,e,f). The second approach involves the introduction of one or two halogens using the ortho-directing effect of commercial aniline, followed by deamination via the diazonium salt (9b,c,g,h). The target compounds 1–4 were obtained using a one-pot modified Sonogashira cross-coupling reaction, in which the trimethylsilylacetylenes 5–8 were desilylated in situ with tetrabutylammonium fluoride (TBAF) and underwent cross-coupling with the phenyl bromide or iodide 9a–j. This reaction is usually carried out at 60 °C, but in the case of the di- or tri-halogenophenyl derivatives (9b,h,i,j) the reaction was performed at room temperature in order to limit formation of bis coupling adduct.
Scheme 1.

Synthesis of compounds 1, 2, 3, and 4. Reagents and conditions: (a) trimethylsilylacetylene, PdCl2(PPh3)2, Et3N; (b) 9a–j, PdCl2(PPh3)2, Et3N, TBAF, DMF, 60 °C or rt; (c) see Ref. 20.
Scheme 2.

Synthesis of compounds 9a–h. Reagents and conditions: (a) (i) SOCl2, reflux, (ii) NH4OH; (b) SOCl2, reflux; (c) Br2, AcOH, 50 °C; (d) isoamyl nitrite, DMF, 50 °C.
The recently disclosed compounds F-MTEB and F-PEB24,25,28 (Fig. 1) represented a major step in the development of highly potent and selective mGluR5 antagonists. We hypothesized that the increase in activity of those compounds could be attributed to the favorable presence of the 3,5-disubstitution on the phenyl ring. In order to test this hypothesis, we synthesized disubstituted compounds in four series previously shown to give potent mGluR5 antagonist: thiazolyl series 1, 6-methylpyridyl series 2, 5-methylpyridyl series 3, and pyridyl series 4. The choice of 3,5-substitution was dictated by our previous work showing good antagonist activity for compounds monosubstituted at position 3 bearing a nitro, halo or cyano group.29,30 Compounds were evaluated for antagonism of glutamate-mediated mobilization of intracellular calcium31 in cultured human embryonic kidney cells (HEK-293) expressing rat mGluR5 receptors (Table 1). All compounds had full antagonist properties at the mGluR5 receptor, with good to excellent activity (3b = 7.3 nM to 3d = 0.32 nM), indicating tolerance for the different 3,5-disubstitution patterns. The combination of cyano and nitro (group a) as 3,5-disubstitents seems to be less favorable, with values in all four series averaging 2.5 nM, followed by iodo/nitro (group h) in series 1 and 4 averaging 1.9 nM, followed by cyano/chloro (group c) averaging 1.27 nM in all four series. No simple trend appeared in the four series; the average activities gave the following group ranking: 4 (0.97 nM) > 1 (1.42 nM) ≥ 2 (1.49 nM) > 3 (2.51 nM). However the classification of group 4 the best and 3 the worst is not totally satisfying, since series 3 contained both the most potent antagonist (3d, IC50 = 0.32 nM) and the least potent (3b IC50 = 7.36 nM). Comparison of groups b versus e (NO2/Br vs CN/Br), g versus d (NO2/F vs CN/F) as well as h versus f (NO2/I vs CN/I) indicate the feasibility of substitution of the cyano group by a nitro, leading to compounds with generally better or equal activity (except 4h and 3b). Compounds of series 1 were further evaluated in a competition binding assay32 with [3H]methoxy-PEPY on rat mGluR5-HEK293A cells membrane (Table 2). All tested compounds had binding affinity in the low nanomolar to subnanomolar range. The best compounds in this series bore a cyano group (1c Ki = 0.12 nM and 1e Ki = 0.10 nM) but subnanomolar binding affinity was also observed without the presence of the cyano group, as exemplified by compounds 1b, 1g, and 1j (Ki = 0.3–0.9 nM); all those compounds have sufficient affinity for potential application as mGluR5 brain imaging agents (defined as Bmax/Kd >>1). Substitution with an iodo group led in both cases (CN/I 1f, NO2/I 1h) to compounds with the lowest binding affinity. The calculated lipophilicity of those compound is relatively high, ranging from c Log P = 2.72 (4a) to 5.47 (1j), however those values are only estimates of the lipophilicity; the experimental value of compound 2a Log D = 2.30 is well below its calculated value c Log P = 5.16 and we therefore estimate that most of these compounds exhibit a lipophilicity appropriate for brain imaging application. None of the compounds have been evaluated as P-gp substrate but their structural resemblance to MPEP, MTEP, F-PEB, and F-MTEB (which are not P-gp substrates) is a good indicator of low chances of being a P-gp substrate for the new compounds.
Table 1.
| a (CN/NO2) | b (NO2/Br) | c (CN/Cl) | d (CN/F) | e (CN/Br) | |
|---|---|---|---|---|---|
| 1 | |||||
| IC50 | 2.63 ± 0.26 | 0.73 ± 0.22 | 1.61 ± 0.49 | 1.89 ± 0.8 | 1.06 ± 0.36 |
| c Log P | 3.47 | 4.85 | 4.40 | 4.00 | 4.67 |
| 2 | |||||
| IC50 | 3.68 ± 1.66 | 0.63 ± 0.25 | 1.22 ± 0.44 | 1.22 ± 0.14 | |
| c Log P | 3.18 | 4.57 | 4.36 | 3.96 | |
| 3 | |||||
| IC50 | 2.64 ± 0.38 | 7.36 ± 4.17 | 1.22 ± 0.22 | 0.32 ± 0.01 | 1.02 ± 0.61 |
| c Log P | 3.14 | 4.52 | 4.14 | 3.74 | 4.41 |
| 4 | |||||
| IC50 | 1.32 ± 0.03 | 0.39 ± 0.07 | 1.03 ± 0.88 | 0.66 ± 0.22 | |
| c Log P | 2.72 | 4.10 | 3.65 | 3.25 | |
| f (CN/I) | g (NO2/F) | h (NO2/I) | i (Br/F) | j (Br/Br) | |
| 1 | |||||
| IC50 | 1.05 ± 0.24 | 0.61 ± 0.22 | 1.75 ± 0.33 | 1.11 ± 0.19 | 1.45 ± 0.32 |
| c Log P | 5.20 | 4.10 | 5.26 | 4.80 | 5.47 |
| 2 | |||||
| IC50 | 0.71 ± 0.16 | ||||
| c Log P | 5.16 | ||||
| Log Dc | 2.30 | ||||
| 4 | |||||
| IC50 | 0.66 ± 0.31 | 0.66 ± 0.32 | 2.09 ± 0.22 | ||
| c Log P | 4.45 | 3.35 | 4.51 | ||
| Literature values | MPEP | MTEP | F-PEB | F-MTEB | |
|
| |||||
| IC50 | 2.024 | 5.024 | 0.66 ± 0.228 | 1.45 ± 0.528 | |
| c Log P | 3.06 | 2.47 | 3.25 | 4.00 | |
IC50 values are expressed as mean ± S.E.M (nM) of at least three independent experiments. In this assay MPEP IC50 = 2.93 nM.
c Log P values are calculated using ChemDraw Ultra 10.0.
Log D is the partition coefficient of [123I]2f in octanol/phosphate buffer pH 7.4.
Table 2.
Competition binding affinity (Ki, nM) of compounds 1a–j using [3H]methoxy-PEPY. Values are expressed as mean ± S.E.M. (nM) of at least three independent experimentsa
| 1 | a(CN/NO2) | b (NO2/Br) | c (CN/Cl) | d (CN/F) | e (CN/Br) |
| 0.36 ± 0.01 | 0.93 ± 0.02 | 0.127 ± 0.038 | 0.36 ± 0.09 | 0.106 ± 0.023 | |
| 1 | f(CN/I) | g (NO2/F) | h (NO2/I) | i (Br/F) | j (Br/Br) |
| 2.14 ± 0.82 | 0.30 ± 0.02 | 1.84 ± 0.47 | 1.06 ± 0.90 | 0.69 ± 0.28 | |
| Literature values |
MPEP | MTEP | F-PEB | F-MTEB | |
|
| |||||
| K i | 1224 | 1624 | 0.20 ± 0.0128 | 0.08 ± 0.0228 | |
In this assay MPEP Ki = 18.7 nM.
In summary, we synthesized 3,5-disubstituted phenylethynyl compounds in four series. All compounds are potent mGluR5 full antagonists. We demonstrated the apparent equivalency between the cyano and nitro group as one of the 3,5-substituents. Specific trends are difficult to draw since each series seems to lead to different favorable 3,5-substitutents profile (compounds 1g, 2b, 3d, and 4b were the best ligands in each series). The high-affinity compound 1e might find application for imaging with 77Br or 76Br, but this would not be as widely applicable as a radioiodinated ligand. Among the compounds bearing an iodine 1h, 1f, 2f, 4f have promising in vitro potency, but the preliminary binding results (1f, 1h) shows a relatively low affinity which might impair their use as SPECT imaging agents.
Supplementary Material
Acknowledgment
This work was supported by a grant from the National Institutes of Health (DA16180) and a grant from NARSAD to G.D.T.
Footnotes
Supplementary data Supplementary data (experimental details for the synthesis and characterization of 7; 8; 9a–j; 1a,b,c,d,e,g,h,i,j; 2a,b,c,d,f; 3a,b,c,d,e and 4a,b,c,d,f,g,h) associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2011.04.047.
References and notes
- 1.Kew JNC, Kemp JA. Psychopharmacology. 2005;179:4. doi: 10.1007/s00213-005-2200-z. [DOI] [PubMed] [Google Scholar]
- 2.Oswald RE, Ahmed A, Fenwick MK, Loh AP. Curr. Drug Targets. 2007;8:573. doi: 10.2174/138945007780618526. [DOI] [PubMed] [Google Scholar]
- 3.Pin JP, Duvoisin R. Neuropharmacology. 1995;34:1. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 4.Conn PJ, Pin JP. Annu. Rev. Pharmacol. Toxicol. 1997;37:205. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 5.Chen N, Luo T, Raymond LA. J. Neurosci. 1999;19:6844. doi: 10.1523/JNEUROSCI.19-16-06844.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Nat. Neurosci. 2001;4:1079. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- 7.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Science. 1997;273:114. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 8.Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, Kuhn R. Neuropharmacology. 1999;38:1493. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 9.Aguirre JA, Kehr J, Yoshitake T, Liu FL, Rivera A, Fernandez-Espinola S, Andbjer B, Leo G, Medhurst AD, Agnati LF, Fuxe K. Brain Res. 2005;1033:216. doi: 10.1016/j.brainres.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 10.Breysse N, Amalric M, Salin P. J. Neurosci. 2003;23:8302. doi: 10.1523/JNEUROSCI.23-23-08302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mela F, Marti M, Dekundy A, Danysz W, Morari M, Cenci MA. J. Neurochem. 2007;101:484. doi: 10.1111/j.1471-4159.2007.04456.x. [DOI] [PubMed] [Google Scholar]
- 12.Wieronska JM, Branski P, Szewczyk B, Palucha A, Papp M, Gruca P, Moryl E, Pilc A. Pol. J. Pharmacol. 2001;53:659. [PubMed] [Google Scholar]
- 13.Li X, Need AB, Baez M, Witkin JM. J. Pharmacol. Exp. Ther. 2006;319:254. doi: 10.1124/jpet.106.103143. [DOI] [PubMed] [Google Scholar]
- 14.Belozertseva IV, Kos T, Popik P, Danysz W, Bespalov AY. Eur. Neuropsychopharmacol. 2007;17:172. doi: 10.1016/j.euroneuro.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Neugebauer V. Pain. 2002;98:1. doi: 10.1016/s0304-3959(02)00140-9. [DOI] [PubMed] [Google Scholar]
- 16.Zhu CZ, Hsieh G, El-Kouhen O, Wilson SG, Mikusa JP, Hollingsworth PR, Chang R, Moreland RB, Brioni JD, Decker MW, Honore P. Pain. 2005;114:195. doi: 10.1016/j.pain.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 17.Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, Corsi M, Orzi F, Conquet F. Nat. Neurosci. 2001;4:873. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- 18.Lee B, Platt DM, Rowlett JK, Adewale AS, Spealman RD. J. Pharmacol. Exp. Ther. 2005;312:1232. doi: 10.1124/jpet.104.078733. [DOI] [PubMed] [Google Scholar]
- 19.Hodge CW, Miles MF, Sharko AC, Stevenson RA, Hillmann JR, Lepoutre V, Besheer J, Schroeder JP. Psychopharmacology. 2006;183:429. doi: 10.1007/s00213-005-0217-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Besheer J, Stevenson RA, Hodge CW. Eur. J. Pharmacol. 2006;551:71. doi: 10.1016/j.ejphar.2006.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGeehan AJ, Olive MF. Synapse. 2003;47:240. doi: 10.1002/syn.10166. [DOI] [PubMed] [Google Scholar]
- 22.Paterson NE, Semenova S, Gasparini F, Markou A. Psychopharmacology. 2003;167:257. doi: 10.1007/s00213-003-1432-z. [DOI] [PubMed] [Google Scholar]
- 23.Popik P, Wrobel M. Neuropharmacology. 2002;43:1210. doi: 10.1016/s0028-3908(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 24.Hamill TG, Krause S, Ryan C, Bonnefous C, Govek S, Seiders TJ, Cosford NDP, Roppe J, Kamenecka T, Patel S, Gibson RE, Sanabria S, Riffel K, Eng W, King C, Yang X, Green MD, O’Malley SS, Hargreaves R, Burns HD. Synapse. 2005;56:205. doi: 10.1002/syn.20147. [DOI] [PubMed] [Google Scholar]
- 25.Wang J-Q, Tueckmantel W, Zhu A, Pellegrino D, Brownell AL. Synapse. 2007;61:951. doi: 10.1002/syn.20445. [DOI] [PubMed] [Google Scholar]
- 26.Adam MJ, Wilbur DS. Chem. Soc. Rev. 2005;34:153. doi: 10.1039/b313872k. [DOI] [PubMed] [Google Scholar]
- 27.Patel S, Krause SM, Hamill T, Chaudharyb A, Burns HD, Gibson RE. Life Sci. 2003;27:371. doi: 10.1016/s0024-3205(03)00272-8. [DOI] [PubMed] [Google Scholar]
- 28.Cosford NDP, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, Rao S, Washburn M, Varney MA. J. Med. Chem. 2003;46:204. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 29.Alagille D, Baldwin RM, Roth BL, Wroblewski JT, Grajkowska E, Tamagnan GD. Bioorg. Med. Chem. Lett. 2005;15:945. doi: 10.1016/j.bmcl.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 30.Alagille D, Baldwin RM, Roth BL, Wroblewski JT, Grajkowska E, Tamagnan GD. Bioorg. Med. Chem. 2005;13:197. doi: 10.1016/j.bmc.2004.09.042. [DOI] [PubMed] [Google Scholar]
- 31.Rat mGluR5-HEK293A cells were loaded with calcium-sensitive dye according to the manufacturer’s instructions (Calcium 3 kit; Molecular Devices, Sunnyvale, CA) after incubation in glutamate/glutamine-free Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen) containing 10% dialyzed fetal bovine serum for 5 h. Compound A (1 ml) from Calcium 3 kit was dissolved in 20 ml of 1× Hanks’ balanced salt solution (HBSS; Invitrogen) containing 2.5 mM probenecid (Sigma), adjusted to pH 7.4. Cells were loaded for 50 min at 37 °C with 5% carbon dioxide. Dye was then carefully removed and cells were washed with HBSS containing probenecid. Cells were maintained in the same buffer at room temperature for the following assay. Test compound was added 5 min before the manual addition of glutamate. Glutamate was added at a speed of 52 μL/s and calcium flux was measured using a Flexstation II (Molecular Devices) at 25 °C. All of the peaks of the calcium response were normalized to the maximum response to a saturated dose of glutamate (10 μM). 1 μM glutamate was used to generate EC80 response, allowing for a response varying from 70% to 90% of the maximum. EC50 values were calculated from nonlinear curve fitting using GraphPad Prism v. 4.01 and of three separate experiments, presented as the mean ± standard error of the mean (SEM).
- 32.mGluR5 binding assay was determined by measuring the displacement by test compounds of [3H]methoxy-PEPY from rat mGluR5 receptor on membranes prepared from stable rat mGluR5-HEK293A cells. 2 nM [3H]methoxyPEPY was incubated with membrane (10 μg/well) in the binding buffer (50 mM Tris/0.9% NaCl, pH 7.4) with the presence or absence of test compound at room temperature for 1 h with shaking. The membrane-bound ligand was separated from free ligand by filtration through 96-well glass–fiber filter plates (Unifilter-96 GF/B; PerkinElmer Life and Analytical Sciences, Boston, MA) and washed three times with binding buffer (Brandel Cell Harvester; Brandel Inc., Gaithersburg, MD). Scintillation fluid (30 μL) was added to each well and the membrane-bound radioactivity was measured by liquid scintillation counting (TopCount; PerkinElmer Life and Analytical Sciences). Nonspecific binding was estimated using 5 μM MPEP. The inhibition constant Ki was calculated from IC50, which was obtained from nonlinear curve fitting by GraphPad Prism v. 4.01 from three separate experiments in duplicates.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.