

Abstract
Background
SF1126 is a peptidic pro-drug inhibitor of pan-PI3K/mTORC. A first-in-human study evaluated safety, dose limiting toxicities (DLT), maximum tolerated dose (MTD), pharmacokinetics (PK), pharmacodynamics (PD) and efficacy of SF1126, in patients with advanced solid and B-cell malignancies.
Patients and methods
SF1126 was administered IV days 1 and 4, weekly in 28 day-cycles. Dose escalation utilised modified Fibonacci 3+3. Samples to monitor PK and PD were obtained.
Results
Forty four patients were treated at 9 dose levels (90–1110 mg/m2/day). Most toxicity was grade 1 and 2 with a single DLT at180 mg/m2 (diarrhoea). Exposure measured by peak concentration (Cmax) and area under the time-concentration curve (AUC0-t) was dose proportional. Stable disease (SD) was the best response in 19 of 33 (58%) evaluable patients. MTD was not reached but the maximum administered dose (MAD) was 1110 mg/m2. The protocol was amended to enrol patients with CD20+ B-cell malignancies at 1110 mg/m2. A CLL patient who progressed on rituximab [R] achieved SD after 2 months on SF1126 alone but in combination with R achieved a 55% decrease in absolute lymphocyte count and a lymph node response. PD studies of CLL cells demonstrated SF1126 reduced p-AKT and increased apoptosis indicating inhibition of activated PI3K signalling.
Conclusion
SF1126 is well tolerated with SD as the best response in patients with advanced malignancies.
Keywords: PI3K/mTORC pathway, SF1126, Pharmacokinetics, Pharmacodynamics, Refractory solid tumours, B-cell malignancies
1. Introduction
The phosphatidylinositol 3-kinase (PI3K) pathway is unique in that all major signalling hotspots have been found to be mutated and/or amplified in a myriad of human malignancies.1 Moreover, PI3K signalling serves integral functions for stromal and endothelial cells within the tumour microenvironment. Receptor tyrosine kinases up-stream of PI3K, p110α and p85α subunits of PI3K, ser/thr kinase AKT and lipid phosphatase PTEN are frequently altered.2 The high frequency of PI3K driver mutations has led to the discovery and development of isoform- and pan-specific as well as dual PI3K/mTOR ATP-site competitive small molecule inhibitors.3 Several of these novel agents are currently in early phase clinical trials and may show promise for the treatment of tumours addicted to PI3K.4
PI3K binds directly or indirectly to and is activated by several upstream receptor and non-receptor protein tyro-sine kinases.5–8 Once activated, PI3K phosphorylates its lipid substrate phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] to PtdIns(3,4,5)P3 [PIP3] a critical intra-cellular lipid second messenger. This process is opposed by the tumour suppressor PTEN which is frequently deleted or mutated in human cancers9 and results in constitutive PI3K activation. PI3K in turn activates AKT, an important down-stream effector through interactions with PtdIns(3,4,5)P3 via its PH domain which mediates cell proliferation, survival, cell cycle progression, apoptosis, angiogenesis and autophagy.10–12
LY294002 is a pan PI3K and mTORC inhibitor with demonstrated anti-tumour and anti-angiogenic activities in vivo.13,14 However, LY294002 is not a viable drug candidate due to insolubility and short half-life. SF1126 is a novel RGDS peptide conjugate of LY294002 pro-drug. For human studies two times per week schedule was determined consistent with efficacy in animal models and toxicity studies.15 Here we describe a phase I first-in-human trial of SF1126 in patients with advanced solid tumours and B-cell malignancies. SF1126 is well tolerated and showed efficacy in several types of human malignancies.
2. Methods
2.1. Patient population
The study was conducted at 4 US sites with institutional review board approval and all patients providing informed consent. Patients were ≥18 years with advanced solid malignancies refractory to standard treatments. The main inclusion criteria were: life expectancy ≥12 weeks; ECOG performance status 0-1; women of non-childbearing potential (post-menopausal or history of bilateral oophorectomy or hysterectomy) or not pregnant or willing to use contraception; left ventricular ejection fraction (LVEF) >45% [ECHO or MUGA]. Exclusion criteria included: chemotherapy or radiotherapy within 4 weeks of study start; inadequate haematologic (neutrophils ≤1.5×109/L, platelets ≤100 × 109/L, haemoglobin ≤9 g/dL), hepatic (serum bilirubin >ULN of reference range or ALT or AST or ALKP >2.5 ULN of reference range or >5 times ULN of reference range with liver metastases) or renal function (serum creatinine >ULN of reference range or CrCl of <50 mL/min or >+2 proteinuria on 2 consecutive dipsticks within 24 h); history of ischaemic heart disease or myocardial infarction within 3 months of the study; severe or uncontrolled systemic conditions or current unstable or uncompensated respiratory or cardiac con ditions. The protocol was amended and IRB approved to treat patients with CD20+ B-cell malignancies with SF1126 alone and/or in combination with rituximab at the maximum administered dose (MAD). The inclusion criteria were relapsed or refractory B-cell malignancy in patients, ≥18 years, relapsed/refractory to purine ana logues or alkylating agents or not amenable to first line therapy.
2.2. Study objectives
This was a Phase I open label, 3+3 modified Fibonacci dose escalation study whose primary objective was to identify a safe dose of SF1126 administered on day 1 and 4 of a 28-day cycle for exploration in future studies. Dose escalation initially began with 1 patient per cohort until grade ≥ 2 toxicity was observed (accelerated phase). Subsequently dose escalation was 3 patients per cohort (non-accelerated phase). Dose limiting toxicities (DLTs) were assessed only during the 1st cycle: febrile neutropenia, grade 4 neutropenia for >4 d, grade 3 or 4 thrombocytopenia, grade 3 or 4 non-haematologic toxicities and >grade 1 neuropathy. Other objectives were to determine pharmacokinetics (PK), pharmacodynamic (PD) in tumour specimens and to assess anti-tumour activity using Response Eval uation Criteria in Solid Tumours (RECIST, version 1.1).16 Efficacy assessment for B-cell NHL was based on IWG response criteria17 while for CLL it was analysed by IWCLL criteria.18
2.3. Drug administration
SF1126 was administered as an intravenous infusion over 90-min in escalating doses (90, 140, 180, 240, 320, 430, 630, 840 and 1110 mg/m2/day) on day 1 and 4, repeated weekly on a 28-day cycle until study with drawal. Patients continued on SF1126 in the absence of disease progression (clinical benefit) or toxicity which could not be managed by dose reduction. For patients with CD20+ B-cell malignancy (CLL and B-NHL), after the completion of cycle 1, Rituximab (375 mg/m2 IV, once per week) was permitted in combination with SF1126 (post-SF1126).
2.4. Study parameters
Screening and baseline assessments included obtain ing relevant history and details of previous cancer treatment. Safety was monitored at baseline and throughout the study by adverse event reporting, physical examination including full neurological assessment, resting 12-lead ECG, vital signs, clinical chemistry, haematology and urinalysis. QTc and QRS intervals were determined from serial triplicate ECGs taken at baseline (prior to treatment), and at the end of infusion (predicted Cmax), and 2 and 4 h after the end of infusion on study Days 1, 8, 15, 22 and 28 of cycle 1. In addition on Day 1 of cycle 1, ECGs were performed 24 h post dose.
2.5. PK analysis
In patients in the accelerated and non-accelerated phases, PK was performed during and following SF1126 infusion. No PK samples were taken from the patients with B-cell malignancies (treated at MAD). Blood samples were collected on Day 1 of cycles 1 and 2, pre-infusion and at 15 and 30 min during infusion and within 10 min before the end of infusion, and within 10 min after the end of infusion, and at 15, 30, 60 and 120 min, and 4, 8, 24 and 48 h post infusion. A single PK trough sample was obtained pre-dose on study days 8, 15 and 22 of cycle 1 and day 1 of all other cycles up to a maximum of 6 cycles. Plasma samples were analysed for the pro-drug SF1126 along with the hydrolysis products SF1101 (LY204002) and SF1174 using validated methods developed and run at MicroConstants, Inc. (San Diego). Summary statistics were tabulated for the PK parameters of SF1126 and SF1101 and SF1174 by dose/cohort and study day. Summary statistics were also tabulated by dose/cohort and study day. To describe the dependency on dose, scatter plots of Cmax and AUC versus dose were provided for each day measured. PK parameters such as time to peak concentration (tmax), peak concentration (Cmax), minimal concentration (Cmin), volume of distribution (Vd), half-life (t1/2), total clearance (Cl) and area under the time-concentration curve (AUC) were estimated for each patient and were derived from plasma concentration versus time.
2.6. PD analysis
PD analyses included 18FDG PET imaging at base line and within 24 h of last dose cycle 2. Additionally skin punch and tumour biopsies (fresh frozen) were obtainable in several patients before and after dosing. IHC analyses were performed at MD Anderson Cancer Center, Houston, Texas (B. Hennessey, G. Mills) for pAkt (Ser473) and pS6K (Ser235, Ser236) [Cell Signal ling Technology, Danvers, MA]. A PD response was considered if it occurred within the first 4 weeks of therapy with a given dose of SF1126 and met at least one of the criteria: 50% decrease in SUV of PET imaging at cycle 2 day 28 and ≥50% change from baseline in tumour biopsy phosphoprotein markers.
Four patients with CLL were treated with SF1126. Whole blood (50 mL) was collected for cells and serum at 6 time points: day 1: pre-, post-1-h, post-4-h, post-6-h; day 4: pre-; day 8: pre-treatment. Mononuclear cells were separated from peripheral blood samples by Ficoll-Hypaque density centrifugation. Using Annexin V staining to detect apoptosis, treated cells were harvested and rinsed with cold PBS once. After centrifugation for 5 min, cells were resuspended in 500 μl of 1 × Annexin V binding buffer (BioVision, Annexin V-FITC Reagent Kit, Cat.#1001-1000) and then added 5 μl of Annexin V-FITC and 5 μl of Propidium Iodide (BioVision, Annexin V-FITC Reagent Kit). After incubation for 5 min at room temperature in the dark, the samples were analysed by flow cytometry. Apoptosis was also analysed by PARP-cleavage [Santa Cruz Biotechnology, Santa Cruz, CA] while dose-dependent de-phosphorylation of Akt (Ser473) [Cell Signalling Technology, Dan vers, MA] was analysed by Western blotting.
3. Results
3.1. Patient demographics and dose escalation
A total of 130 complete cycles of SF1126 were administered to 39 patients with advanced solid and 5 patients with CD20+ B-cell malignancies. The median age was 62-yr (25-84-yr) with 24 (59%) females. The most common tumour types enrolled were colorectal cancer [23%], gastrointestinal stromal tumour [16%] and epithelial ovarian cancer [11%] (Table 1). A total of 9 dose levels (90, 140, 180, 240, 320, 430, 630, 840 and 1110mg/m2/day) were explored (Table 2). The MAD was 1110 mg/m. The MTD was not established.
Table 1.
Summary of patient demographics.
| Patient characteristics | N | % |
|---|---|---|
| Age | ||
| Median (range) | 62 (25–84) | |
| Gender | ||
| Male | 18 | 41 |
| Female | 26 | 59 |
| ECOG performance status | ||
| 0 | 19 | 49 |
| 1 | 20 | 51 |
| Number of prior treatments | 4 (1–14) | |
| Diagnosis | ||
| Breast | 3 | 7 |
| Colorectal | 10 | 23 |
| Endometrial | 1 | 2 |
| Gist | 7 | 16 |
| Non small cell lung | 1 | 7 |
| Ovarian | 5 | 11 |
| Pancreatic | 2 | 4.6 |
| Prostate | 3 | 7 |
| Renal Cell | 3 | 7 |
| Sarcoma | 1 | 2 |
| Squamous cell head/neck | 1 | 2 |
| Unknown | 1 | 2 |
| Urachal | 1 | 2 |
| B-cell malignancies | 5 | 11.4 |
| Total | 44 |
Table 2.
Dose-escalation scheme and response to SF1126.
| Cohort | Dose | Patients | Patients completed ≥1 | Patients with on study | Patients with stable | Duration |
|---|---|---|---|---|---|---|
| mg/m2 | dosed | cycle | ≥2 months | disease | (weeks) | |
| 1 | 90 | 6 | 3 | 1 | Prostate | 20 |
| 2 | 140 | 3 | 3 | 1 | Breast | 8 |
| 3 | 180 | 6 | 6 | 3 | Endometiral | 20 |
| Gist | 20 | |||||
| Ovarian | 12 | |||||
| 4 | 240 | 3 | 3 | 2 | GIST | 12 |
| CRC | 8 | |||||
| 5 | 320 | 3 | 3 | 2 | Pancreas | 15 |
| CRC | 12 | |||||
| 6 | 430 | 4 | 3 | 3 | GIST | 12 |
| Ovarian | 12 | |||||
| CRC | 13 | |||||
| 7 | 630 | 3 | 3 | 3 | CRC | 12 |
| CRC | 13 | |||||
| RCC | 14 | |||||
| 8 | 840 | 3 | 3 | 3 | RCC | 84 |
| 9 | 1110 | 8 | 6 | 6 | Ovarian | 26 |
| TUO | 16 | |||||
| GIST | 76* | |||||
| 10 | 1110 | 5 | 4 | 2 | CLL | 16 |
| DLBCL | 4 | |||||
| Total | 44 | 33 | 24 | 19 | Mean 21 | |
3.2. Adverse events
A total of 577 adverse events were reported for the 44 patients dosed with SF1126 of which 262 were classed as SF1126-related. Thirty five of 44 patients had at least one SF1126-related AE (90%). There was no grade 4 AEs. There were nine grade 3 AEs reported in 5 patients: oedema (90 mg/m2), Alkaline phosphate increase (140 mg/m2), diarrhoea (180 mg/m2), weakness (240 mg/m2), hypoglycaemia (320 mg/m2), anaemia (430 mg/m2), urticarial/pruritus (630 mg/m2), hypokalaemia (840 mg/m2) and hypersensitivity reaction (1110 mg/m2) (Table 3). One grade 3 DLT (diarrhoea) in the second patient on study (180 mg/m2) was noted in the same patient subsequently retreated at 90 mg/m2. The majority of AEs were CTCAE Grade 1 or 2 in severity with the most common treatment-emergent adverse events being nausea (38.5%), fatigue (35.9%), vomiting (30.8%), diarrhoea (28.2%), pyrexia (28.2%), chills (17.9%), anorexia (12.8%), anaemia (12.8%), pruritus (12.8%) and headache (10.3%) (Table 3).
Table 3.
Treatment related adverse events (Grade 3) and common grade 1 and 2 adverse events.
| Grade 3 SF1126-related adverse events | |||
|---|---|---|---|
|
| |||
| AE | Dose (mg/m2) | Grade 3 (n = 9) | |
| Generalised oedema | 90 | 1(11.1%) | |
| Alkaline phosphatase increased | 140 | 1(11.1%) | |
| Diarrhoea | 180 | 1(11.1%) | |
| Muscular weakness | 240 | 1(11.1%) | |
| Hypoglycaemia | 320 | 1(11.1%) | |
| Haemoglobin decreased | 430 | 1(11.1%) | |
| Pruritus/urticaria | 630 | 1(11.1%) | |
| Hypokalaemia | 840 | 1(11.1%) | |
| Hypersensitivity | 1110 | 1(11.1%) | |
| Grade 1 and 2 SF1126-related adverse events | |||||
|---|---|---|---|---|---|
| Nausea | 38.5% | Chills | 17.9% | ||
| Fatigue | 35.9% | Anorexia | 12.8% | ||
| Vomiting | 30.8% | Anaemia | 12.8% | ||
| Diarrhoea | 28.2% | Puritus | 12.8% | ||
| Pyrexia | 28.2% | Headache | 12.8% | ||
3.3. Pharmacokinetics
The PK of SF1101 (active P13K inhibitor), SF1126 (whole conjugate) and SF1174 (targeting moiety of the conjugate) were determined for 65 subject profiles (38 Cycle 1, 27 Cycle 2). There were no consistent differences between PK parameters during cycle 1 and 2. Exposure as measured by Cmax and AUC0–t was generally dose proportional. For SF1101 the mean t1/2 for dose groups ranged from 1.0 to 2.4-h, with no apparent dependence on dose. The mean Cmax ranged from 579 to 7430 ng/mL (Fig. 1A). Mean AUC0–t ranged from 1052 to 25,786 ng × h/mL. The AUC0–t values at doses ≥140 mg/m2exceeded exposure found in mouse studies to be efficacious, represented by the red horizontal line in Fig. 1B. For SF1126, large inter-subject and intra-subject variability was observed in plasma concentrations. The mean Cmax ranged from 67 to 1304ng/mL and the mean AUC0-t ranged from 41 to 1036 ng × h/mL. For SF1174, the mean Cmax ranged from 214 to 7979 ng/mL and the mean AUC0–t ranged from 253 to 25,564 ng × h/mL. The mean t1/2 ranged from 0.3 to 4.4-h. There was no evidence of accumulation as measured by Cmax and AUC0–t, between Day 1 and 4 of cycles 1 and 2 or between dosing in cycles 1 and 2.
Fig. 1.
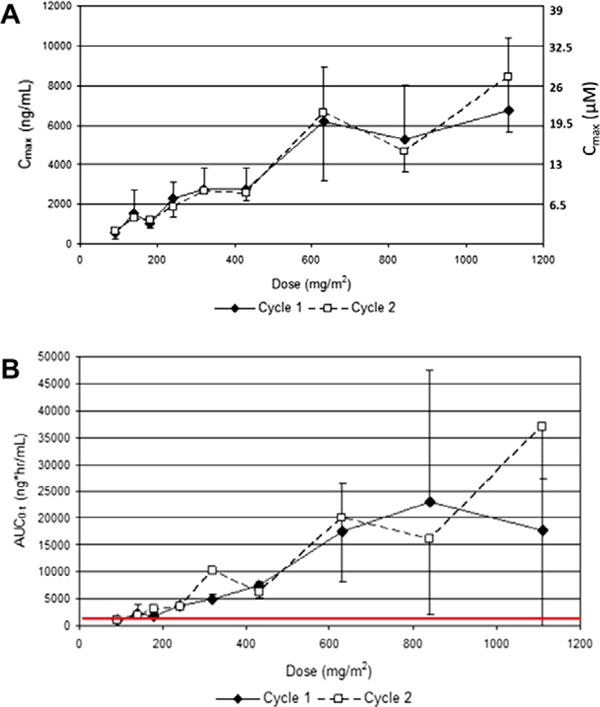
(A) SF1101 PK AUC0–t versus Dose: SF1101 Concentrations for dose cohorts ≥140 mg/m2 achieve higher than targeted exposure levels; (B) SF1101 PK Cmax versus Dose: mean and standard deviation for Cmax versus dose, SF1101 concentrations reach 20–30 μM.
3.4. Pharmacodynamics
Imaging by 18FDG-PET of an ovarian cancer patient treated at 180mg/m showed a >30% decrease in SUV of a pleural-based lesion (Fig. 2). A colon cancer patient treated at 840 mg/m2 showed a 25% decrease in SUV of a lesion in the inferior right lobe of the liver (Fig. 2). A pancreas cancer patient treated at 240 mg/m2 showed that a liver lesion had complete pS6 inhibition at 22.5-h after the 8th dose of SF1126 (Fig. 3A) versus significant pS6 activity at screening (Fig. 3B). In contrast, pS6 in skin punch biopsies in this patient continued to be elevated pre- and post-SF1126 treatment (261 min) (Fig. 3C and D, respectively). These data suggest that the inhibition of PI3K is unaffected in normal tissue compared to tumour tissue. Hence, a dose ≥180mg/m2 may be considered a biologically effective dose for SF1126 consistent with the AUC0–t values at human doses ≥ 140 mg/m exposure found to exceed those shown to be efficacious in mouse xenograft studies. Both 18FDG-PET and skin punch biopsies were performed on the majority of patients on study. Imaging data shown in Fig. 2 were for those patients who had an 18FDG-PET response while the IHC data are consistent with the example shown in Fig 3. No conclusions could be drawn regarding molecular aberrations of the PI3K pathway and response to SF1126.
Fig. 2.
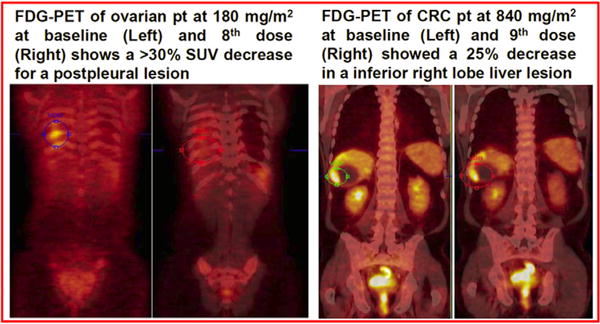
18FDG-PET response in a patient with (A) Metastatic ovarian cancer treated at 180 mg/m2 (SUV >30% decrease from baseline) and (B) Metastatic colon cancer treated at 840 mg/m2 (SUV >25% decreased from baseline).
Fig. 3.
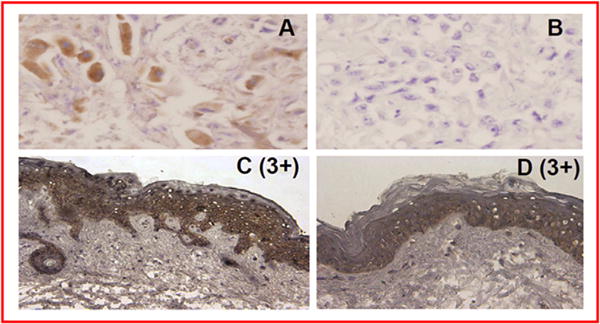
(A and B). A pancreas cancer patient treated at 240 mg/m2 indicated that a liver lesion had a complete pS6 inhibition at 22.5-h after the 8th dose of SF1126 and (C and D). A skin punch of the same patient showed pS6 continued to be elevated pre- and post-SF1126 treatment (261 min).
Time course analysis by flow cytometry of isolated lymphocytes from CLL patients demonstrated consistently increased late apoptosis relative to baseline following treatment with SF1126 (Table 4). In all patients, % late apoptosis increased at 4-h, then declined at 6-h. At 24-h post-SF1126 there was a second peak of late apoptosis. However, at 72-h and 1 week post-SF1126 patient 1 and 2 showed increased % late apoptosis while patient 3 and 4 were back at baseline. Western blotting analysis showed that for patient 1 and 2, pAkt and PARP cleavage matched % late apoptosis: pAkt decreased at 4-6-h, increased at 24-h and then decreased at 72-h and 1 week post-SF1126; PARP cleavage was opposite to that of the pAkt profile (Fig. 4). These results indicate early and late apoptosis match levels of Akt inhibition and induction of PARP-cleavage by SF1126 in a time-dependent manner.
Table 4.
Time course of% apoptosis from baseline for the four CLL patients treated with SF1126.
| Time points (h) | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| 1-h Post-dose | 13 | −3 | 52 | 8 |
| 4-h Post-dose | 38 | 38 | 214 | 54 |
| 6-h Post-dose | 6 | 17 | 38 | ** |
| 24-h Post dose | 44 | ** | 100 | 35 |
| 72-h Post-dose | 56 | 48 | −5 | −4 |
| 1-week Post-dose | 44 | 24 | ** | 15 |
Fig. 4.
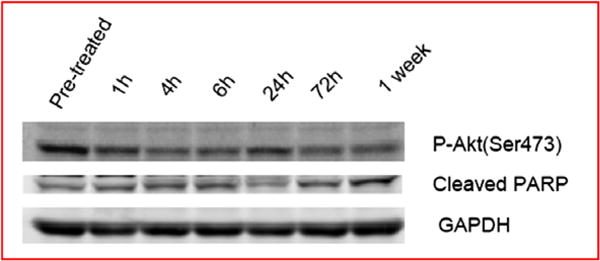
Western blotting analysis of a representative CLL patient showed pAkt and PARP cleavage matched % apoptosis. Phospho-Akt decreased at 4–6-h, increased at 24-h but decreased at 72-h and 1 week post-SF1126; PARP cleavage was opposite to the pAkt profile.
3.5. Solid tumour efficacy
Thirty-nine advanced solid tumour patients were treated and 19 of 33 (58%) were evaluable for response however the remainder were not evaluable due to PD at cycle 1. Stable disease (SD) was the best response with a mean duration of 21 weeks (range 8–84 weeks) (Table 2). Eight patients had SD ≥16 weeks. A receptor tyrosine kinase resistant GIST patient had SD for 48 weeks on the BIW schedule and continued to have SD on a Q1W schedule (per patient's preference) for additional 28 weeks (total 76 weeks). A clear cell renal cancer (RCC) patient had SD stable for 84 weeks (21 cycles). A patient with metastatic colorectal cancer (CRC) with recurring ascites required less frequent paracentesis on study. A patient with metastatic androgen-resistant prostate cancer experienced significantly decreased bone pain for 26 weeks.
3.6. Efficacy in B-cell malignancies
Four patients with CLL and 1 patient with DLBCL were treated with SF1126 at 1110 mg/m2 (Table 5). CLL patient #1 with del17p with no prior therapy completed 16 weeks of SF1126 with SD as best response. CLL patient #2 with 4 prior therapies and bulky lymphadenopathy completed 2 cycles of SF1126 with PD. CLL patient #3 with 2 prior therapies had 2 episodes of hypersensitivity reactions and could not complete cycle 1. CLL patient #4 with 2 prior therapies including rituximab achieved SD after 2 months of SF1126 alone. When rituximab (Q1W x 2) was added to SF1126 there was a significant decrease in absolute lymphocyte count (Fig. 5), lymph node and spleen size. These results suggest that SF1126 plus rituximab may be an active combination in CLL and should be explored in additional patients. Biomarker studies (% apoptosis, PAPR-cleavage and pAkt) performed pre-/post-SF1126 on har vested CLL cells in cycle 1 indicates anti-tumour activity (Table 4 and Fig. 4) but the data cannot be cor related to response.
Table 5.
Summary of CLL and DLBCL patients treated with SF1126.
| Pt# | History | Clinical response |
|---|---|---|
| 1 | 43 years F Del 17p CLL [Rai l] | LN Flare C1D1/C1D4 |
| [No Prior Rx] | Decrease Size of LNs (C1D8/C1D11;WCC (Dec 40%)Stable Disease (16 weeks) as best response | |
| 2 | 63 years M CLL [Rai IV] | LN Flare C1D1/C1D4 |
| [4Prior Rx] | WCC fluctuating | |
| 3 | 41 years F CLL [Rai l] | Severe Hypersensitivity Reactiona (C1D1/C1D4) |
| [2 Prior Rx] | Remarkable improvement in symptomsb | |
| 4 | 82 years M CLL [Rai IV] | LN Flare, Fluctuating in LN size & WCC |
| [2 Prior Rx] | Producing urinec | |
| Haemodialysis | Stable disease (8 weeks) on SF1126 converting to unconfirmed partial response with add'n of Rituximab (see Fig. 3)d | |
| 5 | 78 years M DLBCL (Non-GC) [4 Prior Rx] | >40% LN reductione, LN redness ablated |
Patient has a history of allergies and discontinued after two doses due to grade 2 symptoms resistant to premedication.
Patient suffered from odynophagia, dystonia and dysphagia but after 2nd SF1126 dose patient's oral performance noticeably improved allowing patient to eat and talk.
After four doses of SF1126 patient undergoing haemodialysis 3×/wk began producing new appreciable quantities of urine.
Patient taken off study due to pneumonia related issues, unconfirmed PR by decreased ALC, LN size and spleen size.
Patient taken off study at end of C1 due to a non-drug related mechanical fall requiring hospitalisation.
Fig. 5.
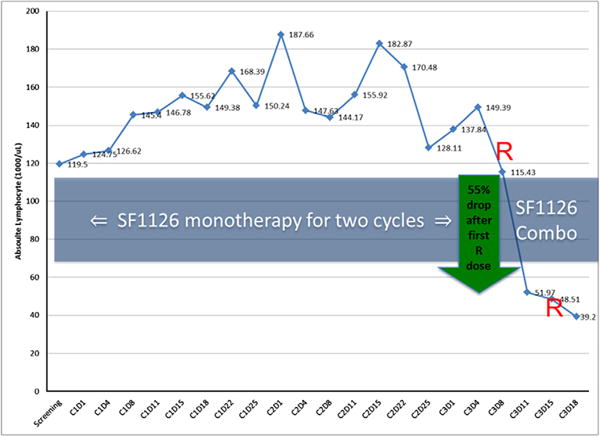
SF1126 plus rituximab is effective in a therapy resistant CLL patient. The patient was treated with SF1126 for 2 cycles with stable disease. Rituximab when added to SF1126 at cycle 3 d 1 and 8 showed significant decrease in the absolute lymphocyte count.
4. Discussion
A first-in-human Phase I study was conducted to determine the safety, tolerability, PK, PD and efficacy of SF1126 in patients with advanced solid tumours and B-cell malignancy. A total of 9 dose levels were explored and there was a dose-dependent increase in exposure with SF1126 measured by Cmax (Fig. 1A) and AUC0–t (Fig. 1B) with a mean plasma half-life ranging from 1 to 2.4-h, with dose levels ≥140 mg/m2 achieving higher than targeted exposure levels.
SF1126 was well tolerated and the most common treatment emergent AEs were gastrointestinal in nature (Table 3) with no myelosuppression or ECG changes. Inhibition of PI3K is known to cause peripheral tissue insulin resistance by enhancing β-cell sensitivity to glucose and produces a compensatory increase in insulin secretion. However, this effect is transient and did not lead to any significant disturbance in glucose homoeostasis. The MTD was not reached but the MAD was 1110 mg/m2 achieving 20–30 μM plasma levels. Twenty four solid tumour patients completed 2 cycles and 6 of 8 patients completed >2 cycles of SF1126 at 1110 mg/m2 (Table 2). Two patients had prolonged clinical benefit, one with metastatic RCC resistant to mTORC1 therapy at the 840 mg/m2 dose (84 weeks) and one with GIST resistant to tyrosine kinase inhibitors at the 1110 mg/m2 dose (76 weeks) (Table 2). SF1126 had no effect on normal skin epidermis S6 phosphorylation status however it potently inhibited tumour pS6 level in a patient with pancreas cancer treated at the 240 mg/m2 dose (Fig. 3).
Pan-PI3K inhibition has been demonstrated in vitro to have desirable effects on anti-tumour chemotaxis and proliferation in CLL cells compared to delta specific inhibition,19 which provides a rationale to explore the therapeutic activity of pan-PI3K inhibition with SF1126. Moreover, SF1126 is an mTORC 1 and 2 inhibitor and rapalogs have shown clinical activity in B-cell malignancies.20 In the four CLL patients treated at the MAD [1110 mg/m2] time course analysis by flow cytometry of purified cells demonstrated significantly increased % late apoptosis. Percentage late apoptosis increased at 4-h, declined at 6-h but a second peak of late apoptosis was observed 24-h post-SF1126 (Table 4). The PK profile of SF1126 and metabolites (SF1101 and SF1174) indicate serum t1/2 varying from 0.3 to 4.4-h that may explain early (4-h) and late (24-h) peaks of late apoptosis. Western blotting analysis showed that pAkt and PARP cleavage matched the % late apoptosis (Fig. 4). Since the mean and standard deviation for Cmax versus dose achieves concentrations of 20–30 μM serum levels for less than 4-h, pAkt inhibition and PARP cleavage are consistent with the PK profile of SF1126.
In one patient with therapy resistant CLL promising preliminary anti-tumour activity alone and in combination with rituximab was observed (Fig. 5). Also initial anti-lymphoma activity was noted in a patient with DLBCL (Table 5 and Fig. 5) and builds on recent pre-clinical data.21 The recommended phase II dose is 1110 mg/m2 is based on complete pathway knockdown of pS6, achieving greater than targeted exposure levels and no trending toxicity observed to indicate approaching MTD level. Also no obvious benefit was observed by increasing dose beyond target inhibition levels and pro vides opportunity for chemoimmunotherapy combinations.
Several PI3K small molecule inhibitors (SMI) have been evaluated in early phase clinical trials.4,22 Phase I data for the dual PI3K/mTOR oral inhibitor, XL765 (Exelixis, CA) reported 11 patients on study for ≥16 weeks and 7 patients for ≥24 weeks23 with path way inhibition of 50–80% in surrogate tissue. A phase I clinical trial of GDC-0941 (Genentech, CA), a pan-PI3K inhibitor reported anti-tumour activity in 3 of 19 patients with relapsed/refractory solid tumours.24 The pan-PI3K inhibitor, XL147 (Exelixis, CA), showed a PR in a NSCLC patient with a confirmed PI3K3CA mutation and SD in 13 of 75 evaluable patients. The major DLT was a skin rash.25 PX-866 (Oncothyreon, WA) an irreversible SMI, showed 60% SD of evaluable patients with 1 reversible grade 3 transminitis.26 A phase I study BKM120 (Novartis, Switzerland) a pan-PI3K SMI demonstrated 1 PR in a triple negative breast can cer (KRAS mutated) patient and 7 patients on study for P8 months. AEs were grade 3 rash, abdominal pain and mood alterations. Two grade 4 hyperglycaemic events were observed at DLT.27 Two dual PI3K/mTOR SMIs BEZ235 (Novartis) [ClinicalTrials.gov] and BGT226 (Novartis) are in phase I studies for advanced solid tumours. BGT226 showed an AE profile of nausea, vomiting and diarrhoea with SD.28
A PI3K p110δ-specific inhibitor CAL-101 evaluated in patients with refractory non-Hodgkin's lymphoma and chronic lymphocytic leukaemia showed durable responses of >6 months in 5 patients.29 Phospho-AKT (T308) in CLL cells from a subset of patients showed reduction by >90% post-dose. SF1126 compares well with other PI3K inhibitors in early phase clinical trials for target inhibition, AEs and anti-tumour activity. Future studies are planned in CD20+ B-cell malignancies of SF1126 plus rituximab.
Acknowledgments
We wish to thank the patients who participated in this clinical study and their families and the clinical re- search support services (CRSS) at the Arizona Cancer Center, Indiana University Melvin and Bren Simon Cancer Center, Emory University and Virginia G. Piper Cancer Center/TGen. We also thank Laurence Cooke for expert technical help in isolating CLL cells. Dr. Durden thanks the NIH for grant support (CA94233).
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
Disclosure: The study was sponsored, monitored and funded by Semafore Pharmaceuticals. JRG and CFS are employees of Semafore Pharmaceuticals.
References
- 1.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu P, Cheng H, Roberts TM, et al. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discovery. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelman JA. Targeting PI3K signaling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 4.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075–83. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 6.Chen HC, Guan JL. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 1994;91(21):10148–52. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Izuhara K, Feldman RA, Greer P, et al. Interleukin-4 induces association of the c-fes proto-oncogene product with phosphatidylinositol-3 kinase. Blood. 1996;88(10):3910–8. [PubMed] [Google Scholar]
- 8.Karnitz LM, Sutor SL, Abraham RT. The Src-family kinase, Fyn, regulates the activation of phosphatidylinositol 3-kinase in an interleukin 2-responsive T cell line. J Exp Med. 1994;179(6):1799–808. doi: 10.1084/jem.179.6.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leslie NR, Downes CP. PTEN: the down side of PI 3-kinase signalling. Cell Signal. 2002;14(4):285–95. doi: 10.1016/s0898-6568(01)00234-0. [DOI] [PubMed] [Google Scholar]
- 10.Duronio V, Scheid MP, Ettinger S. Downstream signalling events regulated by phosphatidylinositol 3-kinase activity. Cell Signal. 1998;10(4):233–9. doi: 10.1016/s0898-6568(97)00129-0. [DOI] [PubMed] [Google Scholar]
- 11.Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253(1):210–29. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- 12.Jiang BH, Zheng JZ, Aoki M, et al. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci U S A. 2000;97(4):1749–53. doi: 10.1073/pnas.040560897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu L, Zaloudek C, Mills GB, et al. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002) Clin Cancer Res. 2000;6(3):880–6. [PubMed] [Google Scholar]
- 14.Su JD, Mayo LD, Donner DB, et al. PTEN and phosphatidylinositol 3′-kinase inhibitors up-regulate p53 and block tumor-induced angiogenesis: evidence for an effect on the tumor and endothelial compartment. Cancer Res. 2003;63(13):3585–92. [PubMed] [Google Scholar]
- 15.Garlich JR, De P, Dey N, et al. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with anti-tumor and antiangiogenic activity. Cancer Res. 2008;68(1):206–15. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 16.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 17.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–86. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 18.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic updating the National Cancer Institute Working Group 1996 Guidelines. Blood. 2008;111(12):5446–56. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niedermeier M, Hennessy BT, Knight ZA, et al. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: a novel therapeutic approach. Blood. 2009;113(22):5549–57. doi: 10.1182/blood-2008-06-165068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witzig TE, Gupta M. Signal transduction inhibitor therapy for lymphoma. Hematology Am Soc Hematol Educ Program. 2010;2010:265–70. doi: 10.1182/asheducation-2010.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kloo B, Nagel D, Pfeifer M, et al. Critical role of PI3K signaling for NF-kappaB-dependent survival in a subset of activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A. 2011;108(1):272–7. doi: 10.1073/pnas.1008969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowles DW, Jimeno A. New phosphatidylinositol 3-kinase inhibitors for cancer. Expert Opin Investig Drugs. 2011;20(4):507–18. doi: 10.1517/13543784.2011.562192. [DOI] [PubMed] [Google Scholar]
- 23.Brana I, LoRusso P, Besalga J, et al. A Phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765 (SAR245409), a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced malignancies. J Clin Oncol. 2010;28:15s. suppl; abstr 3030. [Google Scholar]
- 24.Wagner AJ, Von Hoff DD, LoRusso PM, et al. A first-in-human phase I study to evaluate the pan-PI3K inhibitor GDC-0941 administered QD or BID in patients with advanced solid tumors. J Clin Oncol. 2009;27:146s. suppl; abstr 3501. [Google Scholar]
- 25.Edelman G, Bedell C, Shapiro G, et al. Phase I dose-escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients (pts) with advanced malignancies. J Clin Oncol. 2010;28:15s. suppl; abstr 3004. [Google Scholar]
- 26.Jimeno A, Herbst RS, Falchook GS, et al. Final results from a Phase I, dose-escalation study of PX-866, an irreversible, pan-isoform inhibitor of PI3 kinase. J Clin Oncol. 2010;28:15s. suppl; abstr 3089. [Google Scholar]
- 27.Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–90. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 28.Markman B, Tabernero J, Krop I, et al. Phase I safety, pharmacokinetic, and pharmacodynamics study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol. 2012 doi: 10.1093/annonc/mds011. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 29.Furman RR, Byrd JC, Flinn I, et al. Interim results from a phase I study of CAL-101, a selective oral inhibitor of phosphatidylinositol 3-kinasep110d isoform, in patients with relapsed or refractory hematologic malignancies. J Clin Oncol. 2010;28:15s. abstract 3032. [Google Scholar]