

Abstract
2-cys peroxiredoxins (Prx), a group of anti-oxidative enzyme proteins, act directly on virally-infected cells to inhibit HIV-1 replication, and indirectly through destruction of HIV infected cells by stimulation of Natural Killer (NK) cell-mediated immune responses. We assayed for antibody-dependent NK cell mediated viral inhibition (ADCVI) using plasma from SIV-infected rhesus macaques. We found that Prx-1 strongly increased ADCVI in a dose-dependent manner, suggesting augmentation of NK cell killing. We also investigated the effect of Prx-1 on NK cell-independent HIV-1 and HIV-2 inhibition. We found that primary HIV isolates were potently inhibited at nM concentrations, regardless of viral clade, receptor usage or anti-retroviral drug resistance. During NK cell independent inhibition, we found that Prx-1 reversed the HIV-1 induced gene expression of Heat shock protein 90 kDa alpha (cystolic), class A member 2, (HSP90), a protein of the stress pathway. Prx-1 highly activated Cyclin-dependent kinase inhibitor 2B (CDKN2B), a gene of the TGF-β pathway, and Baculoviral IAP repeat-containing 2 (Birc-2), an anti-apoptotic gene of the NF-κB pathway. We identified gene-expression networks highly dependent on the NFκB and ERK1/2 pathways. Our findings demonstrate that Prx-1 inhibits HIV replication through NK cell-dependent and NK cell-independent mechanisms.
Keywords: Antibody-dependent NK cell mediated viral inhibition, HIV, NK cell independent HIV inhibition peroxiredoxin
I. Introduction
The mammalian peroxiredoxin (Prx) family made up of six proteins, Prx-1 – 6, is widely expressed in several subcellular compartments, including peroxisomes and mitrochodria. Prx possess thioredoxin- (Trx) or gluthione peroxidase- and chaperone-like activities and thereby protect cells from oxidative stress (reviewed [1]). Recent studies reveal additional functions of Prx in modulating gene-expression and inflammation-related biological reactions such as tissue repair, parasite infection and tumor progression (reviewed [1]). In addition, Prx proteins protect cells as part of the Trx system against apoptotic stimuli [2, 3]. Prx proteins operate in conjunction with Trx proteins to scavenge hydrogen peroxide, a product of inflammatory processes. Prx also governs cell cycle arrest and recovery from cell arrest [4]. The Prx family have specific effects on immune function: Prx-1 interacts with or influences the expression of toll-like receptor 4 and B-cell activating factor (reviewed [1], [5]); Prx plays a role in redox-based regulation of signal transduction [6-8]; deletion of Prx results in activation of T lymphocytes and dendritic cells [9]; Prx-1 and Prx-2 augment cytotoxic Natural Killer (NK) cell activity [10].
Several Prx and Trx proteins have shown anti-HIV activity in vitro and expression of these proteins is associated with improved HIV outcomes (reviewed [11]): (1) Prx proteins are part of an innate anti-HIV host-resistance network that is activated during the acute phase response in repeatedly HIV-1-exposed, uninfected individuals [12]. (2) Prx-1 and Prx-2 which have two reactive cysteines (2-cys) are highly transcribed in CD8+ T cells of HIV Long-term Non-progressors (LNPS), individuals who have contained HIV infection for more than 10 years without drug treatment. (3) Furthermore, Prx-1 and Prx-2 protein levels are elevated in the serum of LNPS in contrast to levels found in asymptomatic or symptomatic HIV patients [13]. (4) Finally, Prx-1, Prx-2 and Prx-4 were found to inhibit HIV-1 replication in vitro [13, 14].
More studies are needed to investigate the possible different mechanisms of action of Prx during HIV-1 infection. In this study we further investigate Prx-1 mediated NK cell-dependent and independent inhibition of HIV. We will also investigate the transcriptional networks that may be involved in Prx-mediated NK cell-independent HIV inhibition.
II. Methods
Ethics statement
For the whole blood collection, the study was reviewed and approved by the Human Research Ethics Committee of the Beth Israel Deaconess Medical Center (BIDMC) and Harvard Medical School (IRB 2006-P-000004). Written consent was waived since no personal data were collected.
Rhesus macaques were infected as previously described with SIVmac251 or SIVsmE660 [15, 16]. All animals were cared for in accordance with the American Association for Accreditation of Laboratory Animal Care guidelines and with approval of the Institutional Animal Care and Use Committee of Harvard Medical School.
Protein production and purification
The human Prx-1 gene was cloned into E. coli DH10Bac vector and subcloned between the EcoRI and Not I restriction site into the pFastBacHTA vector (GenScript Corporation, Piscataway, NJ). Sf9 cells were transfected using Cellfectin (Invitrogen, Cat. No. 10362010) according to the manufacturer’s instructions. Cells were incubated in HyQ SFX-insect liquid medium (Hyclone, Logan, UT) for 5-7 days at 27 °C. Supernatant with recombinant virus was collected. High Five cells were infected with virus at a multiplicity of infection [17] of 5 and Prx-1 was produced in the insect cells. Cells were lysed and purified to more than 95% homogeneity as described earlier [18].
Acute HIV infection assay using primary isolates
For the infection assays, human peripheral blood mononuclear cells (PBMC) from HIV-1-seronegative donors were obtained by Ficoll-Hypaque gradient centrifugation of heparinized whole blood from a commercial vendor (Research Blood Components, Brighton, MA). After 3 days of mitogen stimulation (6.25 μg/mL concanavalin A), PBMC were resuspended at a concentration of 1 × 105 cells/ml in RPMI 1640 culture medium (Sigma-Aldrich, St Louis, MO) supplemented with 10% fetal calf serum (Sigma-Aldrich), penicillin (50 U/ml), streptomycin (50 µg/ml), L-glutamine (2 mM), HEPES buffer (10 mM), and 50 U/ml interleukin-2 in 24-well tissue culture plates (Becton Dickinson, San Jose, Ca). An HIV-1 inoculum of 1,000 50% tissue culture infective doses (TCID)/105 cells was added to the PBMC for 2 h at 37 °C and cells were washed extensively. Different concentrations of Prx (in 5-fold increases) were added in serial dilutions at day 0 and day 4. Fifty percent of medium was replaced at day 4. Each condition was tested in triplicate. To determine viral inhibition, cell-free culture supernatants were harvested and analyzed by an enzyme-linked immunosorbent assay (ZeptoMetrix Corporation, Buffalo, NY) for p24 antigen or p27 antigen on day 7 of culture and compared against a vehicle control. Different drug concentrations were used in a virus-specific cell-based assay to measure inhibition. From these data, the IC50, was calculated using the MacSynergy II Software [19]. Controls for inhibition experiments included vehicle buffer, bovine serum albumin (up to 30 μM) and control insect cell protein homogenate. Controls never reached more than 25 % inhibition compared to untreated controls. The new integrase inhibitor 118-D-24, belonging to the family of azido-containing diketo acid derivates, was used as a control as an anti-HIV drug with a known IC50 between 2 and 10 μM [20]. Cell viability was assessed using Neutral Red staining and Tryphan Blue exclusion staining.
ADCVI assay
ADCVI was measured as the ability of human NK cells to inhibit viral replication in Simian immunodeficiency virus-infected human CD4+ T cells in the presence of plasma from a SIV-infected rhesus monkey. The SIVmac251 or SIVsmE660 used in ADCVI assays was derived from the same quasispecies originally used to infect the animals from which plasma was derived for this study. The ADCVI assay and the animals from which plasma was obtained have previously been described [22, 23]. Plasma from four chronically-infected SIVmac251-infected monkeys was pooled for experiments assaying for ADCVI against SIVmac251; and plasma from four SIVsmE660-infected animals was pooled for experiments assaying ADCVI against SIVsmE660. For the ADCVI assay, whole blood from healthy human donors was obtained from a commercial vendor (Research Blood Components). NK cells were isolated using the RosetteSep human NK cell enrichment cocktail (StemCell Technologies, Vancouver, BC, Canada) and maintained in RPMI supplemented with 10% fetal calf serum and 20 IU/ml interleukin-2 (IL-2). By flow cytometry 50 to 70% of purified cells expressed CD16 after purification, compared to less than 5% prior to purification. Furthermore, after purification, all cells were CD3 receptor negative. In parallel, PBMCs were isolated from the same donor by Ficoll-Hypaque gradient centrifugation and stimulated with 6.25 μg/ml concanavalin A and 20 IU/ml of IL-2. After 1 day of stimulation, CD4+ T cells were isolated from total PBMCs using the EasySep human CD4+ T cell enrichment kit (StemCell Technologies). We confirmed by flow cytometry that between 90 and 95% of cells expressed CD4 and CD3 receptor after purification. Purified CD4+ T cells were then infected with SIVmac251 or SIVsmE660 at a multiplicity of infection of 0.01. Three days after isolation from whole blood and 2 days after infection of CD4+ T cells, NK cells and infected CD4+ T cells were washed extensively and resuspended in 96-well plates at 5 × 104 CD4+ T cells and 1 × 105 NK cells per well (2:1 ratio of NK cells to CD4+ target cells). Pooled heat-inactivated rhesus monkey plasma was added at a final dilution of 1:100 or 1:250. Prx-1 was added to the cells at this time. Control wells lacking plasma but containing NK cells (effector control) and viral replication control wells lacking both plasma and NK cells were included on every 96-well plate. Prx vehicle was added to control wells. Supernatants were collected on day 4 for quantification of virus. Viral titers in the supernatant were quantified by p27 ELISA (ZeptoMetrix Corporation, Buffalo, NY). Relative viral inhibition was calculated as the normalized difference between viral titers in control wells compared to the treated sample: % inhibition = 100 % * ((Viral titer control – Viral titer sample)/ viral titer control)). ADCVI independent plasma and Prx inhibition was measured without the addition of NK cells (Prx alone). Experiments were repeated with representative results demonstrated.
Signal transduction pathway profiling
PBMC were infected with the HIV-1 89.6 primary isolate. Cells were harvested after 48 h of Prx-1 treatment and total RNA was recovered using the RNeasy Kit (Qiagen, Valencia, CA) with an on-column DNAse digest (Qiagen) according to the manufacturer’s protocol. Approximately 100 ng RNA was used for cDNA synthesis using the SuperArray RT2 First Strand Kit (SABiosciences, Frederick, MD, cat. no. C-03). cDNA was used for the RT2 Profiler PCR Array Human Signal Transduction PathwayFinder (SABiosciences, Valencia CA, cat. no. PAHS-014A). The genes and their symbols that were investigated can be found at http://www.sabiosciences.com/rt_pcr_product/HTML/PAHS-014A.html. Three arrays of three independent experiments were performed for each treatment condition. Relative levels of transcription were determined by using the ΔCt values for each gene obtained by subtracting the mean threshold cycle [24] of the GAPDH housekeeping gene from the Ct value of the gene of interest. The average ΔCt value for 3 experiments was calculated, for each gene of interest, and the average normalized transcription was calculated as follows: 2(-averageΔCt)−1. Fold increases of gene transcription, before and after treatment was calculated by dividing the average normalized transcription of each gene in the test sample by the corresponding control. Statistical significance in up- or down-regulation of transcription was determined by Student t test (2-sample, equal variance, 2-tailed distribution), comparing the ΔΔCt (ΔΔCt = ΔCt treated - ΔΔCt control). Significant differences were identified when P was less than 0.05.
Analysis of protein interactive networks and statistical analysis
Functional analysis of interacting proteins was determined using a commercial System Biology package, Ingenuity Pathways Analysis (IPA 8.5) (www.ingenuity.com) following the application protocols.
Statistical analysis
The statistical significance of differences between groups was determined using the program GraphPad Prism (San Diego, CA). A P value of <0.05 was considered statistically significant. Statistical analysis was performed using one table T-test or the unpaired T-test. Error bars represent standard error of the mean (S. E.).
III. Results
Direct HIV inhibition by Prx
Prior investigation of the antiviral activity of Prx in immortalized cell lines demonstrated that Prx-1 or Prx-2 had IC50s of approximately 130 nM for inhibiting replication of lab-adapted T-cell tropic or macrophage tropic HIV-1 [13]. No previous experiments have demonstrated antiviral activity of Prx against primary HIV-1 isolates or against HIV-2 strains, or using primary cells as targets. We therefore tested the anti-HIV activity of Prx-1 against an array of primary HIV-1 and HIV-2 isolates from different clades, with various receptor usages and differing drug resistance profiles. After infection of PBMC with the various HIV isolates, we then added Prx-1 at the indicated concentrations (at day 1 and 4 of infection), and quantified viral replication by p24 or p27-antigen ELISA after 7 days. We then used the resulting dose-response curves through day 7 to calculate IC50 values. As controls for Prx-1 treatment, we used whole uninfected insect cell protein. We found that Prx-1 exhibited potent anti-HIV-1 and anti-HIV-2 activity that was independent of viral resistance to antiretrovirals, virus clade or co-receptor usage. Importantly, we found that the anti-viral activity of Prx-1 was observed at therapeutically favorable doses, with IC50 values ranging from 145-450 nM (Table 1).
Table I.
IC50 of Prx-1 for HIV-1 and HIV-2
| HIV isolate | Co-receptor | IC50 (nM) |
|---|---|---|
| HIV-1 10076-4, Nevirapine resistant [36] | R5, X4 | 430 |
| HIV-1G691-6, AZT resistant, post-drug [37] | R5, X4 | 180 |
| HIV-2 7312A [38] | R4, R5, X4, BOP | 150 |
| HIV-2 7924A | R2b, R3, R5, X4, BONZO, BOP | 145 |
HIV-1 induced changes in cellular gene expression without drug treatment
We sought to elucidate the mechanism of action of Prx underlying viral inhibition using gene expression profiling of well-characterized cellular signal transduction pathways. We screened for alterations in gene expression in 18 different signal transduction pathways (totaling 84 independent genes) using a RT-PCR-based gene array. We first investigated the effect of HIV-1 infection alone to establish a baseline of transcriptional profiles that were associated with early viral replication. Compared to vehicle treated PBMC, we found that within 48 h of infection, HIV significantly altered the regulation of 8 genes from 5 signal transduction pathways (Fig. 1A). The stress pathway was prominently altered, with HSP90 expression increased to more than 500-fold over basal levels (P = 0.02). Interestingly, the expression levels of 2 genes from the NF-κB pathway were also altered; TRAF family member-associated NF-κB activator [25] was increased 50-fold, whereas the expression of the anti-apoptotic BIRC2 diminished approximately 10-fold (P = 0.04). We also found that genes Fibronectin 1 (FN1) (P = 0.03) and Myelocytomatoxis viral oncogene homolog (Myc) (P = 0.03) of the Phosphoinositide-3 (PI3) kinase/AKT pathway were down regulated 10-fold (Fig. 1A). Moreover, expression levels of the two Cyclin-dependent kinase inhibitors, CDKN2A and CDKN2B, of the Transforming growth factor- (TGF) pathway were up to 10-fold decreased with P of 0.02 and 0.03, respectively. Finally, we found an up to 5-fold (P = 0.002) down-regulation of the expression of the tumor suppressor and DNA repair gene Breast cancer type 1 susceptibility protein (BRCA1), an element of the estrogen pathway (Fig. 1A).
Figure. 1. The effect of exogenous Prx-1 on cellular signal transduction in HIV infected PBMC.
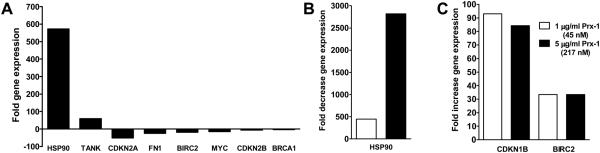
(A) Expression profiling of PBMC infected with HIV-1, without Prx-1 treatment; (B) genes down-regulated after 45 nM Prx-1 treatment of HIV-1 infected PBMC compared to infected untreated control; (C) genes up-regulated after Prx-1 treatment of HIV-1 infected PBMC compared to infected untreated control. For signal transduction gene analysis, 105 human PBMC were infected with a 1000 TCID50 dose of primary isolate HIV-1 89.6 2 hrs at 37 °C. Cells were washed and treated with 45 and 217 nM Prx-1 for 48 h. RNA was purified. A RT-PCR expression array was used to analyze the expression of 84 key genes from 20 different signal transduction pathways (SA Biosciences, Frederick, MD). Genes with significant changes in gene expression (P < 0.05, n = 3) compared to controls, as calculated using the ΔΔCt method, are shown. For Prx-1 treatment only genes with more than 30-fold changes are shown.
Prx-induced alterations in gene expression in HIV-1 infected PBMC
We evaluated the effect of Prx-1 on gene expression in selected signal transduction pathways in HIV-1-infected PBMC. We investigated genes whose expression were affected by treatment with Prx-1 at concentrations of 45 and 217 nM, during HIV infection, concentrations at which significant viral inhibition was observed. We compared alterations in gene expression in HIV-1-infected PBMC treated with the vehicle to those treated with Prx, to determine if Prx treatment affects the same transcriptional pathways as HIV-1.
We found that Prx appeared to have the opposite effect on the expression of several genes as HIV-1 infection. In particular, regulation of 3 genes appeared to be most profoundly altered by Prx exposure at 45 and 217 nM. Expression of the HSP90 molecule, a gene of the stress pathway, which increased almost 600-fold in PBMC infected by HIV-1 and not treated with Prx, compared to uninfected control. This was reversed almost completely by a 500-fold decrease after treatment with 45 nM Prx-1 (P = 0.03). This was even more profound with 217 nM Prx which led to a 3000-fold decrease of HSP90 gene expression (Fig. 1B). Expression of both, CDKN1B, a gene of the TGF-β pathway, and BIRC2, a gene of the NF-κB pathway, increased up to 90- and 30-fold, respectively, after Prx treatment (Fig. 1C).
A functional grouping of the 84 genes in this analysis allowed identification of potential gene pathways affected by Prx treatment. We found that Prx treatment affected several pathways which are known to be activated by HIV-1 infection. The most prominent gene grouping affected by Prx-1 treatment belonged to the hedgehog pathway. Significant changes in the expression of Bone morphogenetic protein 2 (BMP2), BMP4, Engrailed homeobox 1 (EN1), Wingless-type MMTV integration site family, member 1 (WNT1), WNT2 were observed, with P-values of 0.01, 0.01, 0.001, 0.0004, at 45 nM, respectively.
Network analysis of HIV-induced interactomes in PBMC
To gain more insight into the transcriptional networks affected by HIV-1 replication, we performed a biologic network analysis. This analysis method complements data generated from our gene arrays by facilitating the recognition of hierarchical gene clusters. Using this method, we initially analyzed the effect of HIV-1 infection on PBMC in the absence of Prx treatment. We found the NF-κB node of proteins scored significantly during HIV-1 infection, underscoring the well-described role of these proteins in HIV-1 RNA transcription. We identified 6 genes intersecting with the NF-κB node that were significantly altered during HIV-1 infection; Birc2, Myc, FN1, CDKN2A, CDKN2B and BRAC1 (Fig. 2). Network analysis indicated that HSP90 is dependent on the ERK node (Fig. 2).
Figure 2. Interactive network analysis of HIV-1 infected PBMC in the absence of Prx treatment.

Effect of HIV-infection on gene expression in PBMC without Prx treatment. Pathway analysis of significant (P < 0.05) up-regulated (red) and down-regulated (green) genes in HIV-1 infected PBMC using Ingenuity Pathways Analysis Software. Significance was calculated using the ΔΔCt method for three independent experiments.
Network analysis of Prx-induced interactomes during HIV-1 replication
We subsequently examined whether Prx treatment affected the interactome profile in HIV infected cells. Limiting analysis to genes whose expression was changed by at least 2-fold during the 48 h Prx treatment, we identified two potential networks (orange and gray, Fig. 3) affected for Prx treatment. When we synthesized information from both networks, we were able to identify the most significant interactome node. We found that most genes whose expression was influenced by Prx are connected through the NF-κB node (Fig. 3). ERK1/2, Cyclin D1 (CCND1) and CDKN1B nodes also appeared important. Additionally, we found some dependency on the PTGS2 gene (Fig. 3).
Figure. 3. Interactive network analysis of HIV-1 infected PBMC with Prx treatment.

We performed a pathway analysis of significant (P < 0.05) up-regulated (red) and down-regulated (green) genes in PBMC acutely infected with HIV-1 using Ingenuity Pathways Analysis Software. We then combined the two highest scored networks (orange and gray) activated by Prx in HIV-1 infected PBMC to determine the most important nodes. Significance was calculated using the ΔΔCt method for three independent experiments. Genes more than 10-fold altered are shown.
Chemokine/cytokine gene expression after Prx treatment in HIV infected cells
HIV replication is affected by chemokine/cytokine levels. In order to evaluate if induction of chemokine expression by Prx may elicit the antiviral effect, we measured expression of chemokine (C-C motif) ligand 2 (CCL2), chemokine (C-C motif) ligand 20 (CCL20) and chemokine (C-X-X motif) ligand 9 (CXCL9). We also measured expression of cytokines: colony stimulating factor 2 (CSF-2), interleukin 1, alpha (IL-1α), IL-2, IL-4, IL-8 and tumor necrosis factor (TNF, member 2). We indirectly measured the expression of interferons by investigating activation of the interferon regulatory factor 1 (IRF1). We found that the effect of Prx on chemokine/cytokine expression at 45 nM (when significant inhibition occurred) was only limited: the only significantly (P < 0.05) affected genes in these classes were IL-1α (4.8 up-regulated, P = 0.37), IL-2 (6.0-fold down-regulated, P = 0.0001) and IL-4 (5.1-fold down-regulated, P = 0.0005).
Effect of Prx on ADCVI
Augmentation of antibody-independent NK cell killing by Prx has been demonstrated previously [26]. We now investigated the ability of Prx to augment NK cell mediated ADCVI in which antibodies bound to viral peptides presented on the cell surface are recognized by NK cells. This mode of NK cell recognition of infected cells might be important for controlling progression of chronic viral disease and may also be one contributor to vaccine efficacy.
We studied the effect of Prx on ADCVI by measuring the ability of human NK cells to inhibit SIV replication in human CD4+ T cells in the presence of plasma from SIV-infected rhesus monkeys. We utilized SIV for these experiments to permit the study of virus strain-specific antibody responses, since we have access to the identical viral strains which have infected animals and to which antibody responses have been generated. The SIV strains used in these ADCVI assays originated from the same viral stocks originally used to infect the animals from which plasma was derived for this study. A similar experiment is complicated and technically challenging with HIV as standard laboratory strains of HIV are genetically significantly different from circulating patient strains, and in vitro activity of patient plasma against laboratory strains may not accurately represent activity against the patient’s infecting strain of virus.
We have previously used human PBMCs and NK cells to study ADCVI directed against SIV in plasma from SIV-infected rhesus and have demonstrated robust, reproducible, SIV-strain specific ADCVI of human effector cells against human target cells [22, 23]. Similar results have been obtained by others demonstrating the utility of human effector and target cells along with SIV and rhesus plasma in studying ADVCI [27].
We infected purified human CD4+ T cells with SIVmac251. Two days later, we washed the infected cells and co-incubated them simultaneously with pooled plasma from SIV infected rhesus macaques and human NK cells, at an effector to target ratio of 2:1. Plasma was heat-inactivated and added at dilutions of 1:100 and 1:250; similar results were observed at either dilution. These plasma dilutions were previously shown to optimally distinguish between ADCVI and NK-independent antibody inhibition of virus; and these E:T ratios optimally distinguished between ADCVI and antibody-independent NK killing [22]. Prx-1 was added at the indicated concentrations (Fig. 4) at the time of effector:target cell mixing. Viral replication was quantified four days later using p27 ELISA, and viral inhibition was quantified in comparison to viral replication controls containing no Prx and no plasma. Control wells for Prx assays included NK cells as well were termed effector controls; other control wells did not include NK cells and did not include effectors.
Figure. 4. Prx augmentation of ADCVI dependent and independent HIV inhibition.
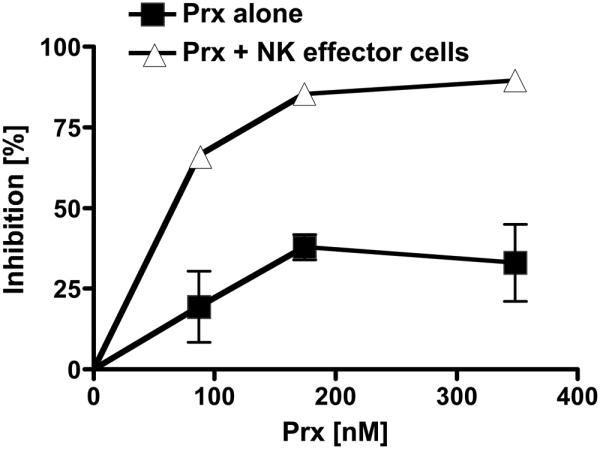
Purified human CD4+ T cells were infected with SIVmac251 at a multiplicity of infection of 0.01. Human NK cells were simultaneously purified from the same donor and cultured separately. Two days after infection, CD4+ T cells and purified NK cells were washed extensively and resuspended in 96-well plates at 5 × 104 CD4+ T cells and 1 × 105 NK cells per well. Pooled heat-inactivated plasma from 4 rhesus monkeys chronically-infected with SIVmac251 was added at a final dilution of 1:100 or 1:250. Prx was added at the indicated concentrations. Four days later, viral titers in culture supernatants were quantified by p27 ELISA. Inhibition was calculated from controls wells lacking Prx. ADCVI independent inhibition was measured without the addition of NK cells (Prx alone). Experiments were repeated multiple times in separate donors with representative results from a 1:250 dilution of plasma demonstrated. Error bars represent standard error of the mean (S. E.). Some are too small to show.
In the absence of NK cells or Prx and the presence of plasma, there was negligible inhibition of viral replication compared to control wells without added plasma, suggesting that at this dilution, plasma alone has no inhibitory effect on replication. Furthermore, the addition of Prx to SIV-infected CD4+ cells with plasma from SIV-infected rhesus did very modestly inhibit viral replication; we found that after a one-time treatment, inhibition was between 19 – 38 % (Fig. 4, dark squares). This suggests a modest NK-independent inhibition of viral replication, when Prx was added only once compared to the above experiments when Prx was added twice. However, we found that viral inhibition greatly augmented in a dose dependent manner in the presence of NK cells. We observed a 85.3 ± 2.2% increase of SIVmac251 inhibition at 174 nM (P = 0.007) and 89.5% ± 0.8 (P < 0.0001) increase of SIVmac251 inhibition at 348 nM when compared to controls with the same dilutions of plasma and ratio of NK cells, but without Prx (Fig. 4, light triangles). This suggests that Prx substantially enhances ADCVI. We found similar results when experiments were repeated with SIVsmE660 infected cells.
IV. DISCUSSION
The Prx family of proteins plays a role in the innate and adaptive immune response. The direct effect of Prx on NK cell cytotoxicity has previously been demonstrated [10, 26]. We extended these studies to investigate whether Prx might also have an effect on ADCVI, a mechanism of viral inhibition targeted by anti-viral antibodies and effected by NK cells that might augment the adaptive immune response to chronic infection and conversely provide an anamnestic property to the rapid early innate immune response [22]. We observed that Prx enhanced ADCVI activity in vitro by more than 3-fold suggesting that Prx might modulate ADCVI during natural viral infection. In particular, this observation might have implications for the role of Prx in either containing HIV replication through ADCVI during late infection, or for blocking acquisition of infection in the presence of pre-existing anti-HIV antibodies and a very early innate immune response. Supporting these hypotheses, Prx has previously been shown to participate in an innate anti-HIV-1 host-resistance network in HIV-1 exposed but uninfected individuals [12]. In addition, Prx levels were increased in the plasma and in CD8+ T-cells of HIV LTNP compared to progressors [13]. It has been presumed that Prx binds to TLR4, inducing activation of NK cells through production of chemokines and IFN-γ. It is also possible that Prx binds to macrophage migration inhibitory factor [28], inhibiting MIF; subsequent release of MIF into the plasma is restricted resulting in down-regulation of NKG2D expression, a receptor important for the perforin-mediated cytolytic response of NK cells (reviewed [1]).
In addition to influencing the innate immune response, as part of the thioredoxin system Prx might also have additional effects on retroviral infection (reviewed [11]), [29]. Over-expression of Prx was shown to induce resistance to HIV infection in T-cell lines exposed to lab-adapted HIV-1 [13]. Furthermore, reduced levels of Prx in T-cell lines correlated with higher levels of HIV-1 replication [14]. Prx was shown to inhibit HIV transcription through inactivation of the HIV-1 long terminal repeat (LTR) [14]. In this study, we extended these prior findings, demonstrating that Prx was able to inhibit replication of primary isolates of HIV-1, that it is able to inhibit HIV-2, and that this inhibition is relevant in primary T-cells, not just cell lines.
We further explored the transcriptional networks by which Prx might directly inhibit HIV replication. We found that Prx down-regulated the stress pathway protein HSP90, which was conversely highly up-regulated after HIV-1 infection. The up-regulation of HSP90 by HIV is well described as is the down-regulation of HSPs through Prx [1], and the inhibition of HIV by HSP90 inhibitors [30-32]. Our data also indicate that HIV inhibition by Prx is not mediated by effects on cytokines/chemokines because of the comparable lack of effects of Prx on expression of these classes of genes. Furthermore, our network analysis describing gene expression relationships in HIV infected cells before and after Prx treatment, exposed transcriptional nodes that may be involved in Prx-mediated virus inhibition. Our data indicate that viral inhibition by Prx is achieved through down-regulation of NF-κB (reviewed [33]) and ERK1/2 pathways, pathways which have previously been demonstrated to be instrumental in HIV-1 inhibition [34, 35]. Similarly, we observed that Prx down-regulated PTGS2 another modulator of viral replication [34].
V. Conclusions
In summary, our data describe multiple mechanisms by which Prx may be able to inhibit HIV replication. We confirm previous findings that Prx appears to have a direct anti-viral effect, and then extend this finding to elaborate the transcriptional pathways which underlie this inhibition of HIV. Beyond a direct anti-viral effect, however, we provide evidence that Prx may augment the innate immune system by several mechanisms to enhance killing of HIV infected cells. Interestingly, we show that Prx may play a role in NK function through ADCVI. The pleiotropic effects of Prx on HIV-1 replication and the host response raise the possibility of harnessing this pathway in future HIV therapeutics.
VI. Acknowledgments
The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: Nevirapine-resistant HIV-1 isolate N119, AZT-resistant HIV and post-drug isolate G691-6 was from Dr. Douglas Richman (University of California); HIV-2 7312A and HIV-2 7924A isolate were from John Moore (Weill Medical College of Cornell University).
Biography

Ralf Geiben-Lynn received the PhD. degree from the Justus-Liebig University in Giessen, Germany. He is currently Group leader at the Harvard Medical School and Beth Israel Deaconess Medical Center, Department of Viral Pathogenesis, Boston, MA, USA.
He was Post-doctoral Research Fellow at the Harvard Medical School and Massachusetts General Hospital under Dr. Bruce Walker at the AIDS Research Center, Boston, MA, USA. His research interest includes the development of immunomodulatory anti-viral drugs for HIV/HCV coinfection, HSV and the Influenza Virus H1N1. He is also interested in mechanisms of action studies to understand the efficacies of different vaccine vector modalities.
Footnotes
VII. CONFLICT OF INTEREST STATEMENT
We have no conflict of interest to declare.
References
- 1.Ishii T, Warabi E, Yanagawa T. Novel roles of peroxiredoxins in inflammation, cancer and innate immunity. J Clin Biochem Nutr. 2012;50:91–105. doi: 10.3164/jcbn.11-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Y, Kok KH, Chun AC, Wong CM, Wu HW, Lin MC, Fung PC, Kung H, Jin DY. Mouse peroxiredoxin V is a thioredoxin peroxidase that inhibits p53-induced apoptosis. Biochem Biophys Res Commun. 2000;268:921–927. doi: 10.1006/bbrc.2000.2231. [DOI] [PubMed] [Google Scholar]
- 3.Kim H, Lee TH, Park ES, Suh JM, Park SJ, Chung HK, Kwon OY, Kim YK, Ro HK, Shong M. Role of peroxiredoxins in regulating intracellular hydrogen peroxide and hydrogen peroxide-induced apoptosis in thyroid cells. J Biol Chem. 2000;275:18266–18270. doi: 10.1074/jbc.275.24.18266. [DOI] [PubMed] [Google Scholar]
- 4.Phalen TJ, Weirather K, Deming PB, Anathy V, Howe AK, van der Vliet A, Jonsson TJ, Poole LB, Heintz NH. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol. 2006;175:779–789. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moon EY, Kang JS, Han SH, Yang KH, Pyo S, Lee MY, Lee HK, Yu DY. Differential role of peroxiredoxin II (PrxII) on the expression of toll-like receptor 4 (TLR4) and B-cell activating factor (BAFF) in ovalbumin (OVA)-induced mouse asthma. Int Immunopharmacol. 2008;8:935–944. doi: 10.1016/j.intimp.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 6.Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med. 2008;45:1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, et al. Regulation of PDGF signaling and vascular remodeling by peroxiredoxin II. Nature. 2005;435:347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 8.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 9.Moon EY, Noh YW, Han YH, Kim SU, Kim JM, Yu DY, Lim JS. T lymphocytes and dendritic cells are activated by the deletion of peroxiredoxin II (Prx II) gene. Immunol Lett. 2006;102:184–190. doi: 10.1016/j.imlet.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Shau H, Gupta RK, Golub SH. Identification of a natural killer enhancing factor (NKEF) from human erythroid cells. Cell Immunol. 1993;147:1–11. doi: 10.1006/cimm.1993.1043. [DOI] [PubMed] [Google Scholar]
- 11.Masutani H, Ueda S, Yodoi J. The thioredoxin system in retroviral infection and apoptosis. Cell Death Differ. 2005;12(Suppl 1):991–998. doi: 10.1038/sj.cdd.4401625. [DOI] [PubMed] [Google Scholar]
- 12.Misse D, Yssel H, Trabattoni D, Oblet C, Lo Caputo S, Mazzotta F, Pene J, Gonzalez JP, Clerici M, Veas F. IL-22 participates in an innate anti-HIV-1 host-resistance network through acute-phase protein induction. J Immunol. 2007;178:407–415. doi: 10.4049/jimmunol.178.1.407. [DOI] [PubMed] [Google Scholar]
- 13.Geiben-Lynn R, Kursar M, Brown NV, Addo MM, Shau H, Lieberman J, Luster AD, Walker BD. HIV-1 antiviral activity of recombinant natural killer cell enhancing factors, NKEF-A and NKEF-B, members of the peroxiredoxin family. J Biol Chem. 2003;278:1569–1574. doi: 10.1074/jbc.M209964200. [DOI] [PubMed] [Google Scholar]
- 14.Jin DY, Chae HZ, Rhee SG, Jeang KT. Regulatory role for a novel human thioredoxin peroxidase in NF-kappaB activation. J Biol Chem. 1997;272:30952–30961. doi: 10.1074/jbc.272.49.30952. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Permar SR, Buzby AP, Letvin NL. Memory CD4+ T-lymphocyte loss and dysfunction during primary simian immunodeficiency virus infection. J Virol. 2007;81:8009–8015. doi: 10.1128/JVI.00482-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeh WW, Jaru-Ampornpan P, Nevidomskyte D, Asmal M, Rao SS, Buzby AP, Montefiori DC, Korber BT, Letvin NL. Partial protection of Simian immunodeficiency virus (SIV)-infected rhesus monkeys against superinfection with a heterologous SIV isolate. J Virol. 2009;83:2686–2696. doi: 10.1128/JVI.02237-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shirahata T, Shimoi A, Kanda H, Goto H, Nakane A. Importance of early gamma interferon production in Propionibacterium acnes-induced resistance to Toxoplasma gondii infection in mice. J Vet Med Sci. 1994;56:293–297. doi: 10.1292/jvms.56.293. [DOI] [PubMed] [Google Scholar]
- 18.Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr Purif. 1999;15:34–39. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- 19.Drusano GL, Prichard M, Bilello PA, Bilello JA. Modeling combinations of antiretroviral agents in vitro with integration of pharmacokinetics: guidance in regimen choice for clinical trial evaluation. Antimicrob Agents Chemother. 1996;40:1143–1147. doi: 10.1128/aac.40.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Svarovskaia ES, Barr R, Zhang X, Pais GC, Marchand C, Pommier Y, Burke TR, Jr., Pathak VK. Azido-containing diketo acid derivatives inhibit human immunodeficiency virus type 1 integrase in vivo and influence the frequency of deletions at two-long-terminal-repeat-circle junctions. J Virol. 2004;78:3210–3222. doi: 10.1128/JVI.78.7.3210-3222.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casimiro DR, Wang F, Schleif WA, Liang X, Zhang ZQ, Tobery TW, Davies ME, McDermott AB, O’Connor DH, Fridman A, et al. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with dna and recombinant adenoviral vaccine vectors expressing Gag. J Virol. 2005;79:15547–15555. doi: 10.1128/JVI.79.24.15547-15555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asmal M, Sun Y, Lane S, Yeh W, Schmidt SD, Mascola JR, Letvin NL. Antibody-dependent cell-mediated viral inhibition emerges after simian immunodeficiency virus SIVmac251 infection of rhesus monkeys coincident with gp140-binding antibodies and is effective against neutralization-resistant viruses. J Virol. 2011;85:5465–5475. doi: 10.1128/JVI.00313-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Y, Asmal M, Lane S, Permar SR, Schmidt SD, Mascola JR, Letvin NL. Antibody-dependent cell-mediated cytotoxicity in simian immunodeficiency virus-infected rhesus monkeys. J Virol. 2011;85:6906–6912. doi: 10.1128/JVI.00326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaston B, Singel D, Doctor A, Stamler JS. S-nitrosothiol signaling in respiratory biology. Am J Respir Crit Care Med. 2006;173:1186–1193. doi: 10.1164/rccm.200510-1584PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thivierge M, Le Gouill C, Tremblay MJ, Stankova J, Rola-Pleszczynski M. Prostaglandin E2 induces resistance to human immunodeficiency virus-1 infection in monocyte-derived macrophages: downregulation of CCR5 expression by cyclic adenosine monophosphate. Blood. 1998;92:40–45. [PubMed] [Google Scholar]
- 26.Sauri H, Ashjian PH, Kim AT, Shau H. Recombinant natural killer enhancing factor augments natural killer cytotoxicity. J Leukoc Biol. 1996;59:925–931. doi: 10.1002/jlb.59.6.925. [DOI] [PubMed] [Google Scholar]
- 27.Forthal DN, Landucci G, Cole KS, Marthas M, Becerra JC, Van Rompay K. Rhesus macaque polyclonal and monoclonal antibodies inhibit simian immunodeficiency virus in the presence of human or autologous rhesus effector cells. J Virol. 2006;80:9217–9225. doi: 10.1128/JVI.02746-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Mari JF, Saada JI, Mifflin RC, Valentich JD, Powell DW. HETEs enhance IL-1-mediated COX-2 expression via augmentation of message stability in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol. 2007;293:G719–728. doi: 10.1152/ajpgi.00117.2007. [DOI] [PubMed] [Google Scholar]
- 29.Kraft-Terry S, Gerena Y, Wojna V, Plaud-Valentin M, Rodriguez Y, Ciborowski P, Mayo R, Skolasky R, Gendelman HE, Melendez LM. Proteomic analyses of monocytes obtained from Hispanic women with HIV-associated dementia show depressed antioxidants. Proteomics Clin Appl. 2010;4:706–714. doi: 10.1002/prca.201000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Higashi C, Saji C, Yamada K, Kagawa H, Ohga R, Taira T, Fujimuro M. The effects of heat shock protein 90 inhibitors on apoptosis and viral replication in primary effusion lymphoma cells. Biol Pharm Bull. 2013;35:725–730. doi: 10.1248/bpb.35.725. [DOI] [PubMed] [Google Scholar]
- 31.Roesch F, Meziane O, Kula A, Nisole S, Porrot F, Anderson I, Mammano F, Fassati A, Marcello A, Benkirane M, Schwartz O. Hyperthermia stimulates HIV-1 replication. PLoS Pathog. 2012;8:e1002792. doi: 10.1371/journal.ppat.1002792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vozzolo L, Loh B, Gane PJ, Tribak M, Zhou L, Anderson I, Nyakatura E, Jenner RG, Selwood D, Fassati A. Gyrase B inhibitor impairs HIV-1 replication by targeting Hsp90 and the capsid protein. J Biol Chem. 2010;285:39314–39328. doi: 10.1074/jbc.M110.155275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Lint C, Bouchat S, Marcello A. HIV-1 transcription and latency: an update. Retrovirology. 2013;10:67. doi: 10.1186/1742-4690-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asmal M, Whitney JB, Luedemann C, Carville A, Steen R, Letvin NL, Geiben-Lynn R. In Vivo Anti-HIV Activity of the Heparin-Activated Serine Protease Inhibitor Antithrombin III Encapsulated in Lymph-Targeting Immunoliposomes. PLoS One. 2012;7:e48234. doi: 10.1371/journal.pone.0048234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montes M, Tagieva NE, Heveker N, Nahmias C, Baleux F, Trautmann A. SDF-1-induced activation of ERK enhances HIV-1 expression. Eur Cytokine Netw. 2000;11:470–477. [PubMed] [Google Scholar]