

Abstract
Cysteine is commonly used to attach peptides onto gold surfaces. In this work, we show that the inclusion of an additional linker of four residues in length (-PPPPC) of a rigid, hydrophobic nature, is a better choice for forming peptide self-assembled monolayers (SAMs) with well-ordered structure and high surface density. We compare the structure and function of the nonfouling peptide EKEKEKE-PPPPC-Am with EKEKEKE-C-Am. Circular dichroism (CD), attenuated total internal reflection Fourier transform infrared spectroscopy (ATR-FTIR), and molecular dynamics (MD) results show that EKEKEKE-PPPPC-Am forms a secondary structure while EKEKEKE-C-Am has a random structure. Surface plasmon resonance (SPR) sensor results show that protein adsorption on EKEKEKE-PPPPC-Am/gold is very low with small variation while protein adsorption on EKEKEKE-C-Am/gold is high with large variation. X-ray photoelectron spectroscopy (XPS) results show that both peptides have strong gold-thiol binding onto a gold surface, indicating that their difference in protein adsorption is due to their assembled structures. Further experimental and simulation studies were performed to show that -PPPPC is a better linker than -PC, -PPC, and -PPPC. Finally, we extend EKEKEKE-PPPPC-Am with the cell-binding sequence RGD and demonstrate control over specific vs. non-specific cell adhesion without using poly (ethylene) glycol (PEG). Adding a functional peptide to the nonfouling EK sequence avoids complex chemistries that are used for its connection to synthetic materials.
INTRODUCTION
A nonfouling peptide was formed by alternating negatively charged glutamic acid (E) and positively charged lysine (K) amino acid residues1. This corresponds with our recent analysis of over a thousand proteins that indicates E and K are the two most prevalent amino acids on the surfaces of proteins. The EK sequence forms a strong hydration layer similar to zwitterionic materials2. Natural materials based on nonfouling peptides have many biomedical applications. A non-peptide synthetic thiol anchor1,3,4 is often used to attach functional peptides to gold surfaces. Polypeptide mimics have also been used as water-resistant anchors to surfaces and display fouling resistance.5 EK peptide-based SAMs containing synthetic butanethiol anchors have been previously shown to have high surface packing densities and ultra-low fouling properties on gold surfaces.1 It is, however, highly desirable to replace these synthetic linkers with a natural peptide for surface attachment to form an all-peptide based material. The amino acid cysteine (C), which contains a thiol side chain,6 has been commonly used to attach peptides onto gold surfaces. Although cysteine binds strongly to gold, unlike synthetic thiol anchors, a peptide using cysteine alone as its surface anchoring group was not able to achieve well-ordered structure and high surface density necessary for ultra-low fouling surfaces7.
In addition to low fouling properties, it is beneficial to achieve specific interactions for biomedical applications. One such application is mimicking extracellular matrix proteins using the well-established arginine-glycine-aspartate (RGD) sequence, which can mimic fibronectin, vitronectin, fibrinogen, osteopontin, and bone sialoprotein8. This and other functional sequences are commonly conjugated onto a nonfouling synthetic polymer, such as PEG9, to prevent nonspecific protein adsorption which can undermine sequence specificity. Such conjugation processes are complex10. Polydispersity of polymers, identification of appropriate conjugation sites, non-uniformly distributed functional groups, and difficult post conjugation evaluation11,12 are all drawbacks to various conjugation methods. Integrating the nonfouling EK peptide sequence with RGD allows replacement of nonfouling synthetic materials and avoids the complex chemistries used in bioconjugation.
In this work, we investigate the impact of the length and the nature of a short linker to form a well-packed, ultra-low fouling peptide SAM, specifically the additional inclusion of one to four proline linker residues which provide hydrophobicity and helical secondary structure. We compare structures of the non-fouling sequence EKEKEKE-PPPPC-Am and EKEKEKE-C-Am in solution and on a gold surface. The low fouling segment of the peptide contains four negative glutamic acid residues (E) and three positive lysine residues (K). Overall charge neutrality of the peptide is maintained by leaving the N-terminus as a free amine, which contributes an extra positive charge to the peptide. The C-terminus of all peptides was amidated to cap the negative charge of the carboxylic acid which could create a negative charge near the gold binding thiol that might impede packing of the SAM through electrostatic repulsion.
The secondary structures of these two sequences were characterized using circular dichroism (CD) and attenuated total internal reflection Fourier transform infrared spectroscopy (ATR-FTIR) in a solution and on a surface, respectively. Molecular dynamics simulations were also performed to further elucidate secondary structure differences. X-ray photoelectron spectroscopy (XPS) was used to evaluate surface attachment. Nonspecific protein adsorption was measured via surface plasmon resonance (SPR) sensors. Furthermore, RGD was added to the N-terminus of the EKEKEKE-PPPPC-Am sequence to gain specific cell adhesion against an ultra-low fouling background of the EK peptide, eliminating the need for conjugation of biomolecular recognition peptides to nonfouling synthetic polymers, such as PEG. The peptide sequence RGD-EKEKEKE-PPPPC-Am used in this work is illustrated in Figure 1. This all-in-one peptide contains biomolecular recognition, ultra-low fouling, and surface anchoring functions.
Figure 1.
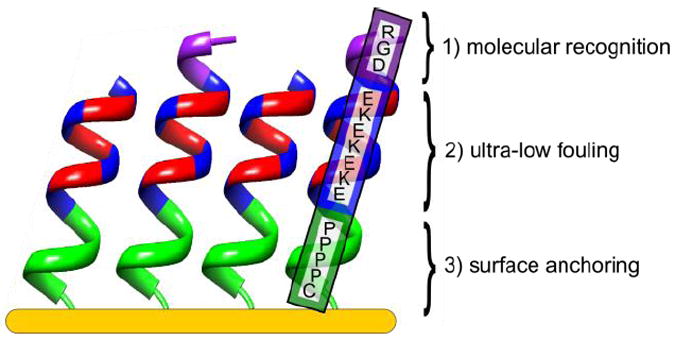
All-in-one natural peptide SAM on gold with three distinct functions incorporated into the peptide sequence: 1) biomolecular recognition; RGD 2) ultra-low fouling; EK 3) surface anchoring composed of four rigid, hydrophobic proline (P) residues and a cysteine (C) residue with a secondary structure to achieve well-ordered structure and high surface density.
RESULTS AND DISCUSSION
Impact of nature and length of linker on SAM formation
Surface plasmon resonance (SPR) shows that simply using a cysteine residue containing a thiol side chain for surface anchoring is insufficient to form a low fouling peptide SAM (Figure 2a). The fouling to fibrinogen and lysozyme for the linker-free peptide SAM (EKEKEKE-C-Am) is 38.25 ± 28.96 ng/cm2 and 5.25 ± 4.40 ng/cm2, respectively. In addition to high fouling, there is a large distribution in the fibrinogen protein adsorption values (Figure 2b).
Figure 2.
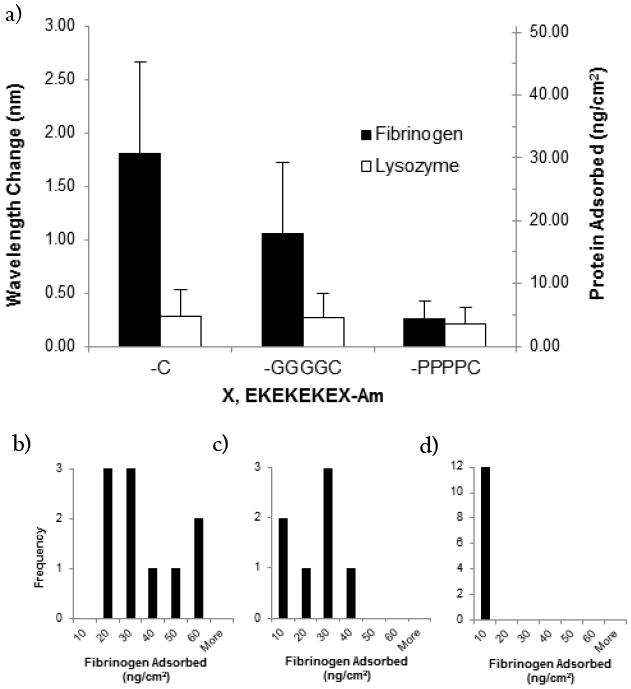
(a) Protein adsorption on peptide SAMs composed of the linker-free peptide (EKEKEKE-C-Am), the glycine linker peptide (EKEKEKE-GGGGC-Am), and the proline linker peptide (EKEKEKE-PPPPC-Am). SPR results are in the unit of wavelength shift (nm) or converted surface concentration (ng/cm2). (Black: fibrinogen, White: lysozyme). Each data point represents an average value ± standard deviation from at least three independent measurements. Lower Panel: Histograms of adsorbed fibrinogen protein (ng/cm2) for (b) linker-free peptide (c) glycine linker peptide and (d) proline linker peptide.
Here, we consider the effect of including a proline (P) linker and a glycine (G) linker in the EKEKEKE-C-Am peptide studied. Proline was selected based on its hydrophobicity, its ability to act as an α-helix destabilizing residue13, and its rigid structure which all should promote packing for SAM formation. Additionally, the covalently constrained backbone of proline reduces the entropy loss upon adsorption of the peptide to the surface, whereas other amino acids lose mobility upon adsorption14. Glycine was selected based on its hydrophilicity and its flexibility for comparison with proline.
The additional inclusion of four prolines as a linker resulted in an ultra-low fouling SAM, defined as having an adsorption of less than 5 ng/cm2 of fibrinogen15. The fouling to fibrinogen and lysozyme for the proline linker peptide SAM (EKEKEKE-PPPPC-Am) is 4.35 ± 2.85 ng/cm2 and 3.52 ± 2.57 ng/cm2, respectively, which is comparable to the previously studied butanethiol containing peptide SAM1. Furthermore, the proline linker peptide has a narrow distribution for fibrinogen protein adsorption, as opposed to the large distribution seen for the linker-free peptide (Figure 2d).
As a comparison to the rigid, hydrophobic proline residue, the flexible, hydrophilic glycine residue was also used as a linker. SPR indicates that the inclusion of a glycine linker (EKEKEKE-GGGGC-Am) results in a high fouling SAM, with fouling to fibrinogen and lysozyme of 17.94 ± 11.35 ng/cm2 and 4.51 ± 3.82 ng/cm2, respectively. It is not surprising that the flexible linker compromised the fouling properties of the SAM as this would disrupt close packing on the surface, just as in the linker-free peptide. The glycine linker peptide also has a large distribution for fibrinogen protein adsorption (Figure 2c). In contrast, the inclusion of the rigid, hydrophobic proline linker allows for favorable hydrophobic interactions between chains, enabling better packing. These hydrophobic groups may be analogous to the role of the methyl groups found in synthetic butanethiol anchors.1
To determine how many linker residues are needed to promote close packing of the surface, the effect of linker length on the fouling properties of the SAMs was examined. Backbone and side-chain Monte Carlo simulations were performed using the Rosetta scoring function16 on peptide sequences containing one to four prolines in the linker. These simulations predict peptide conformations in solution. Because these peptides contain so few residues, they lack a deep free energy minimum (fold) and are expected to shift between several low energy conformations. A sample of the lowest energy structures predicted by simulations for peptide sequences containing one to four prolines in the linker shows that the addition of proline favors an extended, rigid conformation for the peptide sequence (Supporting Figure S1). We hypothesize that the extended, rigid conformation allows for closer packing of peptide chains, thus enabling the formation of a uniform, low fouling surface. As expected, SPR indicates an increase in proline number corresponds to a decrease in the amount of adsorbed protein (Supporting Figure S1). The linker containing four prolines is the threshold for achieving an ultra-low fouling surface and thus four peptide residues was chosen as the linker length. Overall, simply using a cysteine residue for surface anchoring does not provide a peptide capable of self-assembly into an ultra-low fouling surface. Thus, selection of an appropriate peptide linker that will allow close-packing of chains is essential. Here, we have identified a linker of four residues in length and of a rigid, hydrophobic nature as one possible robust option. Other strategies have been considered using natural peptides to improve peptide self-assembly on surfaces17, on nanoparticles18, and for membrane formation19.
Impact of secondary structure on SAM formation
In order to further investigate the large differences in fouling between the linker-free peptide (EKEKEKE-C-Am) and the proline linker peptide (EKEKEKE-PPPPC-Am), we examine secondary structure in solution via CD and molecular simulations, and on the surface via ATR-FTIR. CD spectra were collected in 10 mM potassium phosphate, 50 mM sodium sulfate buffer, pH 7.4 (Figure 3a). The CD spectrum for the linker-free peptide is indicative of a disordered structure with very low ellipticity above 210 nm and negative bands near 195 nm20. In contrast, the CD spectrum for the proline linker peptide designates an extended polyproline helix conformation with a strong negative band near 200 nm and a weaker positive band around 225 nm21.
Figure 3.
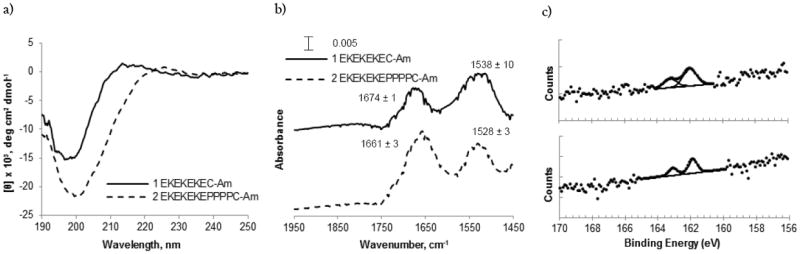
Secondary structure comparison of the linker-free peptide (EKEKEKE-C-Am) and the proline linker peptide (EKEKEKE-PPPPC-Am). (a) CD spectra of peptide sequences EKEKEKE-C-Am (1, Solid) and EKEKEKE-PPPPC-Am (2, Dashed) in 10 mM potassium phosphate, 50 mM sodium sulfate buffer, pH 7.4. (b) Representative ATR-FTIR spectra for peptide SAMs EKEKEKE-C-Am (1, Solid) and EKEKEKE-PPPPC-Am (2, Dashed) on Au (111) coated mica substrate. Average maximum intensities for amide I (left) and amide II (right) bands are listed above peaks in the spectrum from at least three independent measurements. (c) XPS S2p spectra for EKEKEKE-C-Am (top) and EKEKEKE-PPPPC-Am (bottom) peptide SAMs adsorbed on Au (111) coated mica substrate. To properly fit the experimental peptide SAM spectrum a S2p doublet with 2:1 area ratio and splitting of 1.2 eV was used.
Replica-exchange molecular dynamics simulations were performed in solution to supplement CD data for the linker-free peptide, the proline linker peptide, and the glycine linker peptide. Figure 4 shows the lowest energy secondary structure at each residue position for the three peptides in the top panel. The linker-free and the glycine linker peptide lack secondary structure. The proline linker peptide displays helical structure most likely due to the rigidity imparted on the backbone from the proline linker. The lower panel of Figure 4 shows the C-α to C-α end to end distance as a probability distribution with the vertical lines showing the median. The proline linker peptide is indeed more extended than both the linker-free and the glycine linker control peptides.
Figure 4.
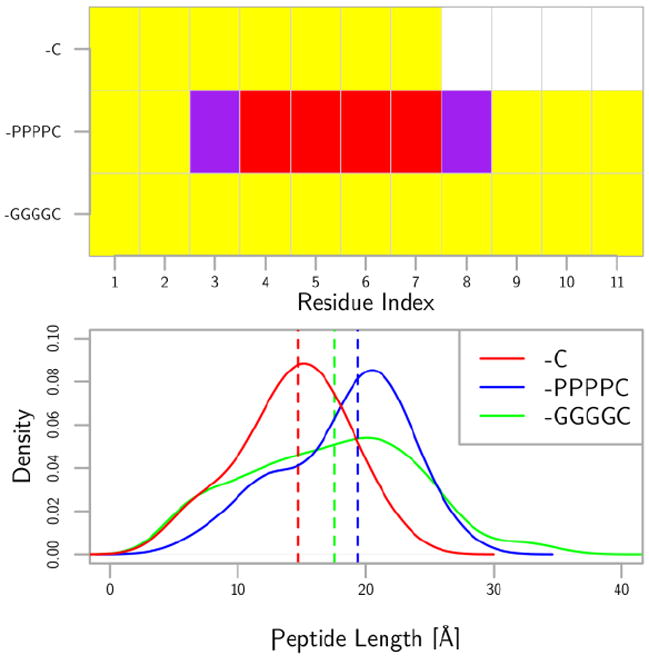
The top panel shows the secondary structure mode for each residue on the three different peptides. The secondary structure was determined using DSSP on the trajectories obtained from tempered annealing molecular dynamics simulations of dilute peptides. The yellow is no secondary structure, the purple is a turn or bend with a hydrogen bond, and red is alpha-helix. The bottom panel shows the probability distribution of the Cα-Cα distance between the initial and final residues in the peptide. The probability distribution is taken from a kernel density estimator with a bandwidth of 3. The vertical lines are the medians of the data. The proline linker peptide is the most extended among the three.
We examined the linker-free and proline linker peptide SAMs on a gold surface using ATR-FTIR. Evaluation of ATR-FTIR amide I bands provides surface sensitive information about the secondary structure of peptide monolayers. The ATR-FTIR spectrum for the linker-free peptide resulted in maximum intensities for amide I and amide II bands at 1674 ± 1 and 1538 ± 10 cm-1, whereas the ATR-FTIR spectrum for the proline linker peptide resulted in maximum intensities for amide I and amide II bands at 1661 ± 3 and 1528 ± 3 cm-1 (Figure 3b). The higher shifted intensity of the amide I band for the linker-free peptide at 1674 cm-1 is not characteristic of an α-helix or a β-sheet, suggesting that the structure of this peptide is unordered 22,23. The amide I band for the proline linker peptide at 1661 cm-1 is shifted slightly higher than regular alpha helical structures typically with amide I bands at 1650-1658 cm-1. This most likely represents the presence of distorted and/or 310 helices as expected for a peptide containing several proline residues24. Furthermore, the major amide I band is considerably broad, indicating a larger distribution of slightly different helical structures.
Results from CD, molecular simulations, and ATR-FTIR all support a disordered, random structure for the linker-free peptide and an extended, helical structure for the proline linker peptide. This secondary structure characterization provides a possible explanation for the differences in fouling between the two surfaces. The disordered structure of the peptide containing no linker (EKEKEKE-C-Am) could impede packing of the peptide SAM, resulting in non-uniform surface coverage leading to high fouling and large variations in the results. At the same time, the defined, extended conformation of the proline linker peptide (EKEKEKE-PPPPC-Am) allows for closer packing of the monolayer and a more uniform surface with consistent ultra-low fouling behavior. These results indicate that it is preferable to have a well-defined secondary structure to form a closely-packed, uniform monolayer.
Finally, XPS data were collected for the linker-free and proline linker peptide SAMs to confirm that their difference in performance is due to their secondary structure rather than their surface binding characteristics. The binding energy of the S2p3/2 peak was 161.9 eV for both surfaces, consistent with sulfur atoms bound to the gold surface as a thiolate species (Figure 3c)25. The presence of only bound thiolate species in the high resolution sulfur spectra indicate that the thiol side chain of the cysteine anchor is bound to the gold and is not simply physically adsorbed on the surface. XPS data also provide information about the surface coverage and lateral packing density of a SAM, determined by measurement of the attenuation of the metal substrate photoelectrons (Au4f)26. The gold signal for the proline linker peptide is lower than the gold signal for the linker-free peptide, indicating that the proline linker peptide SAM surface is more densely packed (Supporting Figure S2). However, the longer length of the proline linker peptide compared to the linker-free peptide may also partially account for the attenuation of the gold signal. The higher surface coverage of the proline linker peptide SAM indicates improved packing of the peptide chains, corroborating the secondary structure data from CD, molecular simulations, and ATR-FTIR.
Several parameters influence monolayer architecture and thus low fouling behavior, including chemical moieties, secondary structure formation, peptide length, and peptide packing density. The inclusion of the four proline residue linker introduced in this work induces helical structure at the base of the peptide as evidenced by ATR-FTIR and molecular simulations. The helical formation reduces the mobility of the chains. Thus, this rigid secondary structure results in more uniform peptide molecular conformations, allowing chains to pack tighter, increasing packing density and reducing fouling. As seen in Supporting Figure S1, the number of proline residues in the linker has a profound effect on fouling, due to its importance in maintaining the rigid structure necessary for well-packed monolayers. The transition from three to four proline residues, however, shows that the effect of the peptide length lessens beyond three linker residues. Similarly, as seen in Figure 4, the EK residues more than four units beyond the linker are no longer influenced, adopting the random conformations seen in the linker-free and glycine linker sequences. Unlike mixed charge alkanethiol SAMs, where regular crystalline structures are observed1, we expect that the solvent-exposed surface of peptide SAMs is a uniform display of mixed charge E and K residues. Our previous work on mixed E and K random coplymerization studies1 show that even with amorphous conformations, non-fouling is achieved.
Specific biomolecular recognition on ultra-low fouling background
In this experiment, we extend EKEKEKE-PPPPC-Am with the cell-binding sequence RGD. Cell adhesion onto peptide SAMs composed of mixtures of RGD-EKEKEKE-PPPPC-Am and EKEKEKE-PPPPC-Am was evaluated to display the biomolecular recognition capabilities of our all-in-one peptide biomaterial. The molar percentage of RGD-EKEKEKE-PPPPC-Am was increased from 0 to 100%. Peptide SAMs were seeded with NIH-3T3 fibroblast cells and incubated in supplemented medium for a period of 24 hours and then evaluated for cell adhesion by phase contrast microscopy. As expected, the number of adhered cells increases proportionally to the molar percentage of the RGD-EKEKEKE-PPPPC-Am peptide (Figure 5). The SAM composed entirely of EKEKEKE-PPPPC-Am had no cell attachment, corresponding with the ultra-low fouling results for protein adsorption. The scrambled sequence RDG-EKEKEKE-PPPPC-Am was mixed with EKEKEKE-PPPPC-Am at molar percentages of 50% and 75% and evaluated for cell adhesion as a control. Surfaces containing scrambled RDG showed significantly less cell adhesion than peptide SAMs containing RGD (Supporting Figure S3). Furthermore, cells adhered to scrambled RDG surfaces displayed rounded morphology, whereas cells adhered to RGD surfaces were well spread out. These results show that interactions seen for the RGD sequence are specific and that the EK portion of the peptide maintains an ultra-low fouling background.
Figure 5.
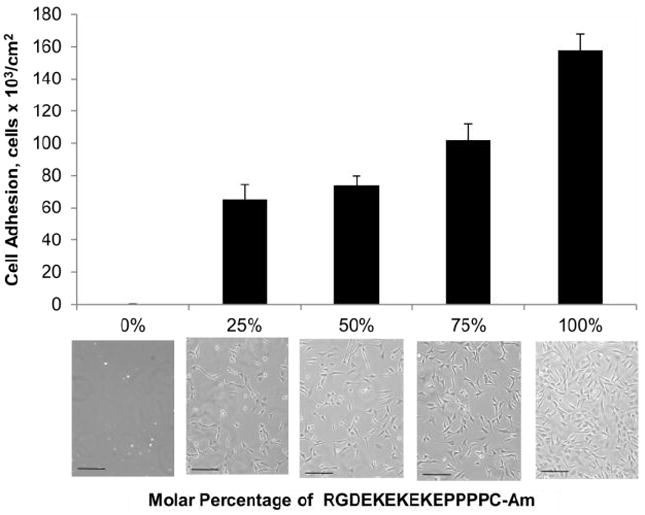
Cell adhesion results after 24 hour incubation in supplemented medium for peptide SAMs composed of mixtures of RGD-EKEKEKE-PPPPC-Am and EKEKEKE-PPPPC-Am. The molar percentage of RGD-EKEKEKE-PPPPC-Am was increased from 0 to 100%. The scale bar represents 100μm. Cell adhesion for the scrambled peptide sequence RDG-EKEKEKE-PPPPC-Am was evaluated as a control (see Supporting Information).
CONCLUSIONS
Overall, simply using a cysteine residue for surface anchoring does not provide a peptide capable of self-assembly into an ultra-low fouling surface on gold surfaces. Selection of an appropriate peptide linker that will allow close-packing of chains is essential. Here, we have identified a linker of four residues in length together with the amino acid cysteine and of a rigid, hydrophobic nature as one possible robust linker. This provides a well-defined secondary structure needed to promote closely packed monolayers with ultra-low fouling properties. The linker -PPPPC is an effective surface anchoring sequence for a wide range of applications beyond nonfouling involving peptide assembly onto a gold surface. The tri-amino acid sequence RGD was added as an example for the inclusion of specific interactions on a nonfouling background, eliminating the need for bioconjugation to synthetic materials such as PEG. This strategy is applicable to any other functional peptides beyond RGD. The concept of this all-in-one natural peptide can be easily adapted to introduce biorecognition, nonfouling and surface binding functions into all natural materials.
EXPERIMENTAL SECTION
Preparation of peptide SAMs
Peptides were ordered from Synthetic Biomolecules (San Diego, CA) at a purity of >95%. Peptides were synthesized using Fmoc chemistry and purified by reverse phase high performance liquid chromatography (RP-HPLC). Gold coated chips were cleaned by rinsing with Millipore water, ethanol (Decon Labs, Inc., King of Prussia, PA), and then drying with filtered air. They were placed in the UV cleaner for 20 minutes. Cleaned gold chips were incubated in a phosphate buffered saline (PBS) solution (pH 7.4 and Ionic Strength of 150 mM, Sigma Aldrich, St. Louis, MO) containing 0.14 mM peptide for 24 hours. Once removed, the gold chips were rinsed with Millipore water and dried by filtered air. This procedure was followed for all peptide SAM surfaces prepared for SPR, ATR-FTIR, XPS, and cell adhesion experiments.
Protein adsorption by SPR sensor
A four-channel SPR sensor was used to measure protein adsorption. Samples were rinsed with Millipore water, dried by filtered air, and mounted to the device. The temperature controller was set to 25 ± 0.01 °C. Protein adsorption was measured by flowing PBS over the SAM at 40 μL/min for 10 minutes, followed by 1 mg/mL protein solutions of fibrinogen (from bovine plasma, Sigma) or lysozyme (from chicken egg white, Sigma) for 10 minutes, and finally a PBS rinse for 10 minutes. The wavelength shift between baselines before protein injection and after rinsing was used to quantify the total amount of protein adsorbed. A reference channel containing PBS was flown for each chip and used to correct for baseline drift. A 1 nm wavelength shift from 750 nm corresponds to 17 ng/cm2 adsorbed proteins27. The detection limit for the SPR is 0.3 ng/cm2.
CD of peptide solutions
CD spectra were recorded between 190 and 270 nm (step resolution of 0.2 nm) of 0.1 mg/mL peptide in 10 mM potassium phosphate (JT Baker, Austin, TX), 50 mM sodium sulfate buffer (EMD, Darrnstadt, Germany), pH 7.4 at 25 °C with a JASCO J-720 CD spectropolarimeter using an optical cell of 1.0 mm path length. Each experiment was repeated three times. Buffer spectra were recorded and subtracted from the sample spectra.
Secondary structure simulations
Molecular dynamics were conducted using the GROMACS 4.5.3 simulation engine28 and the AMBER99sb-ildn force field29. Replica exchange with 100 replicas, exchange attempts every 50 fs, and a time step of 2 fs in the NVT ensemble was used for the simulations. The temperature distribution was from 300 K to 450 K and can be seen in the SI. Particle-mesh Ewald sums was used to treat electrostatics30. The Van der Walls cut-off was 1 nm with an appropriate shifting function. The “v-rescale” thermostat31 was used, a stochastic thermostat not to be confused with velocity rescaling. The thermostat time constant was 0.5 ps. The simulations were run for 20 ns and the secondary structure was characterized at the 300 K replica using the DSSP program32 and the per-residue modes are shown in Figure 4. Further information on the preparation of the systems and information on the Monte Carlo simulations of the proline linker peptide are found in the Supporting Information.
ATR-FTIR of peptide SAMs
Peptide SAMs were assembled on gold coated mica substrates. After incubation the chips were dried in a desiccator overnight before evaluation by ATR-FTIR. The ATR-FTIR spectra were acquired using Harrick’s GATR single-angle reflection accessory in conjunction with a Bruker Tensor spectrometer. Each spectrum was collected with a minimum of 80 scans, at 4 cm-1 resolution, and 65° incident angle. Optimum contact between the germanium crystal and the sample were maintained throughout measurements. Bare gold on mica was recorded as the background spectra and subtracted from the sample spectra. Three replicates of each SAM were analyzed.
XPS of peptide SAMs
Peptide SAMs were assembled on gold coated mica substrates. XPS experiments were performed on a Kratos Axis Ultra DLD spectrometer using a monochromatic Al K α x-ray source (hν = 1486.6 eV) operated at 10 mA at 15 kV. The analysis area was approximately 300 × 700 μm. Survey spectra were acquired with an analyzer pass energy of 80 eV. The high resolution S2p and C1s spectra were acquired with an analyzer pass energy of 20 eV. All XPS data were acquired at a nominal photoelectron takeoff angle of 0°, where the takeoff angle is defined as the angle between the surface normal and the axis of the analyzer lens. Three spots on two replicates of each SAM were examined. The compositional data are averages of the values determined at each analysis spot.
Cell seeding of peptide SAMs
Prior to cell adhesion experiments, samples were sterilized by soaking and rinsing with copious amounts of sterilized PBS. NIH-3T3 fibroblasts (ATCC, Manassas, VA) were plated at 4 × 104 cells/mL in 3 mL Dulbecco’s Modified Eagle Medium (DMEM) with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin-streptomycin (PS) (Invitrogen Corp, Carlsbad, CA) and incubated for 24 hours at 37 °C in 5% CO2 and 100% relative humidity. Cell seeding density was determined using a hemocytometer. Cell adhesion and morphology was observed under a phase contrast microscope (Nikon Eclipse TE2000-U). The number of cells adhered was determined by visually counting cells present in at least three microscope images (100x objective magnification).
Supplementary Material
Acknowledgments
This work was made possible by financial support from the National Science Foundation (CBET-0854298) and Office of Naval Research (N00014-10-1-0600). A.K.N. acknowledges the National Science Foundation for fellowship support. XPS samples were analyzed by the NESAC/BIO facility at the University of Washington (NIH Grant EB-002027). The authors thank Lara Gamble and Jeanette Stein for useful discussions relating to this project.
Footnotes
Supporting Information
Simulations and SPR data for effect of peptide linker length, XPS surface composition data, control cell adhesion experiment, and more detailed simulation methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chen S, Cao Z, Jiang S. Biomaterials. 2009;30:5892–5896. doi: 10.1016/j.biomaterials.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Chen S, Zheng J, Li L, Jiang S. J Am Chem Soc. 2005;127:14473–14478. doi: 10.1021/ja054169u. [DOI] [PubMed] [Google Scholar]
- 3.Chelmowski R, Köster D, Kerstan A, Prekelt A, Grunwald C, Winkler T, Metzler-Nolte N, Terfort A, Wöll C. J Am Chem Soc. 2008;130:14952–14953. doi: 10.1021/ja8065754. [DOI] [PubMed] [Google Scholar]
- 4.Bolduc OR, Pelletier JN, Masson JF. Anal Chem. 2010;82:3699–3706. doi: 10.1021/ac100035s. [DOI] [PubMed] [Google Scholar]
- 5.Statz AR, Meagher RJ, Barron AE, Messersmith PB. J Am Chem Soc. 2005;127:7972–7973. doi: 10.1021/ja0522534. [DOI] [PubMed] [Google Scholar]
- 6.Boncheva M, Vogel H. Biophys J. 1997;73:1056–1072. doi: 10.1016/S0006-3495(97)78138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostuni E, Chapman RG, Holmlin RE, Takayama S, Whitesides GM. Langmuir. 2001;17:5605–5620. doi: 10.1021/la0015258. [DOI] [PubMed] [Google Scholar]
- 8.Hynes RO. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 9.VandeVondele S, Vörös J, Hubbell JA. Biotechnol Bioeng. 2003;82:784–790. doi: 10.1002/bit.10625. [DOI] [PubMed] [Google Scholar]
- 10.Veronese FM. Biomaterials. 2001;22:405–417. doi: 10.1016/s0142-9612(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 11.Perlin L, MacNeil S, Rimmer S. Soft Matter. 2008;4:2331–2349. [Google Scholar]
- 12.Niu X, Wang Y, Luo Y, Xin J, Li Y. J Mater Sci Technol. 2005;21:571–576. [Google Scholar]
- 13.MacArthur MW, Thornton JM. J Mol Biol. 1991;218:397–412. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 14.Creighton TE. Science. 1990;247:1351–1352. doi: 10.1126/science.247.4948.1351-a. [DOI] [PubMed] [Google Scholar]
- 15.Tsai WB, Grunkemeier JM, Horbett TA. J Biomed Mater Res. 1999;44:130–139. doi: 10.1002/(sici)1097-4636(199902)44:2<130::aid-jbm2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 16.Rohl CA, Strauss CEM, Misura KMS, Baker D. Method Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 17.Zhang S, Yan L, Altman M, Lässle M, Nugent H, Frankel F, Lauffenburger DA, Whitesides GM, Rich A. Biomaterials. 1999;20:1213–1220. doi: 10.1016/s0142-9612(99)00014-9. [DOI] [PubMed] [Google Scholar]
- 18.Lévy R, Thanh NTK, Doty RC, Hussain I, Nichols RJ, Schiffrin DJ, Brust M, Fernig DG. J Am Chem Soc. 2004;126:10076–10084. doi: 10.1021/ja0487269. [DOI] [PubMed] [Google Scholar]
- 19.Nam KT, Shelby SA, Choi PH, Marciel AB, Chen R, Tan L, Chu TK, Mesch RA, Lee B, Connolly MD, Kisielowski C, Zuckermann RN. Nat Mater. 2010;9:454–460. doi: 10.1038/nmat2742. [DOI] [PubMed] [Google Scholar]
- 20.Greenfield NJ. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woody RW. J Am Chem Soc. 2009;131:8234–8245. doi: 10.1021/ja901218m. [DOI] [PubMed] [Google Scholar]
- 22.Surewicz WK, Mantsch JHH, Chapman D. Biochemistry. 1993;32:389–394. doi: 10.1021/bi00053a001. [DOI] [PubMed] [Google Scholar]
- 23.Sakurai T, Oka S, Kubo A, Nishiyama K, Taniguchi I. J Peptide Sci. 2006;12:396–402. doi: 10.1002/psc.740. [DOI] [PubMed] [Google Scholar]
- 24.Singh BR, editor. Infrared Analysis of Peptides and Proteins: Principles and Applications. American Chemical Society; Washington, DC: 2000. [Google Scholar]
- 25.Castner DG, Hinds K, Grainger DW. Langmuir. 1996;12:5083–5086. [Google Scholar]
- 26.Herrwerth S, Eck W, Reinhardt S, Grunze M. J Am Chem Soc. 2003;125:9359–9366. doi: 10.1021/ja034820y. [DOI] [PubMed] [Google Scholar]
- 27.Wolfbeis OS, Homola J, editors. Surface Plasmon Resonance Based Sensors. Vol. 4 Springer Series on Chemical Sensors and Biosensors, Springer; New York, U.S.: 2006. [Google Scholar]
- 28.Hess B, Kutzner C, van der Spoel D, Lindahl E. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 29.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 31.Bussi G, Donadio D, Parrinello M. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.