

Abstract
Background
Eosinophils rapidly undergo apoptosis unless exposed to prosurvival cytokines such as interleukin 5 (IL-5) or granulocyte-macrophage colony stimulating factor (GM-CSF). In vivo, eosinophils are exposed to TGF-β 1 which can induce apoptosis suggesting it may function to counteract the effects of IL-5 or GM-CSF and limit, in vivo tissue eosinophilia.
Objective
The objective of this study was to investigate the proapoptotic effects of TGF-β alone and in combination with IL-5 on eosinophils.
Methods
Peripheral blood eosinophil (PBEos) viability was assessed using flow cytometry after exposure to TGF-β1 and IL-5. Calpain-1 activation was determined in cell extracts by western blot analysis of endogenous substrates and with a fluorogenic α-spectrin substrate. Molecular interactions between calpain1 and calpastatin were assessed by immunoprecipitation and western blotting.
Results
Physiologic concentrations of TGF-β1 significantly antagonized the prosurvival effects of IL-5. TGF-β1-induced apoptosis was suppressed by inhibitors of calpain, or its downstream target, caspase 3. TGF-β1 signaling through Smad3 was unaffected by IL-5 and was required for the pro-apoptotic effects of TGF-β1. However, IL-5 induced Akt phosphorylation was inhibited by TGF-β1 and was associated with accelerated calpain cleavage and eosinophil death.
Conclusion
TGF-β1 induces calpain-1 activation through antagonism of Akt which induces caspase activation and eosinophil apoptosis.
Keywords: TGF-β1, IL-5, Eosinophils, Calpain-1, Apoptosis, Caspase 3, Intracellular signaling
Introduction
Peripheral blood eosinophils (PBEos) are terminally differentiated, nondividing cells with a life-span of approximately 3 days in the circulation. Eosinophil production and longevity are positively regulated by IL-5 which induces both bone marrow production as well as increased life-span in the blood and tissues [1]. In addition, IL-5 possesses eosinophil chemotactic activity, increases eosinophil adhesion to endothelial cells and enhances eosinophil effector functions [2]. Interference with IL-5 signaling by mepoluzimab dramatically reduces eosinophil counts in the blood and tissues and shows considerable promise for the treatment of asthma as well as eosinophilic gastritis and eosophagitis [3].
IL-5 induces a variety of signaling cascades including Jak-STAT, Ras-Extracellular Signal Regulated Kinase (ERK) and Phosphatidylinositol-3 Kinase (PI3K)/Akt (protein kinase B, PKB) pathways. All have been implicated in IL-5-dependent PBEos survival, proliferation and differentiation in vitro and in vivo [4–6]. Activation of these cascades antagonizes Bax activation, prevents mitochondrial disruption and blocks caspase cleavage and activation [7,8]. Much less clear is how the function and longevity of IL-5 activated PBEos or tissue eosinophils are suppressed in vivo upon cessation of pro-inflammatory stimuli.
One possible mechanism invokes pro-apoptotic co-signaling by anti-inflammatory cytokines such as TGF-β1 [9]. Tissue eosinophils are exposed to autocrine and paracrine sources of TGF-β1, whose expression increases at the sites of allergic inflammation including asthmatic airways [10]. Human eosinophils have intact Smad signaling and respond to the anti-inflammatory properties of TGF-β including acceleration of apoptosis [10–14]. How TGF-β1 antagonizes eosinophil survival in the context of IL-5 as seen in vivo remains incompletely understood.
Calpains are non-lysosomal cysteine proteases that are selectively activated in response to calcium signals [15] and thereby control cellular functions such as cytoskeletal remodeling, cell-cycle progression, gene expression and apoptotic cell death [16–18]. In mammals, calpain-1 (calpain I, μ-calpain) and calpain-2 (calpain II, m-calpain) are ubiquitously expressed and distinguished by their in vitro calcium requirements. Both calpain-1 and calpain-2 are heterodimers consisting of an 80KD, calcium-binding catalytic subunit and a 30-kDa regulatory subunit. The activity of calpains is tightly controlled by the endogenous inhibitor calpastatin. Calpastatin is an intrinsically unstructured protein that in the presence of calcium, reversibly binds to and inhibits four molecules of calpain [19,20]. Calpains have been reported to regulate neutrophil apoptosis [21,22]. Recently we showed that calpain inhibitors reduced PBEos death likely by preventing the cleavage and activation of pro-apoptotic Bax [7].
In this study, we demonstrate that calpain-1 is activated during spontaneous eosinophil apoptosis which is antagonized by IL-5. TGF-β1 can overcome IL-5 mediated suppression of calpain by inducing extracellular calcium entry, blocking Akt signaling and accelerating the cleavage of calpastatin and procalpain-1. These results suggest that TGF-β1 can induce PBEos apoptosis by modulating IL-5 signaling through Akt and calpain dependent mechanisms.
Methods
Reagents
Recombinant human IL-5, purified human TGF-β1 and caspase-3 inhibitor (Z-DEVD-FMK) were purchased from R&D Inc. (Minneapolis, Minn., USA). Calpeptin, LY-294002 (phosphatidylinositol 3-kinase inhibitor), Akt inhibitor IV, BAPTA/AM, fluorogenic alpha-spectrin, CytoBuster™ protein extraction reagent, cocktail set III were purchased from Calbiochem (La Jolla, CA, USA). TGF-β1/activin type I receptor kinase inhibitor (SB431542) was purchased from Tocris (Ellisville, Mo., USA). Antibody to phospho-Erk1/2 and total Erk1/2 were purchased from Promega (Madison, WI, USA). Anti-calpain1/2, anti-phospho-JNK (Thr183/Tyr185), anti-phospho-p38 (Thr180/Tyr182), anti-Akt, anti-phospho-Akt (Ser473), anti-phospho-Smad2 (Ser465/467) and anti-phospho-Smad 3 (Ser423/425) were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-Smad2 and Smad3 were purchased from Abcam (Cambridge, MA, USA). Anti-calpastatin was purchased from Chemicon International (Temecula, CA, USA). Anti-α-spectrin was purchased from Novus Biologicals (Littleton, CO, USA). Monoclonal anti-β-actin (Ab-1) was purchased from Oncogene Research Products (San. Diego, CA, USA). Horseradish peroxidase–conjugated anti-rabbit (secondary antibody; NA934V) and the enhanced chemilumiscence ECL immunoblot detection system were from Amersham Pharmacia Biotech (Piscataway, NJ, USA).
Calpain activity assay
Calpain activity was determined in cytosolic extracts, prepared with CytoBuster Protein Extraction Reagent. Protein concentrations were determined by the Bradford method. Protease activity was determined for 15μg protein after the addition of 100μl reaction buffer (50 mM Tris/HCl, pH 7.5, 50 mM NaCl, 1mM EDTA, 1mM EGTA, 5mM 2-mercaptoethanol, 5 mM CaCl2) and 50μl of the fluorogenic calpain-1 substrate (10μM). The change in fluorescence was measured in a SpectraMax Gemini XPS spectrofluorometer (Molecular Devices Corporation, USA.) at an excitation wavelength of 490 nm and an emission wavelength of 518 nm.
Subjects and eosinophil preparation
Peripheral blood was obtained by venipuncture from healthy or mildly atopic donors. Peripheral blood eosinophils were purified by a negative immunomagnetic procedure as described [23]. Only eosinophil preparations >97% pure were used. After isolation, eosinophils were cultured at 37°C in a humidified atmosphere of 5% CO2 and 95% air at a density of 1×106 cells per ml in RPMI-1640 medium, 10% (vol/vol) FBS and gentamycin (50 mg/ml) all from Life Technologies. All participants have a clinical record at the University of Wisconsin Hospital and informed consent was obtained according to an approved protocol of the University of Wisconsin Hospital Institutional Review Board.
Cell viability and flow cytometry
Eosinophils (1×106 cells per ml) were cultured in 96-well tissue culture plates (BD Biosciences). As needed, cells (1×105) were stained with trypan blue and counted manually or annexin V–fluorescein isothiocyanate and propidium iodide (BD Bioscience) and analyzed by flow cytometry.
Immunoblot and Immunoprecipitation
For immunoblot, equal numbers of untreated or treated eosinophils were pelleted, suspended in cold water plus SDS-PAGE loading buffer and boiled for 5 minutes prior to SDS-PAGE and western blotting. For immunoprecipitation, cells were lysed in NP-40 buffer (50 mM Tris/HCl, 150 mM NaCl, 2 mM EDTA, 1% NP-40, pH 8.0). Protein concentrations were determined by the Bradford method. 2–5 μg antibody was added to each supernatant of cell lysates, followed by incubation for 2–4 h at 4°C. Protein G–agarose beads (Sigma-Aldrich) were added and incubation was continued overnight with shaking. Pellets were washed five times with lysis buffer and beads were dissolved in SDS-PAGE loading buffer prior to immunoblot analysis.
Statistical analysis
Data sets were analyzed with statistics package provided in GraphPad Prism 5 (La Jolla, CA). One way ANOVA was used to comapre groups. All data are reported as the mean ± the standard error of the mean (SEM); n = 3–5. p < 0.05 was considered significant.
Results
Calpain is involved in spontaneous or TGF-β mediated apoptosis
Eosinophils rapidly undergo spontaneous apoptosis in culture in the absence of IL-5 or GM-CSF. Autocrine secretion of TGF-β1 has been reported to reduce eosinophil survival possibly through the activation of protein kinase R (PKR) [12,14]. However, the upstream effectors necessary for TGF-β1 to antagonize IL-5 prosurvival signaling remains poorly understood. Therefore, pure PBEos were cultured with or without TGF-β1 and/or IL-5. After 3 days, cell viability was assessed by propidium iodine (PI) and annexin-V staining by flow cytometry (Figure 1A, 1B) or by western blotting for cleaved PARP and caspase 3 (Figure 1C). We confirmed previous observations that TGF-β1 attenuates the pro-survival effects of IL-5 in a dose-dependent manner (Figure 1A). However, even supraphysiologic concentrations of TGF-β1 failed to fully antagonize the effects of IL-5. In addition, exogenous TGF-β1 had no effect on spontaneous eosinophil apoptosis occuring in the absence of exogenous prosurvival cytokines. These data suggest that IL-5 prosurvival signaling can only be partially antagonized by TGF-β1 and that spontaneous eosinophil cell death is not dependent on TGF-β1.
Figure 1.

Calpain and caspase inhibitors enhance the survival of eosinophils. (A) PBEos were left untreated (0), treated with TGF-β1 (0.5 ng/ml), IL-5 (3pM) or IL-5 (3pM) plus TGF-β1 (as shown) and stained with annexin-V/PI and analyzed by FACS after 72 h. Data presented as a percentage of viable cells at time 0. (B) PBEos were left untreated (−) or treated with TGF-β1 (0.5ng/ml), IL-5 (3pM), calpeptin (CalP, 20μM) or caspase 3 inhibitor (CaspI, 25μM), assessed by FACS as in (A) after 72 h of incubation and presented as a percentage of viability at time 0. (C) PBEos were untreated (C) or treated with TGF-β1, lysed and analyzed by immunoblot for full-length or cleaved PARP (C PARP) and full-length or cleaved caspase 3 (C Casp3). For panels (A) and (B), **; P< 0.01 (one-way ANOVA). Data are representative of at least three experiments with 3 different donors (error bars, SEM of triplicate donors).
Recently we reported that prosurvival signaling prevented Bax cleavage and activation [7]. Calpeptin, a cell permeable calpain-1 inhibitor, blocked Bax cleavage as well as apoptosome activation and eosinophil apoptosis [7]. These data suggested Bax cleavage by calpain-1 was a critical control point in the induction of eosinophil apoptosis. Therefore PBEos were treated with IL-5 or TGF-β1 alone or together with cell permeable inhibitors to calpain-1 and -2 or caspase 3. Inhibitors to caspase 3 or calpain significantly attenuated spontaneous eosinophil apoptosis (Figure 1A,1C), suggesting eosinophil apoptosis requires both calpain and caspase 3. The viability of cells treated with both TGF-β1 and IL-5 and caspase 3 or calpain inhibitors was nearly identical to those cells treated with IL-5 alone. Cells treated with IL-5 plus Z-VAD and/or calpeptin showed slightly greater 3 day survival (75% vs. 82%, n=3 donors, p>0.05) than after IL-5 alone. These results suggest that the proapoptotic effects of TGF-β1 involves the activation of calpain and caspase 3 while IL-5 suppresses both, consistent with prior data. Immunoblot analysis of total eosinophil lysates revealed only calpain-1 was expressed at detectable levels by resting or IL-5 stimulated cells (data not shown). Therefore, we conclude that calpain-1 was the target of calpeptin and this isoform is required for spontaneous or TGF-β mediated apoptosis.
Calpain-1 activation is increased during eosinophil apoptosis
Calpain-1 is expressed as an 80KD (p80) pro-enzyme that associates with an endogenous inhibitor, calpastatin [20,24]. Cleavage to a 76KD (p76) active form is triggered by increased intracellular calcium which reduces binding to calpastatin [20,25]. Given the ability of exogenous calpain inhibitor to attenuate eosinophil apoptosis [Figure 1A,1C], we evaluated the kinetics of calpain cleavage after the addition of IL-5, TGF-β1, neither or both cytokines by immunoblot analysis. As shown in Figure 2A, freshly isolated PBEos contained very little active p76 calpain. In the absence of exogenous cytokines, increasing amounts of p76 appeared over the next 24h. Consistent with its failure to accelerate spontaneous apoptosis, TGF-β1 alone had no discernable effect on the conversion of p80 to p76, which was indistinguishable from untreated controls even after 48 h (not shown). IL-5, however, significantly reduced calpain cleavage which was antagonized by TGF-β1. These results suggest that TGF-β1 may antagonize IL-5 by inducing calpain cleavage.
Figure 2. Calpain-1 is activated by calcium during eosinophil apoptosis.

(A) Calpain-1 immunoblot analysis in PBEos lysed immediately (0), left untreated (R), or treated as shown. The p80 and p76 calpain signals were quantified with Image J software 1.34S (NIH) and ratio calculated and normalized to that observed for control untreated PBEos. (B) Immunoblot analysis of α-spectrin in eosinophils treated as (A). (C) PBEos were treated as shown, lysed and calpain-1 autolysis was determined by immunoblot. ** p<0.01; # p< 0.05 (one-way ANOVA). Data representative of at least 3 independent experiments from different donors and expressed as the mean ± SEM.
Based on these results, we evaluated calpain activity in cells using a fluorescently labeled, model peptide. Extracts were prepared immediately after cell isolation as well as 6 h thereafter. Over this time, there was a significant increase of calpain-1 activity in lysates from untreated eosinophils that was almost entirely suppressed by IL-5 [E2]. Consistent with these results, IL-5 treatment prevented the cleavage of α-spectrin, an endogenous calpain substrate [26] that was increased to untreated levels by TGF-β1 plus IL-5 (Figure 2B). Therefore, calpain-1 activation is a cytokine dependent process that occurs early in the pro-death pathway and likely drives apoptosis in IL-5 deprived or TGF-β1 treated eosinophils.
Extracellular Ca2+ is essential for calpain-1 activation
Calpain-1 is activated after the regulatory, C-terminal EF-hands bind calcium, likely facilitating a conformational change and autolysis [20]. To address if calcium flux was required for calpain activation, cells were untreated or activated with TGF-β1, IL-5 or both in the presence or absence of EGTA or BAPTA/AM. After 6 hrs, BAPTA/AM as well as EGTA alone, or in combination with TGF-β1, reduced or eliminated calpain-1 autolysis, (Figure 2C), suggesting calcium flux from the extracellular to intracellular space was likely necessary for calpain-1 cleavage.
Calpastatin cleavage is induced by TGF-β1
As mentioned above, calpain activation is also regulated by calpastatin, a highly specific endogenous inhibitor [20,27]. Given the above data, we hypothesized that changes in the intracellular levels of calpastatin likely preceded calpain activation. Therefore, freshly isolated PBEos were left untreated or treated with IL-5 alone or together with TGF-β1 prior to immunoblot analysis of calpastatin. In agreement with other cell types [19], multiple calpastatin isoforms are present in eosinophils (Figure 3A), with freshly isolated cells predominantly expressing the 110 kDa form. In the absence of IL-5, calpastatin was rapidly degraded which occurred contemporaneously with calpain autolysis (Figure 3A). Immunoprecipitation/immunoblot of calpain (Figure 3B) revealed that TGF-β1 blocked the interaction between procalpain-1 (80 kDa) and full-length calpastatin. Thus, prosurvival cytokines such as IL-5 prevent apoptosis, in part by stabilizing the calpastatin-calpain interaction which prevents calpain activation.
Figure 3. Calpastatin degradation is induced by TGF-β, blocked by IL-5 and precedes calpain cleavage and activation.
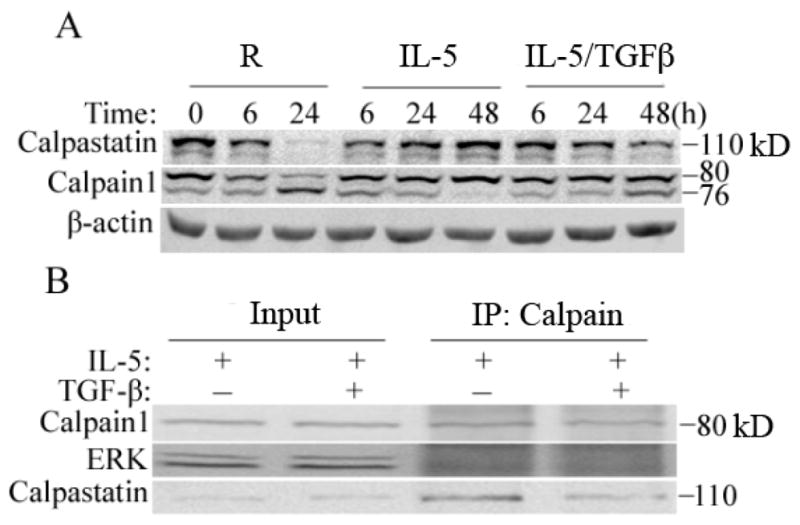
(A) Immunoblot analysis for calpastatin and calpain-1 in PBEos treated as shown. (B) Immunoprecipitation (IP) and immunoblot analysis of PBEos treated as shown for 24h. IP’s analyzed as shown and representative of at least 3 independent experiments from different donors.
TGF-β1 inhibits Akt phosphorylation induced by IL-5 signaling
Our data shows that TGF-β1 functions as a negative modulator of the prosurvival action of IL-5 in eosinophils. In most cell types, TGF-β1 triggers Smad2/3 phosphorylation [11]. Therefore, we analyzed the effect of the TGF-β1 receptor kinase I inhibitor, SB431242 on Smad 2/3 phosphorylation and the survival of untreated cells or those treated with TGF-β1, IL-5 or both cytokines. As shown in Figure 4B, Smad3 was heavily phosphorylated after TGF-β1 or TGF-β1 plus IL-5. Consistent with a Type I receptor mediated event, phosphorylation was blocked by SB431242. Smad2 was constitutively phosphorylated and unaffected by cell treatment with cytokines or Type I Receptor blockade. Therefore, in eosinophils, Smad3 but not Smad2, is responsive to TGF-β1 through Type 1 Receptor signaling. We next assessed if Smad3 signaling mediated TGF-β1 effects on eosinophil survival. As shown in Figure 4A, low concentrations of SB431242 alone had modest, positive effects on eosinophil survival. Notably, Type I Receptor blockade fully restored IL-5 mediated survival despite the presence of TGF-β1. These results indicate that Type I Receptor-Smad3 signaling likely initiates the proapoptotic effects of TGF-β1.
Figure 4. TGF-β1 inhibits Akt phosphorylation induced by IL-5 signaling.

(A) 72 hr viability of PBEos left untreated (0), treated as shown with or without SB431542 (SB). (B, C and D) Immunoblots of PBEos treated with SB, LY294002 (LY) or Akt Inhibitor IV (AktI). (E) and (F) Viability of PBEos treated as shown and presented as a percentage of viability at time 0. **, p< 0.03 (one-way ANOVA). All blots are representative of at least 3 independent experiments with different donors and data are expressed as the mean ± SEM.
IL-5 rapidly induces ERK phosphorylation and is essential for promoting eosinophil survival [21]. Therefore we evaluated the effects of TGF-β1 on ERK as well as p38 and JNK. After 30 minutes incubation, IL-5 induced robust ERK phosphorylation which was unaffected by TGF-β1 (Figure 4C). Neither JNK nor p38 were phosphorylated under these conditions. The PI-3-K/Akt pathway has been reported to be involved in eosinophil survival and allergic inflammation [6]. Thus, Akt phosphorylation was assessed under identical conditions. Like ERK, Akt was phosphorylated in response to IL-5 (Figure 4D, p-Akt:Akt ratio = 2.3 fold +/− 0.3 unstimulated control, n=3 experiments). TGF-β1 alone had no effect but prevented IL-5 induced phosphorylation as did the PI-3-K inhibitor LY294002 or Akt Inhibitor IV. These data suggest that TGF-β1 may antagonize IL-5 signaling by preventing Akt activation via PI-3-K.
We tested this hypothesis by exposing untreated or cytokine treated eosinophils to PI-3-K or Akt inhibitors and measuring survival. As shown [Figure 4E,4F], both PI-3-K or Akt inhibitors individually block the prosurvival effects of IL-5 and are additive with TGF-β1. In aggregate, these results suggest that TGF-β1 antagonizes IL-5 prosurvival signaling through a pathway involving activated Smad3 which prevents Akt phosphorylation. This is consistent with data from other cell types [28,29]. However, immunoprecipitations with anti-Akt failed to pull-down Smad3 or calpastatin/calpain (data not shown). These data suggest that the interactions, if they exist, are transient.
Akt antagonizes calpain activation
The data above implicate Akt as well as calpain in signaling and apoptotic decisions by eosinophils treated with IL-5 and TGF-β1. In order to better characterize the relationship between these regulators, we evaluated survival and calpain activation in the presence of IL-5, calpain and Akt inhibitors. Thus eosinophils were left untreated, treated with IL-5 alone or IL-5 in combination with the calpain inhibitor calpeptin or a caspase 3 inhibitor. As shown in Figure 5A, both calpeptin and caspase 3 inhibitor significantly increased the survival of untreated eosinophils. As seen above (Figure 4), IL-5 mediated survival was antagonized by Akt inhibition. However, this effect was significantly attenuated by either calpain or caspase 3 inhibitors. These data suggest that Akt activation is necessary for suppression of calpain cleavage. In order to test this hypothesis, PBEos were treated for 20 h as above prior to lysis and calpain-1 immunoblot analysis. Despite the presence of IL-5, Akt inhibition was associated with procalpain cleavage (Figure 5B). Calpeptin, but not the caspase 3 inhibitor, blocked calpain-1 cleavage in untreated or IL-5 treated eosinophils. Concordant with these results, calpain activity was suppressed by IL-5 but increased by Akt inhibition (E3). These data together suggest that Akt is activated by IL-5 treatment and must remain active to prevent calpain autolysis and downstream apoptosis.
Figure 5. PI-3-K/Akt (PKB) are upstream of calpastatin/calpain.

(A) Eosinophils were left untreated, or treated as shown for 72h. Cell viability was determined by FACS and presented as a percentage of viability at time 0. **, P< 0.01 (one-way ANOVA). (B) Immunoblot for calpain-1 and calpastatin after 20 hr incubation under the conditions shown. All blots are representative of at least 3 independent experiments with different donors and data are expressed as the mean ± SEM.
Discussion
In this study, we demonstrate that calpain-1 activation is a critical control point in apoptotic decisions by primary, peripheral blood human eosinophils. Blockade of calpain activation delayed spontaneous as well as TGF-β1-facilitated eosinophil apoptosis. Calpain-1 activation was contemporaneous with loss of its endogenous inhibitor calpastatin both of which processes were antagonized by IL-5 signaling. TGF-β1 antagonized IL-5 prosurvival signaling by inhibiting Akt phosphorylation which accelerated calpain activation. Finally we show that eosinophil apoptosis was initiated by increased intracellular free Ca2+ that is most likely mobilized from extracellular stores.
Calpains are highly conserved proteases found in nearly all mammalian cells whose deletion is embryonic lethal [30–32]. Calpain-calpastatin have been implicated in apoptotic decisions by other leukocytes including immature T and B cells [33–36] and neutrophils [21]. While calpain has also been recently implicated in apoptotic decisions by G-CSF treated neutrophils [22], the pathways that lead to and control this decision-point remain poorly understood.
Upon binding calcium, calpains become proteolytically active and associate with intracellular membranes [15,37,38]. Here we show that both extra- and intracellular calcium chelation with EGTA or BAPTA, respectively, suppressed calpain-1 activation, although the effects of EGTA were greater. These results establish that extracellular calcium entry is likely required for calpain activation in eosinophils and is probably a very early event. Our data can not presently determine if intracellular release also participates in this process. Based on these results, and the absence of calpain cleavage in the presence of IL-5, we conclude that prosurvival signaling likely suppresses calcium flux from extracellular sources. Calcium entry is critical for a variety of eosinophil functions including transmigration in response to cytokines and lysophosphatidylcholine induced β2-integrin-mediated adhesion [39]. The relevant calcium channels have yet to be defined for any of these events. We were unable to block calpain-1 activation with L-type calcium channel inhibitors or antagonists of the NMDA receptor (data not shown) both of which are expressed by eosinophils.
Despite being nondividing and terminally differentiated, eosinophils have the capacity to significantly alter gene expression in response to prosurvival signaling. Microarray analysis revealed calpastatin mRNA levels increased after both IL-5 and GM-CSF while calpain-1 mRNA was largely unchanged after IL-5 but modestly elevated after GM-CSF [40]. Consistent with those results, calpastatin protein levels progressively increased over 48 h of IL-5 exposure while calpain levels (80KD) remained essentially constant. These results imply that in addition to suppressing calcium flux, IL-5 treatment also prevented calpain activation by increasing the steady state levels of calpastatin. Interestingly, TGF-β1 antagonized these effects of IL-5, leading to calpain activation and the initiation of apoptosis when eosinophils were exposed to both cytokines. Calpain inhibitors reduced spontaneous and Fas receptor-mediated death of neutrophils [21], while calpastatin knock-down accelerated neutrophil apoptosis [41]. Therefore, the calpastatin-calpain system is clearly an important target of both pro-survival (IL-5) and pro-death (TGF-β1) signaling and thus may be a therapeutic target for eosinophilic diseases such as asthma [42].
Through partially shared receptors, IL-5 and GM-CSF mediated signaling enhances eosinophil survival [7,8,39]. Ligation of the IL-5 receptor recruits several kinases, including JAK2, Lyn, and Raf-1, as well as the phosphatase SHP2 [43] leading to JAK-STAT, Ras-ERK [7,40,43] and PI-3-Kγ-Akt signaling [6,44,45]. Here, using human eosinophils, we show that IL-5 induced serine phosphorylation of Akt and ERK while p38 and JNK were unaffected. PI-3-K and Akt antagonists attenuated IL-5-induced eosinophil survival, which was partially restored by calpain inhibitors. Therefore, PI-3-K/Akt is upstream of the calpain/calpastatin system. How Akt modulates calpain activation remains unclear. We were unable to identify a direct interaction between these 2 proteins by immunoprecipitation (data not shown).
TGF-β1 is a multifunctional cytokine that controls the proliferation, differentiation, and function of many cells of the immune system. Most inflammatory cells including eosinophils secrete TGF-β [46] and are responsive to its signaling via canonical SMAD dependent pathways [10–13]. TGF-β1 levels increase in the BAL of patients with allergic asthma and correlates well with airway and parenchymal remodeling [49]. TGF-β1 has been shown to inhibit Jak-STAT signaling pathway induced by IL-5, although the exact mechanisms remain unknown. Here, we confirmed the proapoptotic effects of TGF-β1/Smad signaling in eosinophils. Interestingly, the effects of TGF-β1 were restricted to cells also treated with IL-5, suggesting TGF-β1 prevented IL-5 dependent signaling. Based on its differential phosphorylation after combined stimulation, the likely target was Akt. This conclusion was strengthened by the observation that Akt inhibitors antagonized IL-5 prosurvival signaling in the absence of TGF-β1 and are consistent with a recent report that Akt signaling maintains calpastatin levels in retinal photoreceptors [47]. As TGF-β1 induced p-Smad3 which was blocked by a Type I Receptor antagonist (SB431542), we also conclude that pro-death TGF-β1 1 signaling occurred through canonical Smad pathways. This observation is consistent with the recent finding by Kanzaki et al. [11].
Conclusions
We have showed that calpain-1 plays an important and early role in spontaneous or TGF-β1 mediated eosinophil apoptosis. Conversely, IL-5 antagonizes calpain activation and in doing so, prevents cell death. Eosinophils possess at least partially intact TGF-β1/Smad signaling, Our data will be important in understanding the role of TGF-β1 in the induction of eosinophil apoptosis, asthma and airway remodeling [48].
E1.
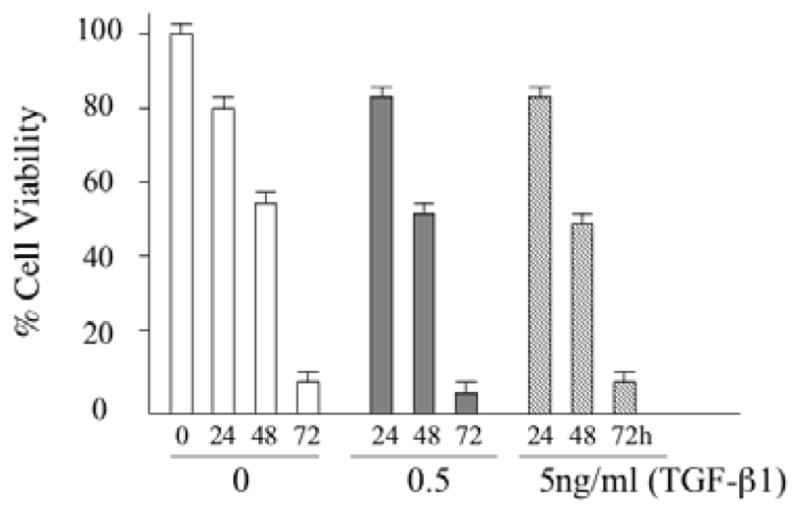
PBEos were stimulated with various concentration of TGF-β1 for the indicated times followed by determination of cell viability by trypan blue exclusion. The results shown are representative of 3 independent experiments.
E2.
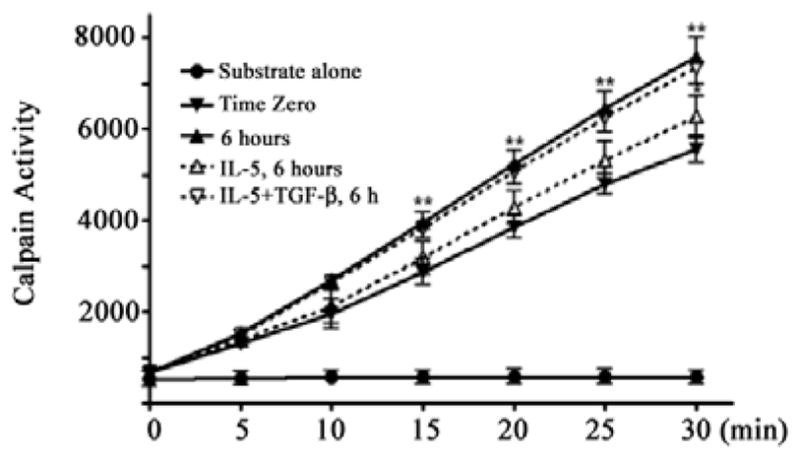
Calpain activity was measured in cell extracts with fluorogenic α-spectrin substrate. Fresh eosinophils were immediately lysed (time zero), left untreated (6 hr) or treated with IL-5 alone or with IL-5 plus TGF-β1 for 6 hr as shown. Substrate alone without cell lysate is the negative control.
E3.
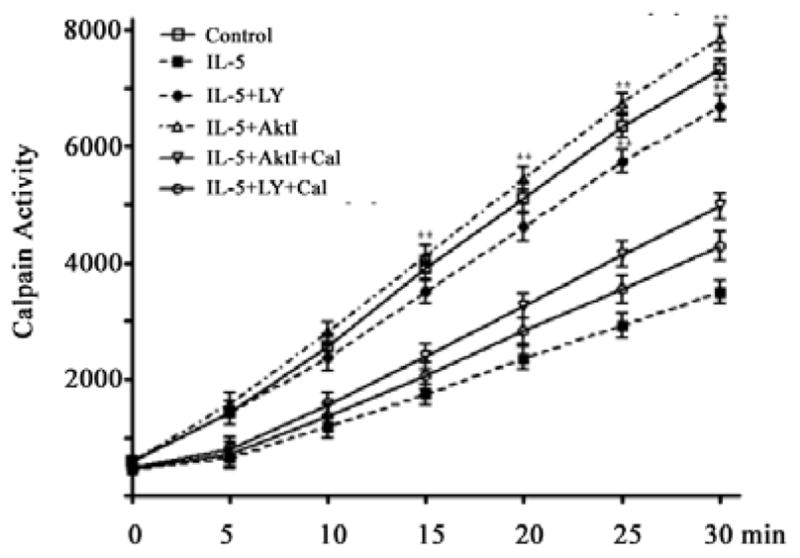
Calpain activity was measured in cell extracts with fluorogenic α-spectrin substrate after 6 hr incubation under the conditions shown.
Key Messages.
TGF-β1 antagonizes prosurvival signaling but does not accelerate apoptosis in the absence of IL-5.
TGF-β1 leads to extracellular calcium flux, triggering activation of calpain-1 and caspase 3.
IL-5 signaling is antagonized by TGF-β1 through effects on Akt.
Acknowledgments
The authors thank members of the lab and the University of Wisconsin Asthma Working Group for suggestions. R01HL087950, P01HL088594 and P30HD03352.
Abbreviations
- IL-5
Interleukin 5
- GM-CSF
Granulocyte-Macrophage colony stimulating factor
- TGF-β1
Transforming growth factor-beta 1
- PBEos
Peripheral blood eosinophils
- SB
TGF-β receptor kinase I inhibitor, SB431242
- PI
Propidium iodine
- ERK
Extracellular regulated kinase
- PI-3-K
Phosphatidylinositol-3 Kinase
- Akt/PKB
Protein kinase B
- PARP
Poly ADP ribose polymerase
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Author Contributions
Q. X. designed and did most experiments and wrote the initial manuscript; Z-J. S. and J. O. designed, performed experiments and helped with data interpretation; H. C. provided help in experimental design and critically read the manuscript and J. S. M. conceived the project, helped to design the experiments, interpreted data and wrote and edited the manuscript.
References
- 1.Takatsu K, Nakajima H. IL-5 and eosinophilia. Curr Opin Immunol. 2008;20:288–294. doi: 10.1016/j.coi.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Arm JP, Lee TH. The pathobiology of bronchial asthma. Adv Immunol. 1992;51:323–382. doi: 10.1016/s0065-2776(08)60491-5. [DOI] [PubMed] [Google Scholar]
- 3.Gleich GJ. Anti-interleukin-5 therapy and severe asthma. N Engl J Med. 2009;360:2577. [PubMed] [Google Scholar]
- 4.Yousefi S, Hoessli DC, Blaser K, Mills GB, Simon HU. Requirement of Lyn and Syk tyrosine kinases for the prevention of apoptosis by cytokines in human eosinophils. J Exp Med. 1996;183:1407–1414. doi: 10.1084/jem.183.4.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pazdrak K, Olszewska-Pazdrak B, Stafford S, Garofalo RP, Alam R. Lyn, Jak2, and Raf-1 kinases are critical for the antiapoptotic effect of interleukin 5, whereas only Raf-1 kinase is essential for eosinophil activation and degranulation. J Exp Med. 1998;188:421–429. doi: 10.1084/jem.188.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinho V, Souza DG, Barsante MM, Hamer FP, De Freitas MS, et al. Phosphoinositide-3 kinases critically regulate the recruitment and survival of eosinophils in vivo: importance for the resolution of allergic inflammation. J Leukoc Biol. 2005;77:800–810. doi: 10.1189/jlb.0704386. [DOI] [PubMed] [Google Scholar]
- 7.Shen ZJ, Esnault S, Schinzel A, Borner C, Malter JS. The peptidylprolyl isomerase Pin1 facilitates cytokine-induced survival of eosinophils by suppressing Bax activation. Nat Immunol. 2009;10:257–265. doi: 10.1038/ni.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dewson G, Cohen GM, Wardlaw AJ. Interleukin-5 inhibits translocation of Bax to the mitochondria, cytochrome c release, and activation of caspases in human eosinophils. Blood. 2001;98:2239–2247. doi: 10.1182/blood.v98.7.2239. [DOI] [PubMed] [Google Scholar]
- 9.Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Shen ZJ, Esnault S, Rosenthal LA, Szakaly RJ, Sorkness RL, et al. Pin1 regulates TGF-beta1 production by activated human and murine eosinophils and contributes to allergic lung fibrosis. J Clin Invest. 2008;118:479–490. doi: 10.1172/JCI32789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanzaki M, Shibagaki N, Hatsushika K, Mitsui H, Inozume T, et al. Human eosinophils have an intact Smad signaling pathway leading to a major transforming growth factor-beta target gene expression. Int Arch Allergy Immunol. 2007;142:309–317. doi: 10.1159/000097500. [DOI] [PubMed] [Google Scholar]
- 12.Alam R, Forsythe P, Stafford S, Fukuda Y. Transforming growth factor beta abrogates the effects of hematopoietins on eosinophils and induces their apoptosis. J Exp Med. 1994;179:1041–1045. doi: 10.1084/jem.179.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pazdrak K, Justement L, Alam R. Mechanism of inhibition of eosinophil activation by transforming growth factor-beta. Inhibition of Lyn, MAP, Jak2 kinases and STAT1 nuclear factor. J Immunol. 1995;155:4454–4458. [PubMed] [Google Scholar]
- 14.Goplen N, Gorska MM, Stafford SJ, Rozario S, Guo L, et al. A phosphosite screen identifies autocrine TGF-beta-driven activation of protein kinase R as a survival-limiting factor for eosinophils. J Immunol. 2008;180:4256–4264. doi: 10.4049/jimmunol.180.6.4256. [DOI] [PubMed] [Google Scholar]
- 15.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 16.Glading A, Lauffenburger DA, Wells A. Cutting to the chase: calpain proteases in cell motility. Trends Cell Biol. 2002;12:46–54. doi: 10.1016/s0962-8924(01)02179-1. [DOI] [PubMed] [Google Scholar]
- 17.Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci. 2005;118:3829–3838. doi: 10.1242/jcs.02562. [DOI] [PubMed] [Google Scholar]
- 18.Abe K, Takeichi M. NMDA-receptor activation induces calpain-mediated beta-catenin cleavages for triggering gene expression. Neuron. 2007;53:387–397. doi: 10.1016/j.neuron.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 19.Hanna RA, Campbell RL, Davies PL. Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature. 2008;456:409–412. doi: 10.1038/nature07451. [DOI] [PubMed] [Google Scholar]
- 20.Moldoveanu T, Gehring K, Green DR. Concerted multi-pronged attack by calpastatin to occlude the catalytic cleft of heterodimeric calpains. Nature. 2008;456:404–408. doi: 10.1038/nature07353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altznauer F, Conus S, Cavalli A, Folkers G, Simon HU. Calpain-1 regulates Bax and subsequent Smac-dependent caspase-3 activation in neutrophil apoptosis. J Biol Chem. 2004;279:5947–5957. doi: 10.1074/jbc.M308576200. [DOI] [PubMed] [Google Scholar]
- 22.van Raam BJ, Drewniak A, Groenewold V, van den Berg TK, Kuijpers TW. Granulocyte colony-stimulating factor delays neutrophil apoptosis by inhibition of calpains upstream of caspase-3. Blood. 2008;112:2046–2054. doi: 10.1182/blood-2008-04-149575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansel TT, De Vries IJ, Iff T, Rihs S, Wandzilak M, et al. An improved immunomagnetic procedure for the isolation of highly purified human blood eosinophils. J Immunol Methods. 1991;145:105–110. doi: 10.1016/0022-1759(91)90315-7. [DOI] [PubMed] [Google Scholar]
- 24.Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stifanese R, Averna M, De Tullio R, Pedrazzi M, Beccaria F, et al. Adaptive modifications in the calpain/calpastatin system in brain cells after persistent alteration in Ca2+ homeostasis. J Biol Chem. 2010;285:631–643. doi: 10.1074/jbc.M109.031674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris AS, Morrow JS. Proteolytic processing of human brain alpha spectrin (fodrin): identification of a hypersensitive site. J Neurosci. 1988;8:2640–2651. doi: 10.1523/JNEUROSCI.08-07-02640.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wendt A, Thompson VF, Goll DE. Interaction of calpastatin with calpain: a review. Biol Chem. 2004;385:465–472. doi: 10.1515/BC.2004.054. [DOI] [PubMed] [Google Scholar]
- 28.Conery AR, Cao Y, Thompson EA, Townsend CM, Jr, Ko TC, et al. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol. 2004;6:366–372. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- 29.Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–365. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- 30.Saido TC, Sorimachi H, Suzuki K. Calpain: new perspectives in molecular diversity and physiological-pathological involvement. FASEB J. 1994;8:814–822. [PubMed] [Google Scholar]
- 31.Squier MK, Cohen JJ. Calpain and cell death. Cell Death Differ. 3(196):275–283. [PubMed] [Google Scholar]
- 32.Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, et al. Conditional disruption of ubiquitous calpains in the mouse. Genesis. 2006;44:297–303. doi: 10.1002/dvg.20216. [DOI] [PubMed] [Google Scholar]
- 33.Squier MK, Cohen JJ. Calpain, an upstream regulator of thymocyte apoptosis. J Immunol. 1997;158:3690–3697. [PubMed] [Google Scholar]
- 34.Yang Y, Liu ZH, Ware CF, Ashwell JD. A cysteine protease inhibitor prevents activation-induced T-cell apoptosis and death of peripheral blood cells from human immunodeficiency virus-infected individuals by inhibiting upregulation of Fas ligand. Blood. 1997;89:550–557. [PubMed] [Google Scholar]
- 35.Ruiz-Vela A, Gonzalez de Buitrago G, Martinez AC. Implication of calpain in caspase activation during B cell clonal deletion. EMBO J. 1999;18:4988–4998. doi: 10.1093/emboj/18.18.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruiz-Vela A, Serrano F, Gonzalez MA, Abad JL, Bernad A, et al. Transplanted long-term cultured pre-BI cells expressing calpastatin are resistant to B cell receptor-induced apoptosis. J Exp Med. 2001;194:247–254. doi: 10.1084/jem.194.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pasini EM, Kirkegaard M, Mortensen P, Lutz HU, Thomas AW, Mann M. In-depth analysis of the membrane and cytosolic proteome of red blood cells. Blood. 2006;108:791–801. doi: 10.1182/blood-2005-11-007799. [DOI] [PubMed] [Google Scholar]
- 38.Croall DE, Ersfeld K. The calpains: modular designs and functional diversity. Genome Biol. 2007;8:218. doi: 10.1186/gb-2007-8-6-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu L, Ridefelt P, Hakansson L, Venge P. Regulation of human eosinophil migration across lung epithelial monolayers by distinct calcium signaling mechanisms in the two cell types. J Immunol. 1999;163:5649–5655. [PubMed] [Google Scholar]
- 40.Bates ME, Liu LY, Esnault S, Stout BA, Fonkem E, et al. Expression of interleukin-5- and granulocyte macrophage-colony-stimulating factor-responsive genes in blood and airway eosinophils. Am J Respir Cell Mol Biol. 2004;30:736–743. doi: 10.1165/rcmb.2003-0234OC. [DOI] [PubMed] [Google Scholar]
- 41.Squier MK, Sehnert AJ, Sellins KS, Malkinson AM, Takano E, et al. Calpain and calpastatin regulate neutrophil apoptosis. J Cell Physiol. 1999;178:311–319. doi: 10.1002/(SICI)1097-4652(199903)178:3<311::AID-JCP5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 42.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 43.Asquith KL, Ramshaw HS, Hansbro PM, Beagley KW, Lopez AF, et al. The IL-3/IL-5/GM-CSF common receptor plays a pivotal role in the regulation of Th2 immunity and allergic airway inflammation. J Immunol. 2008;180:1199–1206. doi: 10.4049/jimmunol.180.2.1199. [DOI] [PubMed] [Google Scholar]
- 44.Ogata N, Kouro T, Yamada A, Koike M, Hanai N, et al. JAK2 and JAK1 constitutively associate with an interleukin-5 (IL-5) receptor alpha and betac subunit, respectively, and are activated upon IL-5 stimulation. Blood. 1998;91:2264–2271. [PubMed] [Google Scholar]
- 45.Duan W, Aguinaldo Datiles AM, Leung BP, Vlahos CJ, Wong WS. An anti-inflammatory role for a phosphoinositide 3-kinase inhibitor LY294002 in a mouse asthma model. Int Immunopharmacol. 2005;5:495–502. doi: 10.1016/j.intimp.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 46.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 47.Arroba AI, Wallace D, Mackey A, de la Rosa EJ, Cotter TG. IGF-I maintains calpastatin expression and attenuates apoptosis in several models of photoreceptor cell death. Eur J Neurosci. 2009;30:975–986. doi: 10.1111/j.1460-9568.2009.06902.x. [DOI] [PubMed] [Google Scholar]
- 48.Kariyawasam HH, Robinson DS. The role of eosinophils in airway tissue remodelling in asthma. Curr Opin Immunol. 2007;19:681–686. doi: 10.1016/j.coi.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 49.Duvernelle C, Freund V, Frossard N. Transforming growth factor-beta and its role in asthma. Pulm Pharmacol Ther. 2003;16:181–196. doi: 10.1016/S1094-5539(03)00051-8. [DOI] [PubMed] [Google Scholar]