

Although cholera toxin is a nonubiquitinated substrate that undergoes retro-translocation to the cytosol, this study identifies a deubiquitinase that controls toxin retro-translocation.
Abstract
Endoplasmic reticulum (ER) membrane–bound E3 ubiquitin ligases promote ER-associated degradation (ERAD) by ubiquitinating a retro-translocated substrate that reaches the cytosol from the ER, targeting it to the proteasome for destruction. Recent findings implicate ERAD-associated deubiquitinases (DUBs) as positive and negative regulators during ERAD, reflecting the different consequences of deubiquitinating a substrate prior to proteasomal degradation. These observations raise the question of whether a DUB can control the fate of a nonubiquitinated ERAD substrate. In this study, we probed the role of the ERAD-associated DUB, YOD1, during retro-translocation of the nonubiquitinated cholera toxin A1 (CTA1) peptide, a critical intoxication step. Through combining knockdown, overexpression, and binding studies, we demonstrated that YOD1 negatively controls CTA1 retro-translocation, likely by deubiquitinating and inactivating ubiquitinated ERAD components that normally promote toxin retro-translocation. YOD1 also antagonizes the proteasomal degradation of nonglycosylated pro-α factor, a postulated nonubiquitinated yeast ERAD substrate, in mammalian cells. Our findings reveal that a cytosolic DUB exerts a negative function during retro-translocation of nonubiquitinated substrates, potentially by acting on elements of the ERAD machinery.
INTRODUCTION
The endoplasmic reticulum (ER) is an intracellular organelle endowed with robust protein-folding capacities to ensure proper folding of secretory and transmembrane proteins before they exit the ER. However, when misfolding occurs, an ER quality control system, known as ER-associated degradation (ERAD), recognizes and retro-translocates these aberrantly folded polypeptides to the cytosol for proteasomal degradation (Smith et al., 2011; Brodsky, 2012). The central component of the ERAD machinery is a transmembrane E3 ubiquitin ligase connected to multiple ER lumenal and cytosolic adaptors. Coordinated actions of the E3 ligases with these adaptors cause the ejection of the misfolded substrate to the cytosol for efficient proteasomal degradation.
A decisive event during ERAD is polyubiquitination of the misfolded substrate as it becomes exposed to the cytosol. Addition of this molecular tag, which is generally required to direct the substrate to the proteasome for degradation, is catalyzed by membrane-bound E3 ubiquitin ligases that also function as the central component of the ERAD machinery by connecting to multiple ER lumenal and cytosolic adaptors (Hirsch et al., 2009). Coordinated actions of the E3 ligases with these adaptors eject the misfolded substrate to the cytosol for efficient proteasomal degradation. To date, there are at least nine membrane-bound E3 ubiquitin ligases in mammals known to be implicated in ERAD, including Hrd1 and gp78 (Claessen et al., 2012), with each linked to a different combination of lumenal and cytosolic adaptors, presumably to serve a unique subset of misfolded substrates.
While the importance of polyubiquitination in promoting ERAD is well understood, the function of deubiquitination in this process is less clear. For deubiquitinases (DUBs) associated with the core proteasome, there is evidence that they play positive (Verma et al., 2002; Yao and Cohen, 2002) and negative (Lam et al., 1997; Hanna et al., 2006) roles during the degradation process. Likewise, recent evidence suggests that DUBs physically linked to the central ERAD machinery facilitate (Wang et al., 2006; Ernst et al., 2009; Sowa et al., 2009) and antagonize (Zhong and Pittman, 2006; Hassink et al., 2009; Blount et al., 2012) proteasome-dependent degradation of the misfolded substrate. The apparent opposing functions of these DUBs likely reflect the different consequences of deubiquitination: in principle, robust removal of the ubiquitin chain should divert the substrate from the degradative fate, whereas simply trimming or editing the polyubiquitin chain may allow favorable interaction with the proteasome or the cytosolic AAA ATPase p97 (Cdc48 in yeast; Ernst et al., 2009) that normally extracts substrates from the ER membrane into the cytosol (Ye et al., 2001).
Regardless of their specific functions, these DUBs are believed to directly deubiquitinate the ubiquitinated misfolded substrate, raising the question of whether DUBs might regulate a nonubiquitinated substrate. Cholera toxin A1 peptide (CTA1) is a well-established nonubiquitinated ERAD substrate (Rodighiero et al., 2002; Kothe et al., 2005). To intoxicate host cells, CT is transported in a retrograde manner from the plasma membrane of intestinal epithelial cells to the ER, where the catalytic CTA1 subunit is generated. CTA1 subsequently disguises as a misfolded protein to co-opt the ERAD machinery and retro-translocates into the cytosol (Lencer and Tsai, 2003). In the cytosol, however, CTA1 escapes proteasomal destruction (Rodighiero et al., 2002) due to its paucity of lysine ubiquitination sites (Hazes and Read, 1997). Instead, the toxin activates a signaling cascade, leading to massive water secretion across the plasma membrane that typifies cholera disease (Spangler, 1992). Importantly, we previously demonstrated that the catalytic activities of Hrd1 and gp78 support CTA1 retro-translocation (Bernardi et al., 2010), suggesting that ubiquitination is important during ER-to-cytosol membrane transport of the toxin. Whether specific DUBs associated with the ERAD machinery control CTA1 retro-translocation is unknown.
Here we combined small interfering RNA (siRNA)-mediated knockdown and overexpression approaches in the context of a cell-based retro-translocation system, coupled with biochemical interaction studies, to demonstrate that the ERAD-associated DUB called YOD1 negatively regulates CTA1 retro-translocation. Because the toxin is not ubiquitinated even when YOD1 activity is perturbed, the action of this DUB is likely toward a ubiquitinated trans-regulatory molecule that normally stimulates retro-translocation. YOD1 also acts as a negative regulator during proteasomal degradation of the postulated nonubiquitinated, nonglycosylated pro-α factor, a yeast ERAD substrate (Werner et al., 1996). The ability of an ERAD-associated DUB to control retro-translocation of nonubiquitinated substrates should direct our efforts to clarify how ubiquitination and deubiquitination of trans-regulatory components of the ERAD machinery might impact this quality control process.
RESULTS
The YOD1 deubiquitinase binds to the Hrd1 E3 ubiquitin ligase
The Hrd1 and gp78 E3 ubiquitin ligases were previously shown to promote CTA1 retro-translocation (Bernardi et al., 2010). As YOD1 is a DUB implicated in ERAD (Ernst et al., 2009), we asked whether it binds to these E3 ligases. We first tested a potential interaction between YOD1 and Hrd1. A C-terminally Myc-tagged wild-type (WT) Hrd1 construct (WT Hrd1-Myc) was transfected with or without an N-terminally FLAG-tagged WT YOD1 construct (FLAG-WT YOD1) in 293T cells. The resulting whole-cell lysate (WCL) was subjected to immunoprecipitation using FLAG antibody–conjugated beads. The immunoprecipitates were subjected to SDS–PAGE followed by immunoblotting with the indicated antibodies. WT Hrd1-Myc coprecipitated with FLAG-WT YOD1 only when FLAG-WT YOD1was expressed in cells (Figure 1A, top and second panels, compare lane 2 with lane 1), indicating that Hrd1 binds to YOD1. Hrd1 contains six transmembrane domains and a C-terminal cytosolic domain that harbors the catalytically active site (Kikkert et al., 2004). For determination of whether Hrd1’s transmembrane or cytosolic domains mediate interaction with YOD1, cells expressing FLAG-WT YOD1 were cotransfected with either WT Hrd1-Myc, a construct containing the cytosolic Hrd1 domain (cyt Hrd1-Myc), or the six transmembrane domains of Hrd1 (TM1-6 Hrd1-Myc). Both WT Hrd1-Myc and cyt Hrd1-Myc but not TM1-6 Hrd1-Myc coprecipitated with FLAG-WT YOD1 (Figure 1B, top three panels, compare lanes 1 and 2 with lane 3), demonstrating that Hrd1’s cytosolic domain interacts with YOD1. Hrd1’s catalytic site within the cytosolic domain does not affect its interaction with YOD1, as a catalytically inactive Hrd1 mutant (C291A Hrd1-Myc) engages FLAG-WT YOD1 with a similar efficiency as WT Hrd1-Myc (Figure 1C, top panel, compare lane 3 with lane 1).
FIGURE 1:

The YOD1 deubiquitinase binds to the Hrd1 E3 ubiquitin ligase. (A) 293T cells were transfected with WT Hrd1-Myc alone or with FLAG-WT YOD1 and lysed in a buffer containing 1% NP-40. The resulting WCLs were incubated with FLAG antibody–conjugated beads, and the immunoprecipitates were subjected to SDS–PAGE followed by immunoblotting with the indicated antibodies. The corresponding WCLs were analyzed by SDS–PAGE and immunoblotting with the indicated antibodies. (B) Cells were transfected with FLAG-WT YOD1 and either WT Hrd1-Myc, cyt Hrd1-Myc, or TM1-6 Hrd1-Myc, and processed as in (A). *, An unidentified band that cross-reacts with the Myc antibody. (C) Cells were transfected with FLAG-WT YOD1 and WT Hrd1-Myc, C291A Hrd1-Myc alone, or C291A Hrd1-Myc and FLAG-WT YOD1, and processed as in (A). (D) Cells were transfected with WT Hrd1-Myc alone or with FLAG-WT YOD1 or FLAG-WT Atx3, and processed as in (A). (E) Cells were transfected with WT Hrd1-Myc alone, WT Hrd1-Myc and FLAG-WT YOD1, WT gp78-Myc alone, or WT gp78-Myc and FLAG-WT YOD1, and processed as in (A). At least three independent experiments were performed in each condition.
As Atx3 is another ERAD machinery–associated DUB that binds to Hrd1 (Wang et al., 2006), we compared the Atx3-Hrd1 and YOD1-Hrd1 binding efficiencies. A similar WT Hrd1-Myc level was detected among the samples (Figure 1D, top panel, compare lane 4 with lane 2) even though more FLAG-WT Atx3 was precipitated when compared with FLAG-WT YOD1 (Figure 1D, second panel, compare lane 4 with lane 2), suggesting that YOD1 is at least as efficient as Atx3 in binding to Hrd1.
Because gp78 also facilitates CTA1 retro-translocation, we assessed its interaction with YOD1. Whereas WT Hrd1-Myc was found in the FLAG-WT YOD1 precipitate (Figure 1E, top and second panels, lane 2), WT gp78-Myc was essentially undetected (Figure 1E, top and second panels, lane 4). This finding indicates that gp78 does not engage YOD1, and demonstrates specificity of the YOD1-Hrd1 interaction.
YOD1 knockdown stimulates CTA1 retro-translocation
We next assessed whether YOD1 regulates CTA1 retro-translocation. Accordingly, cells were transfected with either a scrambled siRNA or two independent siRNAs directed against endogenous YOD1 (i.e., YOD1 #1 and #2 siRNAs). When compared with scrambled siRNA, YOD1 #1 and #2 siRNAs efficiently down-regulated YOD1 (Figure 2A, top panel, compare lanes 2 and 3 with lane 1) without affecting the Atx3 level (Figure 2A, second panel, compare lanes 2 and 3 with lane 1). No up-regulation of the ER-resident Hsp70 binding immunoglobulin protein (BiP) was observed under the knockdown conditions (Figure 2A, third panel, compare lanes 2 and 3 with lane 1). In addition, no splicing of the XBP1 transcription factor mRNA was observed when YOD1 was knocked down, in contrast to cells treated with the well-established ER stress-inducers tunicamycin and dithiothreitol (DTT; Figure 2B, compare lanes 3 and 4 with lanes 1 and 5). These findings indicate that YOD1 can be effectively silenced without triggering significant ER stress.
FIGURE 2:

YOD1 knockdown stimulates CTA1 retro-translocation. (A) WCLs from 293T cells transfected with a scrambled, YOD1 #1, or YOD1 #2 siRNA were analyzed by SDS–PAGE and immunoblotting with the indicated antibodies. (B) Reverse transcription–PCR analysis of the unspliced (u) and spliced (s) forms of the XBP1 mRNA derived from cells transfected with a scrambled siRNA treated with or without tunicamycin or DTT, or from cells transfected with YOD1 #1 or #2 siRNA. (C) Cells were incubated with digitonin and centrifuged. The resulting supernatant and pellet fractions were analyzed for the presence of the cytosolic Hsp90 and ER-resident PDI markers. This protocol is the fractionation procedure utilized in the retro-translocation assay. (D) Cells transfected with a scrambled, YOD1 #1, or YOD1 #2 siRNA were incubated with CT (10 nM) for 90 min and subjected to the retro-translocation assay as in (C). The pellet and supernatant fractions were subjected to SDS–PAGE and were analyzed by immunoblotting with the indicated antibodies. (E) The supernatant CTA1 band intensity generated in (D) was quantified with ImageJ. Data represent the mean of at least three independent experiments. Error bars: ±SD. (F) Cells transfected with a scrambled, YOD1 #1, or YOD1 #2 siRNA were intoxicated with CT (10 nM) for 90 min and harvested, and the resulting WCLs were analyzed with nonreducing SDS–PAGE followed by immunoblotting with the indicated antibodies. (G) WCLs derived from cells transfected with a scrambled, YOD1 #1, or YOD1 #2 siRNA were analyzed by immunoblotting using the indicated antibodies. (H) WCLs derived from cells transfected with a scrambled or YOD1 #1 siRNA and incubated with or without the EDAC cross-linker were analyzed by immunoblotting with the Hrd1 antibody.
To evaluate whether CTA1’s arrival to the cytosol from the ER is affected by the knockdown conditions, we used a previously established cell-based retro-translocation assay designed to examine toxin ER-to-cytosol membrane transport (Forster et al., 2006; Bernardi et al., 2010; Williams et al., 2013). Other laboratories have similarly used this assay to evaluate toxin retro-translocation (Taylor et al., 2010; Wernick et al., 2010; Nery et al., 2011). Briefly, intoxicated cells are treated with a low digitonin concentration to permeabilize the plasma membrane without damaging internal membranes. The samples are subjected to high-speed centrifugation to generate supernatant and pellet fractions. The supernatant fraction represents cytosolic proteins and toxin that retro-translocated from the ER to the cytosol, while the pellet fraction contains membranes, including the ER membrane and toxin that remains in the ER or other membranous compartments. With this protocol, the cytosolic marker Hsp90 is found predominantly in the supernatant fraction (Figure 2C, top panel, compare lane 1 with lane 2), while the ER marker protein disulfide isomerase (PDI) is present exclusively in the pellet fraction (Figure 2C, bottom panel, compare lane 2 with lane 1), confirming the integrity of the fractionation method.
Using this semipermeabilized system, we found that YOD1 knockdown increased the CTA1 level in the supernatant fraction (Figure 2D, top panel, compare lanes 2 and 3 with lane 1; quantified in Figure 2E), suggesting that YOD1 normally exerts a negative function during ER-to-cytosol transport of the toxin. CTA1 formation, which occurs in the ER, is not disrupted or enhanced by YOD1 knockdown (Figure 2F, top panel, compare lanes 2 and 3 with lane 1), indicating that YOD1 does not control toxin transport from the plasma membrane to the ER. YOD1 knockdown also does not affect the steady-state levels of Hrd1, Sel1L, and Derlin-1 (Figure 2G, second through fourth panels, compare lanes 2 and 3 with lane 1), membrane proteins previously demonstrated to regulate toxin retro-translocation (Bernardi et al., 2008, 2010; Dixit et al., 2008; Williams et al., 2013). Despite not affecting Hrd1’s steady-state level, YOD1 knockdown enhanced Hrd1 cross-links (Figure 2H, compare lane 2 with lane 1), suggesting that YOD1’s absence leads to either preferential Hrd1 oligomerization or recruitment of cellular factors to Hrd1.
Knockdown of Atx3 using two different siRNAs (Supplemental Figure S1A, top panel, compare lanes 2 and 3 with lane 1) did not increase the CTA1 level in the supernatant when compared with scrambled siRNA (Figure S1B, top panel, compare lanes 2 and 3 with lane 1; quantified in Figure S1C), in contrast to the YOD1 knockdown (Figure S1B, top panel, compare lanes 2 and 3 with lane 4). Similarly, knockdown of USP14 (Figure S1D, top panel, compare lane 2 with lane 1), a proteasome-associated DUB that negatively regulates proteasome activity (Lee et al., 2010), did not enhance the toxin level in the supernatant (Figure S1E, top panel, compare lane 2 with lane 1; quantified in Figure S1F), while YOD1 knockdown did (Figure S1E, top panel, compare lane 2 with lane 3). We conclude that YOD1, but not Atx3 and USP14, specifically antagonizes CTA1 retro-translocation.
Catalytically inactive YOD1 overexpression decreases CTA1 retro-translocation
We next used an overexpression strategy to further clarify YOD1’s role during toxin retro-translocation. Cells were transfected with the control construct yellow fluorescent protein (YFP), FLAG-WT YOD1, or a catalytically inactive form of YOD1 (FLAG-C160S YOD1) previously shown to block retro-translocation of ERAD substrates (Ernst et al., 2009; Figure 3A, top panel, lanes 2 and 3). Overexpressing WT or mutant YOD1 did not alter the endogenous BiP levels (Figure 3A, second panel, compare lanes 2 and 3 with lane 1), again indicating no induction of severe ER stress under these conditions. Whereas FLAG-WT YOD1 overexpression did not affect the CTA1 level in the supernatant fraction when compared with YFP expression (Figure 3B, top panel, compare lane 2 with lane 1; quantified in Figure 3C), expression of the catalytically inactive mutant significantly blocked appearance of the toxin in the supernatant fraction (Figure 3B, top panel, compare lane 3 with lane 1; quantified in Figure 3C).
FIGURE 3:
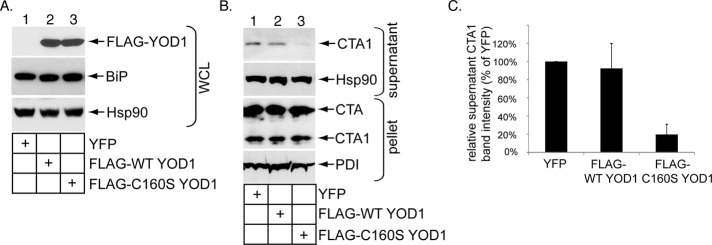
Catalytically inactive YOD1 overexpression decreases CTA1 retro-translocation. (A) Cells were transfected with YFP, FLAG-WT YOD1, or catalytically inactive FLAG-C160S YOD1. The resulting WCLs were subjected to SDS–PAGE followed by immunoblotting with the indicated antibodies. (B) Cells transfected with YFP, FLAG-WT YOD1, and FLAG-C160S YOD1 were incubated with CT (10 nM) for 90 min and subjected to the retro-translocation assay as in Figure 2C. (C) The supernatant CTA1 band intensity in (B) was analyzed as in Figure 2D. Mean of at least three independent experiments. Error bars: ±SD.
Because overexpressing FLAG-C160S YOD1 blocks retro-translocation of numerous ERAD substrates (Ernst et al., 2009), the observed decrease in CTA1 arrival to the cytosol under this condition may simply reflect a general disruption of flux across the ERAD machinery. However, this is an unlikely explanation, because other conditions known to block retro-translocation of ERAD substrates, such as perturbing p97 or proteasome activities, do not affect toxin retro-translocation (Rodighiero et al., 2002; Kothe et al., 2005; Forster et al., 2006). Therefore we contend that expression of this YOD1 mutant must specifically control a critical step during toxin retro-translocation.
Disrupting YOD1 activity does not promote cholera toxin polyubiquitination
Although CTA1 is a nonubiquitinated retro-translocation substrate (Rodighiero et al., 2002; Kothe et al., 2005), it is possible that rapid and robust deubiquitination accounts for why ubiquitinated CTA1 has not been isolated. If so, blocking the deubiquitination reaction may reveal a ubiquitinated toxin intermediate. We therefore asked whether ubiquitinated CTA1 might be detected when YOD1 is down-regulated. Cells transfected with scrambled or YOD1 #1 siRNA were intoxicated with or without CT. The proteasome inhibitor epoxomicin was added to cells to further increase the chance of trapping ubiquitinated CTA1. For detection of potentially ubiquitinated CTA1, cells were lysed in a RIPA buffer; this was followed by addition of a CTA antibody to precipitate the toxin. This mild denaturing condition ensures that any ubiquitinated signal resulting from the precipitated toxin reflects ubiquitinated toxin and not toxin-associated ubiquitinated proteins. All cells in these experiments were transfected with green fluorescent protein–ubiquitin (GFP-Ub), as the GFP antibody was more specific than the pan-ubiquitin antibody in revealing ubiquitination of immunoprecipitated proteins. Using this approach, no GFP-positive signal above the nonintoxicated samples was detected when the toxin was precipitated from cells transfected with either scrambled or YOD1 #1 siRNA (Figure 4A, top panel, compare lanes 3 and 4 with lanes 1 and 2), indicating the absence of CTA1 ubiquitination. In contrast, polyubiquitination of the canonical ERAD substrate, C-terminally hemagglutinin-tagged T-cell receptor α (TCRα−HA), was detected when the proteasome was inhibited with epoxomicin (Figure 4A, top panel, compare lane 5 with lane 6). Similarly, when His-ubiquitin (His-Ub) was used instead of GFP-Ub, no His-Ub–positive signal above the nonintoxicated samples was detected when CTA was immunoprecipitated from cells transfected with either scrambled or YOD1 #1 siRNA (Figure S2, top panel, compare lanes 3 and 4 with lanes 1 and 2), although more background signal was readily observed using this modified ubiquitin construct. In the overexpression studies, no GFP-positive signal above the nonintoxicated samples was detected when the toxin was precipitated under the same denaturing condition from cells transfected with either FLAG-WT YOD1 or FLAG-C160S YOD1 (Figure 4B, top panel, compare lanes 3 and 4 with lanes 1 and 2), whereas polyubiquitination of TCRα-HA was detected (Figure 4B, top panel, lane 5). We conclude that disrupting YOD1 activity does not promote CTA1 ubiquitination, and we propose the mechanism by which YOD1 controls CTA1 retro-translocation is through deubiquitinating factors other than the toxin.
FIGURE 4:

Disrupting YOD1 activity does not promote CT polyubiquitination. (A) Cells transfected with scrambled siRNA, YOD1 #1 siRNA, or TCRα-HA were intoxicated with or without CT (10 nM) for 90 min and lysed in a RIPA buffer containing 0.1% SDS. The resulting WCLs were incubated with a CTA antibody (lanes 1–4) or HA antibody–conjugated beads (lanes 5–6). The immunoprecipitates were subjected to reducing SDS–PAGE followed by immunoblotting with the indicated antibodies. WCLs were also analyzed by immunoblotting with the appropriate antibodies. All cells were transfected with GFP-Ub and incubated with epoxomicin. (B) As in (A), except cells were transfected with FLAG-WT YOD1 or FLAG-C160S YOD1 instead of the siRNAs.
Perturbing YOD1 increases cellular K11-, K48-, and K63-linked polyubiquitinated proteins
If YOD1 functions as a DUB in cells, its down-regulation should increase total cellular polyubiquitinated proteins. To this end, lysates derived from cells transfected with scrambled and YOD1 #1 siRNA were subjected to SDS–PAGE followed by immunoblotting with an antibody that recognizes pan-ubiquitin linkages. Indeed, when compared with control lysates, an increase in the overall signal of high-molecular-weight (HMW) species corresponding to polyubiquitinated proteins was observed in lysates derived from YOD1 knockdown cells (Figure 5A, top panel, compare lane 2 with lane 1). A similar increase in the level of HMW species was found when these same lysates were probed with K11-, K48-, and K63-specific ubiquitin antibodies (Figure 5A, second through fourth panels, compare lane 2 with lane 1). These findings establish that YOD1 acts as a DUB in cells and does not discriminate between different ubiquitin linkages. Notably, YOD1 was shown previously to deubiquitinate artificial K48- and K63-linked ubiquitin chains in vitro (Ernst et al., 2009).
FIGURE 5:

Perturbing YOD1 increases cellular K11-, K48-, and K63-linked polyubiquitinated proteins. (A) WCLs derived from cells transfected with scrambled or YOD1 #1 siRNA were subjected to SDS–PAGE and analyzed by immunoblotting using the indicated antibodies. (B) As in (A), except cells were untransfected or transfected with either FLAG-WT YOD1 or FLAG-C160S YOD1.
Confirming previous reports (Ernst et al., 2009, 2011), overexpressing C160S but not WT YOD1 increased the level of HMW polyubiquitinated proteins in cells (Figure 5B, top panel, compare lane 2 with lane 1). This increased HMW signal can also be detected using the K11-, K48-, and K63-specific ubiquitin antibodies (Figure 5B, second through fourth panels, compare lane 2 with lane 1). We attribute accumulation of these polyubiquitinated proteins to failure of C160S YOD1 to properly deubiquitinate cellular ubiquitinated proteins.
Catalytically inactive YOD1 traps polyubiquitinated proteins
We hypothesize that polyubiquitinated proteins are trapped on C160S YOD1, because they are not deubiquitinated. Accordingly, FLAG-WT or C160S YOD1 was precipitated under a native condition using FLAG antibody–conjugated beads, and the isolated proteins were eluted with a FLAG peptide. The eluted material was subjected to SDS–PAGE followed by Coomassie staining (Figure 6A, top panel) or immunoblotting with either a FLAG (Figure 6A, second panel) or pan-ubiquitin antibody (Figure 6A, third panel). An HMW signal corresponding to polyubiquitinated proteins was detected in purified C160S but not in WT YOD1 (Figure 6A, third panel, compare lane 3 with lane 1). Importantly, this HMW signal was absent when FLAG-C160S YOD1 was purified under a denaturing condition (i.e., 1% SDS; Figure 6, third panel, compare lane 4 with lane 3), indicating that the HMW signal is due to polyubiquitinated proteins noncovalently associated with YOD1 and not polyubiquitination of YOD1. Thus the catalytically inactive YOD1 binds to and traps polyubiquitinated cellular proteins.
FIGURE 6:

Catalytically inactive YOD1 traps polyubiquitinated proteins. (A) For purification of YOD1, cells transfected with either FLAG-WT YOD1 or FLAG-C160S YOD1 were lysed in a buffer containing 1% NP-40 with or without 1% SDS. The resulting WCLs were diluted 10-fold and incubated with FLAG antibody–conjugated beads; the precipitated material was eluted with a FLAG peptide and subjected to SDS–PAGE followed by Coomassie staining or immunoblotting with the indicated antibodies. (B) Transfected FLAG-WT YOD1 or FLAG-C160S YOD1 were precipitated, and the samples were immunoblotted with the indicated antibodies. Quantification of the Hrd1 band intensity is as in Figure 2E. Data represent the mean of three independent experiments. Error bars: ±SD.
As Hrd1, gp78, Derlin-1, and Sel1L are membrane components of the ERAD machinery shown to promote CTA1 retro-translocation (Bernardi et al., 2008, 2010; Dixit et al., 2008; Williams et al., 2013), we assessed whether mutant YOD1 preferentially traps these factors. When FLAG-WT and C160S YOD1 were precipitated from cells under a native condition and the precipitated material probed with antibodies against these membrane proteins, endogenous Hrd1 consistently bound to C160S YOD1 with higher affinity than WT YOD1 (Figure 6B, first panel, compare lane 6 with lane 5; quantified in graph on right). Derlin-1 bound to mutant and WT YOD1 with a similar affinity (Figure 6B, third panel, compare lane 6 with lane 5), whereas gp78, Sel1L, and Derlin-2 (another ERAD membrane component) did not interact with either WT or mutant YOD1 (Figure 6B, second, fourth, and fifth panels, lanes 5 and 6). C160S YOD1 thus appears to trap Hrd1.
Perturbing YOD1 activity disrupts ERAD of the nonubiquitinated yeast pro-α factor
We next examined whether YOD1 might influence the retro-translocation of another nonubiquitinated ERAD substrate. Using an in vitro system, a previous study demonstrated that functional E2 ubiquitin conjugation enzymes associated with the ERAD machinery are not required during proteasome-dependent degradation of the yeast ERAD substrate, mutant nonglycosylated pro-α factor (Werner et al., 1996). This finding suggests that nonglycosylated pro-α factor may not be ubiquitinated during ERAD in yeast. Another study demonstrated that even WT glycosylated pro-α factor can be degraded in a pre-Golgi compartment when it is expressed in mammalian cells (Su et al., 1993). These data raise the possibility that nonglycosylated pro-α factor may undergo ubiquitin-independent ERAD in mammalian cells, providing an opportunity to test whether YOD1 might influence this transport process. A FLAG tag was appended after the signal sequence of nonglycosylated pro-α factor, and the resulting construct was inserted into a mammalian expression vector to generate FLAG-pαF. When the proteasome in cells was inhibited by epoxomicin for 2 h, the steady-state level of FLAG-pαF increased (Figure 7A, top panel, compare lane 4 with lane 2), implicating this yeast mutant protein as an ERAD substrate in mammalian cells. Importantly, when YOD1 was knocked down (Figure 7B, second panel, compare lane 2 with lane 1), FLAG-pαF steady-state level decreased (Figure 7B, top panel, compare lane 2 with lane 1; quantified in Figure 7C), indicating enhanced proteasomal degradation under this condition. In the presence of the proteasome inhibitor epoxomicin, we did not find any evidence that FLAG-pαF is polyubiquitinated under either the control or knockdown condition (Figure 7D, top panel, compare lane 4 with lane 3), whereas TCRα-HA polyubiquitination was detected under the control condition (Figure 7D, top panel, lane 5). Thus, similar to toxin retro-translocation, YOD1 antagonizes pαF retro-translocation. By contrast, the FLAG-pαF steady-state level increased markedly only when FLAG-C160S YOD1 but not FLAG-WT YOD1 was overexpressed (Figure 7E, top panel, compare lane 3 with lanes 1 and 2; quantified in Figure 7F). Again, these findings are consistent with the toxin retro-translocation data, demonstrating that this YOD1 mutant disrupts an important step during pαF retro-translocation. No detectable level of FLAG-pαF polyubiquitination was seen when C160S YOD1 was overexpressed (unpublished data). These results suggest that YOD1 likely acts on ERAD components to negatively control retro-translocation of unglycosylated pαF.
FIGURE 7:

Perturbing YOD1 activity disrupts ERAD of the nonubiquitinated yeast pro-α factor. (A) Cells pretreated with or without epoxomicin were transfected with a pcDNA3.1(−) vector or FLAG-pαF. The resulting WCLs were subjected to SDS–PAGE and immunoblotting with the indicated antibodies. (B) WCLs derived from cells transfected with FLAG-pαF and either scrambled siRNA or YOD1 #1 siRNA were immunoblotted with the indicated antibodies. (C) The FLAG-pαF band intensity in (B) was quantified as in Figure 2D. Mean of at least three independent experiments. Error bars: ±SD. (D) Cells transfected with scrambled siRNA, YOD1 #1 siRNA, FLAG-pαF, and/or TCRα-HA were lysed in a RIPA buffer containing 0.1% SDS. The resulting WCLs were incubated with a FLAG antibody (lanes 1–4) or HA antibody–conjugated beads (lanes 5–6). The immunoprecipitates were subjected to reducing SDS–PAGE followed by immunoblotting with the indicated antibodies. WCLs were also analyzed by immunoblotting with the appropriate antibodies. All cells were transfected with GFP-Ub and incubated with epoxomicin. (E) As in (B), except cells were transfected with YFP, FLAG-WT YOD1, or FLAG-C160S YOD1. (F) The FLAG-pαF band intensity in (E) was quantified as in Figure 2D. Mean of at least three independent experiments. Error bars: ±SD.
Disrupting YOD1 activity affects TCRα degradation without altering its ubiquitination level
To determine whether YOD1 exerts a negative role during ERAD of ubiquitinated substrates, we further examined its function for the ubiquitinated substrate TCRα. A cycloheximide chase experiment revealed that overexpression of C160S but not WT YOD1 markedly stabilized TCRα (Figure 8A, compare lanes 7–9 with lanes 1–3 and 4–6), consistent with a previous report (Ernst et al., 2009). However, in cells treated with epoxomicin, immunoprecipitation of this receptor under a denaturing condition revealed that its ubiquitination level was largely unaffected when mutant YOD1 was overexpressed (Figure 8B, top panel, compare lane 4 with lane 2). This finding demonstrates that YOD1 unlikely affects TCRα’s turnover by regulating its ubiquitination state. When YOD1 is down-regulated, degradation of TCRα is modestly enhanced (Figure 8C, compare lanes 4–6 with lanes 1–3; quantified in Figure 8D). The increased degradation implicates YOD1 as a negative regulator of TCRα retro-translocation, consistent with YOD1’s posited function during ER-to-cytosol transport of CTA1 and pαF. Moreover, under this knockdown condition, TCRα polyubiquitination was not affected (Figure 8E, top panel, compare lane 4 with lane 2), further supporting the notion that YOD1 deubiquitinates elements of the ERAD machinery (and not the substrate) to regulate retro-translocation. Notably, not all ubiquitinated substrates are regulated by YOD1, as no significant difference in the degradation rate of the ubiquitinated ERAD substrate HA-tagged α1-antitrypsin null Hong Kong (NHK-HA) was observed when YOD1 was knocked down (Figure 8F, compare lanes 4–6 with lanes 1–3). While further experiments are necessary to clarify why YOD1 down-regulation does not affect degradation of NHK-HA, an Hrd1-dependent substrate (Christianson et al., 2008), this finding nevertheless demonstrates that YOD1 knockdown does not globally and nonspecifically affect all ERAD-dependent processes.
FIGURE 8:

Disrupting YOD1 activity affects TCRα degradation without altering its ubiquitination level. (A) Cells transfected with TCRα-HA and cotransfected with YFP, FLAG-WT YOD1, or FLAG-C160S YOD1 were incubated with cycloheximide for the indicated time. The resulting WCLs were immunoblotted with an HA antibody. (B) As in Figure 5B. (C) As in (A), except cells were transfected with scrambled siRNA or YOD1 #1 siRNA. (D) The TCRα-HA band intensity in (C) was quantified as in Figure 2C. Mean of at least three independent experiments. Error bars: ±SD. (E) As in (B), except cells were transfected with scrambled siRNA or YOD1 #1 siRNA. (F) As in (C), except cells were transfected with NHK-HA.
DISCUSSION
In summary, using a cell-based semipermeabilized membrane transport assay, we identify the YOD1 DUB as a negative regulator of CTA1 retro-translocation. Because CTA1 is not ubiquitinated even when YOD1 activity is disrupted, YOD1 likely acts on ubiquitinated cellular factors to control toxin retro-translocation.
The motivation for this study stems from our observation that the catalytic activities of the Hrd1 and gp78 E3 ubiquitin ligases promote CTA1 retro-translocation (Bernardi et al., 2010). Because CTA1 is not a ubiquitinated substrate, we hypothesize that ubiquitination of cellular components regulates toxin retro-translocation. An extension of this hypothesis is that cytosolic DUBs act in opposition to these E3 ligases to negatively regulate toxin retro-translocation. To identify a DUB that functions in this capacity, we reasoned that it should be in physical proximity to the E3 ligases. Accordingly, we first asked whether YOD1 binds to Hrd1 and gp78, as this DUB was shown to regulate the ERAD process (Ernst et al., 2009). In conjunction, we assessed whether Atx3 also impacts toxin retro-translocation, because this DUB binds to Hrd1 and controls ERAD (Wang et al., 2006; Zhong and Pittman, 2006). Our coimmunoprecipitation experiments revealed that YOD1 binds to Hrd1 via Hrd1’s cytosolic domain. By contrast, we failed to detect any YOD1-gp78 interaction. Whether the YOD1-Hrd1 interaction reflects a direct interaction or an interaction mediated by Hrd1 adaptors is unknown. As YOD1 contains three distinct domains, an N-terminal ubiquitin regulatory X (UBX) domain (that mediates p97 binding), a central otubain domain, and a C-terminal C2H2 zinc finger domain, any of these domains acting alone or in combination with the others may support interaction with Hrd1. At present, we do not know whether YOD1 engages other membrane-bound E3 ligases implicated in ERAD (Claessen et al., 2012).
To assess whether YOD1 plays a physiological role during CTA1 retro-translocation, we used the siRNA-mediated knockdown approach to down-regulate YOD1. Our results revealed that CTA1 ER-to-cytosol transport is significantly enhanced when YOD1 is knocked down, whereas knockdown of Atx3 or the proteasome-associated DUB USP14 does not affect CTA1 retro-translocation. The simplest interpretation of these findings is that YOD1 specifically and negatively regulates CTA1 ER-to-cytosol transport, consistent with our hypothesis that a Hrd1-associated DUB antagonizes CTA1 retro-translocation. Because YOD1 knockdown did not promote CTA1 ubiquitination, despite increasing total cellular polyubiquitinated proteins, we propose that YOD1 imposes its negative role by deubiquitinating components of the ERAD machinery that normally promote retro-translocation when ubiquitinated (Figure 9A). Supporting this idea, we found that YOD1 knockdown increases Hrd1 cross-links. If the cross-links represent Hrd1 oligomers, it is tempting to speculate that ubiquitination of Hrd1 favors its oligomerization; deubiquitination of Hrd1 by YOD1 would therefore prevent Hrd1 oligomerization. And because Hrd1 oligomerization was previously proposed to be crucial for its retro-translocation activity in yeast (Carvalho et al., 2010), this scenario would explain why YOD1 down-regulation enhances retro-translocation. Alternatively, if the Hrd1 cross-links represent cellular components recruited to Hrd1 in YOD1’s absence, the ubiquitination state of these cellular components may control their ability to bind to and regulate Hrd1’s function. Further experiments are required to clarify these possibilities. It should be noted that nonproteolytic roles of ubiquitination have been well documented, including control of protein–protein interactions, protein activities, and protein localization (Komander and Rape, 2012).
FIGURE 9:
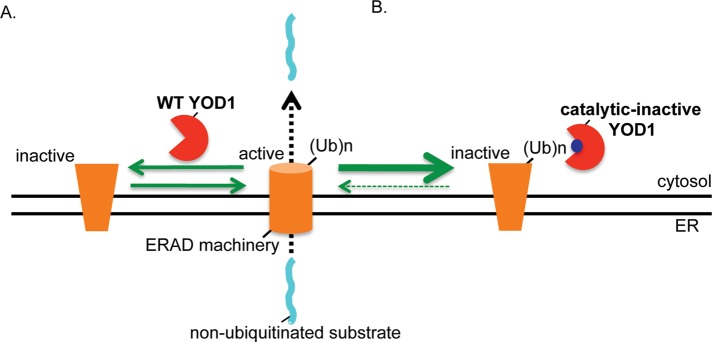
A model of how YOD1 negatively regulates retro-translocation of nonubiquitinated substrates. (A) YOD1 deubiquitinates and inactivates a ubiquitinated component of the ERAD machinery normally important for promoting retro-translocation. Hence, YOD1 down-regulation results in accumulation of the ubiquitinated ERAD component and consequently enhances retro-translocation. (B) By contrast, the catalytically inactive C160S YOD1 mutant binds to and traps the ubiquitinated ERAD component. Trapping effectively inactivates the function of the ubiquitinated ERAD factor, leading to a block in retro-translocation. A potential candidate for the ERAD component trapped by mutant YOD1 is Hrd1 itself.
Our analyses of the steady-state nonglycosylated pro-α factor level under the YOD1 knockdown condition suggests that YOD1 also serves as a negative regulator during ERAD of this postulated, nonubiquitinated, misfolded yeast substrate. Thus YOD1 may function as a general negative regulator during ERAD of nonubiquitinated substrates. By contrast, Atx3 knockdown did not affect CTA1 retro-translocation, despite the fact that it binds to Hrd1. This finding indicates that physical proximity to an E3 ligase is insufficient for a DUB to exert a functional impact. Two reports implicated the DUBs USP19 and USP25 as negative regulators of ERAD by deubiquitinating misfolded substrates (Hassink et al., 2009; Blount et al., 2012). Whether they antagonize CTA1 retro-translocation is unknown.
Interestingly, using cycloheximide chase experiments, we found that YOD1 knockdown also modestly increased TCRα retro-translocation without affecting its ubiquitination level. These findings suggest that YOD1 may even negatively control ER-to-cytosol transport of ubiquitinated substrates via a mechanism that involves deubiquitination of ERAD components rather than substrates. This scenario is consistent with the postulated mechanism by which YOD1 regulates retro-translocation of the nonubiquitinated substrates CTA1 and pro-α factor. Of note, the concept that ubiquitinated trans regulators of the ERAD machinery influence TCRα retro-translocation has been previously proposed (Yu and Kopito, 1999). In contrast to our finding, a prior report found that the steady-state level of TCRα-GFP was unaffected by YOD1 knockdown (Sowa et al., 2009). Factors that may account for this discrepancy are the different assays used to examine ERAD of TCRα and potential variations in the YOD1 knockdown efficiency. YOD1 is unlikely to counter retro-translocation of all Hrd1-dependent substrates, because we find that retro-translocation of NHK, an established Hrd1-dependent substrate (Christianson et al., 2008), is unaffected when YOD1 is down-regulated. This result is consistent with a recent paper demonstrating that, while retro-translocation of Shiga-like A chain in yeast requires catalytically active Hrd1, the closest yeast YOD1 homologue, Otu1, played no evident role (Li et al., 2012).
To further dissect YOD1’s role in CTA1 retro-translocation, we found that overexpression of the catalytically inactive C160S YOD1 but not WT YOD1 markedly decreased CTA1 arrival to the cytosol. Despite an increase in total cellular polyubiquitinated proteins when this YOD1 mutant is expressed, we found no evidence that CTA1 is ubiquitinated under this condition. A similar result was observed for pro-α factor: C160S YOD1 decreased retro-translocation of this yeast ERAD substrate without promoting its ubiquitination. Thus C160S YOD1 is likely affecting cellular components to perturb toxin and pro-α factor retro-translocation. In the case of TCRα, we found that C160S YOD1 expression significantly blocked this receptor's retro-translocation without changing its ubiquitination level. While the C160S YOD1–induced block in TCRα turnover was previously observed (Ernst et al., 2009), its ubiquitination level under this condition was not reported. C160S YOD1 overexpression also stabilized the turnover of another ERAD substrate, RI332 (Ernst et al., 2009). However, although polyubiquitin chains were found to accumulate on immunoprecipitated RI332 when C160S YOD1 was overexpressed (Ernst et al., 2009; Sanyal et al., 2012), it is unclear whether the accumulated polyubiquitin chains reflect increased ubiquitination of RI332 or polyubiquitinated proteins associated with RI332 due to the immunoprecipitation condition. Thus, while YOD1 may directly deubiquitinate an ubiquitinated ERAD substrate, this possibility has yet to be convincingly demonstrated. Regardless, the findings that C160S YOD1 perturbs ERAD of nonubiquitinated substrates suggest that this YOD1 mutant engages ubiquitinated cellular factors to control retro-translocation, similar to our explanation for the YOD1 knockdown results.
To reconcile our model that YOD1 plays a negative role during CTA1 retro-translocation (Figure 9A) with the finding that C160S overexpression blocks toxin retro-translocation, we propose that the catalytically inactive YOD1 mutant binds to and traps ubiquitinated ERAD components critical for retro-translocation (Figure 9B). This reaction renders the ubiquitinated ERAD factor inactive, despite being ubiquitinated, because it remains bound to YOD1 and cannot exert its normal function. In fact, our data indicate that a potential candidate for this factor is Hrd1 itself, as Hrd1 appears to be trapped on mutant YOD1. Trapping could conceivably perturb Hrd1’s retro-translocation activity in several ways, such as blocking Hrd1 oligomerization crucial for its “channel” function or preventing the recruitment of additional host components required for Hrd1 activity.
At present, we do not know whether YOD1 mediates mono- or polydeubiquitination. While there is evidence that monoubiquitination is sufficient to recruit proteins (Hoege et al., 2002), K63-linked polyubiquitin chains can also facilitate protein–protein interactions (Komander and Rape, 2012). Because perturbing YOD1 activity increased cellular K11-, K48-, and K63-linked polyubiquitinated proteins, YOD1 is likely to deubiquitinate any of these linkages. Interestingly, in this regard, K11 linkages were found to be particularly crucial during ERAD in yeast (Xu et al., 2009). Although we assume that YOD1 deubiquitinates ubiquitin attached to a protein, it is also possible that YOD1 deubiquitinates unanchored ubiquitin chains, which have been shown to mediate protein interactions in cells (Xia et al., 2009).
In conclusion, we report the identification of a cellular DUB that antagonizes retro-translocation of the toxic CTA1 subunit of CT. This membrane-translocation event represents a decisive step during toxin host-cell entry. Because all ERAD machinery-associated DUBs identified so far are thought to deubiquitinate misfolded substrates to influence various steps of retro-translocation, we took advantage of the fact that CTA1 is a nonubiquitinated ERAD substrate and investigated whether a DUB might impact its retro-translocation and found that it did. This finding implicates the presence of ERAD components whose activity is controlled by ubiquitination/deubiquitination. As a recent report further supports this model (Wang et al., 2013), the focus now should be to pinpoint these ERAD components and clarify how their ubiquitination state regulates retro-translocation.
MATERIALS AND METHODS
Materials
Monoclonal and polyclonal FLAG, and polyclonal Myc antibodies, as well as FLAG antibody–conjugated beads (M2 affinity gel), were purchased from Sigma-Aldrich (St. Louis, MO). Polyclonal YOD1 antibody was purchased from Abgent (San Diego, CA), polyclonal Atx3 and Hrd1 and monoclonal GFP antibodies from Protein Tech Group (Chicago, IL), monoclonal BiP antibody from BD Biosciences (San Jose, CA), polyclonal Hsp90 and PDI antibodies from Santa Cruz Biotechnology (Santa Cruz, CA), polyclonal Sel1L antibody from Enzo Life Sciences (Farmingdale, NY), monoclonal pan-ubiquitin antibody and protein A and G agarose from Invitrogen (Carlsbad, CA), polyclonal K11 ubiquitin antibody from Genentech (San Francisco, CA), polyclonal K48 and K63 ubiquitin antibodies from Cell Signaling Technology (Danvers, MA), and monoclonal HA antibody from Covance (Princeton, NJ). Polyclonal CTA antibody was produced against denatured CTA purchased from EMD Biosciences (San Diego, CA). Monoclonal Myc (9E10) was a gift from Kristen Verhey (University of Michigan), and polyclonal Derlin-1 antibody was a gift from Tom Rapoport (Harvard Medical School). Purified CT was purchased from EMD Biosciences, epoxomicin from EMD Millipore (Darmstadt, Germany), and cycloheximide from Amresco (Solon, OH).
Constructs
Constructs were gifts from the following: WT, C291A, cyt, and TM1-6 Hrd1-Myc: Emmanuel Wiertz (University Medical Center Utrecht, Netherlands); FLAG-WT and C160S YOD1: Christian Schlieker (Yale University); WT Atx3-FLAG: Yihong Ye (National Institutes of Health [NIH]); WT gp78-Myc: Kazuhiro Nagata (Kyoto University); TCRα-HA: Cezary Wojcik (Indiana University); NHK-HA: John Christianson (University of Oxford); a yeast expression construct containing nonglycosylated pro-α factor: Jeff Brodsky (University of Pittsburgh); peYFP-N1: Kristen Verhey (University of Michigan); and GFP-Ub: Walther Mothes (Yale University). To generate a construct containing yeast nonglycosylated pro-α factor in a mammalian expression vector, the N-terminal signal sequence from human BiP was fused to the nonglycosylated pro-α factor coding sequence. The FLAG sequence was then inserted between the signal sequence and the nonglycosylated pro-α factor coding sequence using overlapping PCR. The resulting PCR product was inserted into pCDNA3.1(−) using standard cloning methods to generate FLAG-pαF.
Retro-translocation assay
The retro-translocation assay was as described previously by Williams et al. (2013).
Cell transfection
The 293T cells were grown to 30% confluency on a 10-cm dish prior to transfection with the Effectene system (Qiagen, Chatsworth, CA). Cells were grown for an additional 24 h prior to experimentation.
siRNA knockdown of YOD1, Atx3, and USP14
Cells were grown to 20–30% confluency on a 10-cm dish prior to a 48-h transfection with the Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA). The sequences of the siRNAs used in this study were YOD1 #1 siRNA (5′-GGGAGGAGCAAUAGAGAUAUU-3′; Invitrogen), YOD1 #2 siRNA (5′-AGUAAGAAUUGAUCGUUUUU-3′; Invitrogen), Atx3 #1 siRNA (5′-GGACCUAUCAGGACAGAGUUU-3′; Invitrogen), Atx3 #2 siRNA: (5′-GGACAGAGUUCACAUCCAUUU-3′, Dharmacon), USP14 siRNA: (5′-GAAACAAGAUGAAUGGAUUUU -3′, Dharmacon). Duplex siRNA (20 nM) was transfected into 293T cells according to the manufacturer's protocol. The control siRNA was scrambled medium GC Stealth RNAi siRNA (Invitrogen).
Coimmunoprecipitation
For detection of FLAG-YOD1 and Hrd1-Myc interaction, 293T cells were transfected with the indicated constructs and lysed in a buffer containing HEPES (pH 7.5, 50 mM), NaCl (150 mM), sucrose (250 mM), MgCl2 (2 mM), NEM (10 mM), and protease inhibitors with 1% NP-40 for 10 min at 4°C. Samples were centrifuged at 16,000 × g for 10 min to generate WCLs used for immunoprecipitation experiments. Where indicated, FLAG antibody–conjugated beads were added to the WCL and incubated at 4°C for 2–4 h. The immune complex was sedimented, washed, and subjected to SDS–PAGE; this was followed by immunoblotting with the appropriate antibodies. For assessment of polyubiquitination of immunoprecipitated proteins, cells were lysed in RIPA buffer containing 0.1% SDS, and the samples were incubated with CTA or FLAG antibodies, or with HA antibody–conjugated beads. The immune complex was processed as above.
Cycloheximide chase experiments
Cells transfected with scrambled, YOD1 #1 siRNA, FLAG-WT YOD1, or FLAG-C160S YOD1 were cotransfected with either TCRα-HA or NHK-HA. During the chase, cells were treated with 100 μg/ml cycloheximide for 0, 1, or 2 h in DMEM/fetal calf serum media. Cells were harvested at the indicated time points and lysed in a buffer containing Tris (pH 7.4, 30 mM), KOAc (150 mM), MgCl2 (4 mM), NEM (10 mM), 1% NP-40, and protease inhibitors for 30 min at 4°C. Cells were centrifuged at 16,000 × g for 15 min, and the resulting supernatant was analyzed by reducing SDS–PAGE followed by immunoblotting with a monoclonal HA antibody.
Purification of WT and C160S YOD1-FLAG
Cells expressing FLAG-WT and C160S YOD1 were lysed in a buffer containing HEPES (pH 7.5, 50 mM), NaCl (150 mM), sucrose (250 mM), MgCl2 (2 mM), NEM (10 mM), and protease inhibitors with 1% NP-40 with or without 1% SDS for 30 min at 4°C. Samples were centrifuged at 16,000 × g for 10 min, and the resulting supernatant was diluted 10-fold with buffer (without detergent) and incubated with FLAG antibody–conjugated beads. The precipitated material was washed, and the bound protein was eluted with a FLAG peptide (0.5 mg/ml). Eluted proteins were subjected to SDS–PAGE followed by Coomassie staining or immunoblotting with either FLAG or pan-ubiquitin antibodies.
XBP1 splicing
XBP1 splicing was as described previously by Bernardi et al. (2010).
Chemical cross-linking
Cells transfected with scrambled or YOD1 #1 siRNA were incubated with 1 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDAC; Sigma-Aldrich) for 30 min at 37°C and quenched with 50 mM Tris HCl (pH 7.5), and the resulting WCL was subjected to immunoblotting.
Supplementary Material
Acknowledgments
We thank Christopher Walczak (University of Michigan) for critical review of the manuscript. B.T. is funded by the NIH (RO1 083252-04).
Abbreviations used:
- CTA1
cholera toxin A1
- DTT
dithiothreitol
- DUB
deubiquitinases
- EDAC
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- GFP-Ub
green fluorescent protein–ubiquitin
- His-Ub
His-ubiquitin
- HMW
high molecular weight
- NHK-HA
HA-tagged α1-antitrypsin null Hong Kong
- PDI
protein disulfide isomerase
- siRNA
small interfering RNA
- TCRα−HA
C-terminally hemagglutinin-tagged T-cell receptor α
- WCL
whole-cell lysate
- WT
wild type
- YFP
yellow fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-06-0332) on September 25, 2013.
REFERENCES
- Bernardi KM, Forster ML, Lencer WI, Tsai B. Derlin-1 facilitates the retro-translocation of cholera toxin. Mol Biol Cell. 2008;19:877–884. doi: 10.1091/mbc.E07-08-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi KM, Williams JM, Kikkert M, van Voorden S, Wiertz EJ, Ye Y, Tsai B. The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol Biol Cell. 2010;21:140–151. doi: 10.1091/mbc.E09-07-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount JR, Burr AA, Denuc A, Marfany G, Todi SV. Ubiquitin-specific protease 25 functions in endoplasmic reticulum-associated degradation. PLoS One. 2012;7:e36542. doi: 10.1371/journal.pone.0036542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151:1163–1167. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22:22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit G, Mikoryak C, Hayslett T, Bhat A, Draper RK. Cholera toxin up-regulates endoplasmic reticulum proteins that correlate with sensitivity to the toxin. Exp Biol Med (Maywood) 2008;233:163–175. doi: 10.3181/0705-RM-132. [DOI] [PubMed] [Google Scholar]
- Ernst R, Claessen JH, Mueller B, Sanyal S, Spooner E, van der Veen AG, Kirak O, Schlieker CD, Weihofen WA, Ploegh HL. Enzymatic blockade of the ubiquitin-proteasome pathway. PLoS Biol. 2011;8:e1000605. doi: 10.1371/journal.pbio.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Mueller B, Ploegh HL, Schlieker C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell. 2009;36:28–38. doi: 10.1016/j.molcel.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster ML, Sivick K, Park YN, Arvan P, Lencer WI, Tsai B. Protein disulfide isomerase-like proteins play opposing roles during retro-translocation. J Cell Biol. 2006;173:853–859. doi: 10.1083/jcb.200602046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Hathaway NA, Tone Y, Crosas B, Elsasser S, Kirkpatrick DS, Leggett DS, Gygi SP, King RW, Finley D. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell. 2006;127:99–111. doi: 10.1016/j.cell.2006.07.038. [DOI] [PubMed] [Google Scholar]
- Hassink GC, Zhao B, Sompallae R, Altun M, Gastaldello S, Zinin NV, Masucci MG, Lindsten K. The ER-resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 2009;10:755–761. doi: 10.1038/embor.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazes B, Read RJ. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry. 1997;36:11051–11054. doi: 10.1021/bi971383p. [DOI] [PubMed] [Google Scholar]
- Hirsch C, Gauss R, Horn SC, Neuber O, Sommer T. The ubiquitylation machinery of the endoplasmic reticulum. Nature. 2009;458:453–460. doi: 10.1038/nature07962. [DOI] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van Voorden S, Thanedar S, Roitelman J, Chau V, Wiertz E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Kothe M, Ye Y, Wagner JS, De Luca HE, Kern E, Rapoport TA, Lencer WI. Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J Biol Chem. 2005;280:28127–28132. doi: 10.1074/jbc.M503138200. [DOI] [PubMed] [Google Scholar]
- Lam YA, Xu W, DeMartino GN, Cohen RE. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature. 1997;385:737–740. doi: 10.1038/385737a0. [DOI] [PubMed] [Google Scholar]
- Lee BH, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer WI, Tsai B. The intracellular voyage of cholera toxin: going retro. Trends Biochem Sci. 2003;28:639–645. doi: 10.1016/j.tibs.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Li S, Spooner RA, Hampton RY, Lord JM, Roberts LM. Cytosolic entry of Shiga-like toxin A chain from the yeast endoplasmic reticulum requires catalytically active Hrd1p. PLoS One. 2012;7:e41119. doi: 10.1371/journal.pone.0041119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery FC, et al. TorsinA participates in endoplasmic reticulum-associated degradation. Nat Commun. 2011;2:393. doi: 10.1038/ncomms1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodighiero C, Tsai B, Rapoport TA, Lencer WI. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002;3:1222–1227. doi: 10.1093/embo-reports/kvf239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S, Claessen JH, Ploegh HL. A viral deubiquitylating enzyme restores dislocation of substrates from the endoplasmic reticulum (ER) in semi-intact cells. J Biol Chem. 2012;287:23594–23603. doi: 10.1074/jbc.M112.365312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler BD. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol Rev. 1992;56:622–647. doi: 10.1128/mr.56.4.622-647.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su K, Stoller T, Rocco J, Zemsky J, Green R. Pre-Golgi degradation of yeast prepro-α-factor expressed in a mammalian cell. Influence of cell type-specific oligosaccharide processing on intracellular fate. J Biol Chem. 1993;268:14301–14309. [PubMed] [Google Scholar]
- Taylor M, Navarro-Garcia F, Huerta J, Burress H, Massey S, Ireton K, Teter K. Hsp90 is required for transfer of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol. J Biol Chem. 2010;285:31261–31267. doi: 10.1074/jbc.M110.148981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Aravind L, Oania R, McDonald WH, Yates JR, III, Koonin EV, Deshaies RJ. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li L, Ye Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J Cell Biol. 2006;174:963–971. doi: 10.1083/jcb.200605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yu YY, Myers N, Hansen TH. Decoupling the role of ubiquitination for the dislocation vs degradation of major histocompatibility complex (MHC) class I proteins during endoplasmic reticulum-associated degradation (ERAD) J Biol Chem. 2013;288:23295–23306. doi: 10.1074/jbc.M113.482018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci USA. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernick NL, De Luca H, Kam WR, Lencer WI. N-terminal extension of the cholera toxin A1-chain causes rapid degradation after retrotranslocation from endoplasmic reticulum to cytosol. J Biol Chem. 2010;285:6145–6152. doi: 10.1074/jbc.M109.062067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JM, Inoue T, Banks L, Tsai B. The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol Biol Cell. 2013;24:785–795. doi: 10.1091/mbc.E12-07-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Yu H, Kopito RR. The role of multiubiquitination in dislocation and degradation of the α subunit of the T cell antigen receptor. J Biol Chem. 1999;274:36852–36858. doi: 10.1074/jbc.274.52.36852. [DOI] [PubMed] [Google Scholar]
- Zhong X, Pittman RN. Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum Mol Genet. 2006;15:2409–2420. doi: 10.1093/hmg/ddl164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.