

Overexpression of the developmentally important T-box transcription factor TBX3 inhibits proliferation but promotes the migration of epithelial cells. TBX3 is transcriptionally up-regulated by the TGF-β1 signaling pathway in a Smad3/4- and JunB-dependent manner and is a key mediator of the antiproliferative and promigratory roles of TGF-β1.
Abstract
The T-box transcription factor, TBX3, plays an important role in embryonic development, and haploinsufficiency of TBX3 causes ulnar–mammary syndrome. Overexpression of TBX3, on the other hand, is associated with several cancers, and preliminary evidence suggests that increased levels of TBX3 may inhibit cell proliferation but promote tumor migration and invasion. Although this suggests that deregulated levels of TBX3 are deleterious in development and promotes disease, very little is known about the signaling pathways that regulate TBX3 expression. Here we show that overexpressing TBX3 inhibits proliferative ability while promoting the migration of breast epithelial cells. We demonstrate that the transforming growth factor β1 (TGF-β1) pathway up-regulates TBX3 protein and mRNA levels and show a detailed transcriptional mechanism by which this occurs. Using in vitro and in vivo assays, we show that Smad3/4 and JunB bind and cooperatively regulate TBX3 promoter activity through a Smad-binding element at −67 base pairs. Further, we show that TBX3 plays a pivotal role in mediating the antiproliferative and promigratory role of TGF-β1 in breast epithelial and skin keratinocytes. This study identifies the TGF-β1 signaling pathway as a potentially important player in the regulation of TBX3 in development and cancer.
INTRODUCTION
The transcription factor TBX3 belongs to the T-box gene family, which is characterized by a conserved DNA-binding domain called the T-box (Bamshad et al., 1997). TBX3 plays an essential role in embryogenesis, and heterozygous mutations in TBX3 result in the autosomal dominant ulnar–mammary syndrome (Bamshad et al., 1997). TBX3 is also overexpressed in several cancers, including breast, pancreatic, liver, bladder, and melanoma, and there is strong evidence linking it to the oncogenic process (Carlson et al., 2001; Fan et al., 2004; Rowley et al., 2004; Ito et al., 2005; Lomnytska et al., 2006; Lyng et al., 2006; Renard et al., 2007). TBX3 functions primarily as a transcriptional repressor, and initial reports suggested that its role in cancer may be related to its capacity to bypass senescence by repressing the cell cycle regulators p19ARF and p21WAF1/CIP1/SDII (Brummelkamp et al., 2002; Hoogaars et al., 2008; Yarosh et al., 2008). More recently, however, we and others reported that TBX3 negatively affects tumor cell proliferation but promotes an invasive cancer phenotype (Renard et al., 2007; Fillmore et al., 2010; Peres et al., 2010; Humtsoe et al., 2012). Of importance, these oncogenic functions appear to be directly related to increased TBX3 levels, but there is a paucity of information regarding the signaling molecules responsible for regulating TBX3 expression.
Transforming growth factor β1 (TGF-β1) is a polypeptide member of the TGF-β superfamily of cytokines. It controls several cellular functions and, like TBX3, plays an important role in mammary morphogenesis and is notoriously activated during breast cancer development (Moses and Barcellos-Hoff, 2011). TGF-β1 also inhibits proliferation and promotes cell migration (Tian et al., 2003), and we therefore hypothesized that it may be upstream of TBX3. Indeed, microarray studies in which normal human breast epithelial cells and keratinocytes were treated with TGF-β1 showed activation of TBX3, but the mechanism involved remains to be elucidated (Kang et al., 2003). The best-defined TGF-β1 signaling cascade involves the Smad proteins (Feng and Derynck, 2005). Binding of TGF-β1 to the type II TGF-β receptor results in recruitment and phosphorylation of the type I receptor, which in turn phosphorylates Smad2 and Smad3 (R-Smad). The phosphorylated Smad2/3 can form a complex with Smad4, which facilitates their nuclear translocation, where they bind the consensus Smad-binding element (SBE) in target genes (Massagué et al., 2005). The affinity of Smads to bind SBE (5′-GTCTAGAC-3′) is low, and therefore multiple SBEs and/or interaction with cofactors are required for them to regulate target genes.
In the present study, we show that ectopically overexpressing TBX3 in normal breast epithelial cells inhibits proliferation, which is accompanied by an increase in migration. Of importance, we demonstrate that stimulation of the TGF-β1 pathway leads to transcriptional up-regulation of endogenous TBX3 in these cells, as well as in skin keratinocytes. Using in vitro and in vivo assays, we find that this occurs by a mechanism involving the cooperation between Smad3/4 and JunB at a SBE at –67 base pairs in the TBX3 promoter. We also demonstrate that TBX3 is a key player in driving the antiproliferative and promigratory roles of TGF-β1.
RESULTS
Overexpressing TBX3 in normal breast epithelial cells inhibits cell growth and promotes migration
There is accumulating evidence that TBX3 plays a direct role in oncogenesis by promoting cell migration and metastasis (Rodriguez et al., 2008; Mowla et al., 2011). Indeed, we previously reported that knocking down TBX3 in MCF-7 breast cancer and vertical growth phase and metastatic melanoma cells resulted in increased proliferation and reduced migration (Peres et al., 2010). Because these studies were performed in already transformed cell lines, we asked whether overexpression of TBX3 in normal breast epithelial cells is sufficient to inhibit cell proliferation while promoting migration. To this end, we overexpressed TBX3 in normal human MCF-12A breast epithelial cells using an inducible adenovirus Tet-off system (Figure 1A) and performed 5-bromo-2-deoxyuridine (BrdU) incorporation and scratch motility assays. The results show that increased levels of TBX3 also resulted in cells with decreased cell proliferation but increased migration, confirming that the TBX3 is sufficient to affect these cellular processes (Figure 1, B and C). We therefore next sought to identify the molecular pathway(s) that up-regulate TBX3 levels and considered TGF-β1 as a candidate, as it is also known to inhibit proliferation but promote cell migration.
FIGURE 1:

TBX3 overexpression inhibits cell proliferation but promotes migration. (A) MCF-12A cells were transduced with an Adeno-X Tet-off expression system, which allows TBX3 expression to be regulated by Dox (10 μg/ml). The efficacy of the system was tested by subjecting protein from the indicated cells to Western blot analysis. (B) MCF-12A cells from A were incubated with BrdU for 3 h and the total number of BrdU-positive nuclei, visualized by fluorescence microscopy, expressed as a percentage of the total number of cells from five fields of view for each condition. (C) MCF-12A cells from A were incubated for 48 h and subjected to the scratch assay. Bars, SD. *p < 0.05, **p < 0.001.
TGF-β1 transcriptionally activates TBX3 expression in MCF-12A breast epithelial cells and HaCaT keratinocytes
To begin to explore the possibility that TBX3 expression is regulated by TGF-β1, we treated MCF-12A and HaCaT keratinocytes with TGF-β1 or vehicle over a time course spanning 30 min to 36 h and performed Western blot analysis. The results show that TBX3 levels increase substantially in response to TGF-β1 treatment in both cell lines. Figure 2, A and B, shows results obtained for four time points between 8 and 36 h. Immunofluorescence experiments also show that TGF-β1–treated cells have an increase in nuclear TBX3 (Figure 2, C and D). Quantitative real-time PCR (qRT-PCR) experiments performed on cells treated with TGF-β1 for 3 and 12 h demonstrate a corresponding increase in TBX3 mRNA levels, suggesting that TGF-β1 may regulate TBX3 transcriptionally (Figure 3, A and B). Indeed, pretreatment of MCF-12A cells with a transcriptional inhibitor, actinomycin D, abolished the TGF-β1–mediated activation of TBX3 mRNA (Figure 3C) and protein (Figure 3D) expression. In summary, these results indicate that TGF-β1 transcriptionally activates TBX3 expression.
FIGURE 2:
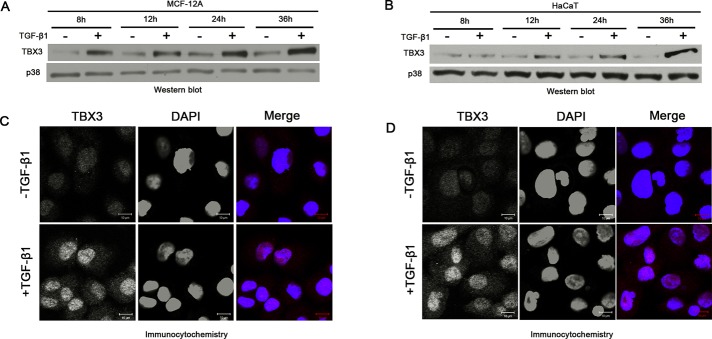
TGF-β1 activates TBX3 protein expression. TBX3 protein from MCF-12A cells (A) or HaCaT cells (B) was prepared after indicated times and examined by Western blot analysis. p38 was used as a loading control. MCF-12A cells (C) or HaCaT cells (D) were treated with TGF-β1 (5 ng/ml) for 12 h, and TBX3 subcellular localization was determined by immunocytochemistry using antibody specific to TBX3. In the merged image the red and blue represent TBX3 and 4′,6-diamidino-2-phenylindole, respectively.
FIGURE 3:

TBX3 is transcriptionally regulated by TGF-β1. Total RNA extracted from MCF-12A cells (A) or HaCaT cells (B) after 3 or 12 h TGF-β1 treatment was reverse transcribed and subjected to qRT-PCR using primers specific to TBX3. mRNA levels were normalized to GUSB. (C) MCF-12A cells were pretreated with vehicle (control) or 5 μg/ml actinomycin D (AD) for 1 h and treated with TGF-β1 for 3 h. (D) RNA and protein were harvested for use in qRT-PCR and Western blotting analysis. Bars, SD. **p < 0.001.
TGF-β1–activated TBX3 expression is mediated by JunB and Smad3/4
To identify the mechanism(s) by which TGF-β1 transcriptionally activates TBX3, we first cotransfected MCF-12A cells with −2186 base pairs of the TBX3 promoter driving a firefly luciferase reporter with increasing concentrations of Smad3/4. Of interest, at all concentrations tested, Smad3/4 had very little effect on basal TBX3 promoter activity, suggesting that the Smads may require another cooperating protein to transactivate TBX3 in the TGF-β1 signaling pathway (unpublished data). On the basis of previous reports, we speculated that JunB may be a cofactor involved in this regulation (Verrecchia et al., 2001). We therefore determined the level of JunB and Smad proteins in response to TGF-β1 over a time course of 0.5 to 4 h. Western blotting showed that TGF-β1 treatment led to an increase in JunB and pSmad3 protein levels, which preceded the increase in TBX3 protein levels. As expected, the total levels of Smad2/3, Smad4, and the p38 loading control were unchanged (Figure 4A).
FIGURE 4:

JunB and Smad proteins mediate the regulation of TBX3 by TGF-β1. (A) MCF-12A cells were serum starved and treated with TGF-β1 at indicated time points and analyzed by Western blotting. (B) MCF-12A cells were either untransfected (UT) or transfected with 30 nM siJunB (top) or 50 nM siSmad4 (bottom) or the equivalent concentration of control siRNA for 24 h, followed by 3 h of TGF-β1 treatment and subjected to Western blotting. (C) RNA from B was subjected to qRT-PCR analysis. Bars, SD. *p < 0.05, **p < 0.001.
To confirm the possible in vivo role of JunB and Smad proteins in TGF-β1–regulated TBX3 expression, we transfected the MCF-12A cells with small interfering RNAs (siRNAs) that specifically target and knock down JunB or Smad4. The rationale for knocking down Smad4 was based on reports that it is the nuclear transporter for Smad2/3 (Feng and Derynck, 2005). As shown in Figure 4, B and C, compared with the untransfected or transfected control siRNA cells, knocking down either JunB or Smad4 did not completely abrogate but did severely compromise TGF-β1–induced activation of TBX3 protein and mRNA levels. Together these results provided compelling evidence that both JunB and Smad proteins are important in the regulation of TBX3 by the TGF-β1 pathway.
JunB and Smad proteins cooperate to activate the TBX3 promoter at SBE-67
To identify the potential cis-acting regulatory element(s) of JunB and Smad proteins, we searched the TBX3 promoter for AP-1–binding and SBE sites. We identified five putative AP-1–binding sites (Figure 5A) and numerous SBEs (not shown) in the −2.1-kb promoter region upstream of the transcription initiation site. To narrow down the region of the TBX3 promoter involved in the TGF-β1–mediated activation, we tested a series of 5’ deletion constructs of the human TBX3 promoter in luciferase reporter assays. Whereas Smad3/4 had no effect on all four constructs, JunB activated all TBX3 promoter deletion constructs. Of interest, the cotransfection of JunB and Smad3/4 demonstrated that they cooperate to activate all four promoters. To identify the site(s) responsible for this activity, we focused on putative sites in the shortest TBX3 promoter construct (−141/+38 base pairs), because it maintained a high level of promoter activity. Given that the ability of Smads to cooperate with their cofactors requires their respective binding sites to be in close proximity, we mutated the two AP-1 sites at −86 base pairs p and the adjacent SBE (−67 base pairs) individually (Figure 5B). These constructs were compared with the wild-type (WT) −141−base pair construct in luciferase reporter assays. Of interest, whereas the activity of the AP-1 mutant was comparable to that of WT, the SBE mutant significantly dampened the JunB-induced activation and abolished the cooperative effect of JunB and Smad3/4 in response to TGF-β1. This is consistent with previous reports that in response to signals from TGF-β1 receptors, Smad proteins can cooperate with other sequence-specific transcription factors to regulate transcription of target genes. Together these data suggest not only that Smad cooperates with JunB to activate the TBX3 promoter, but that this activity is mediated by a SBE. We cannot, however, rule out the possibility that the TBX3 promoter used in this study does not contain all regulatory elements required for TGF-β1-mediated up-regulation of TBX3.
FIGURE 5:

TGF-β1 activation of the TBX3 promoter is mediated by a degenerate SBE at −67 base pairs. (A) Schematic illustration of luciferase reporter constructs containing sequential 5′- deletions of the human TBX3 promoter (400 ng), which were transiently cotransfected into the MCF-12A cells with vectors expressing human JunB and Smad3/4. The arrow indicates the transcription start site at +1, and the asterisk indicates putative AP-1–binding sites. Mean values (± SD) are presented as fold activity over that of an empty firefly luciferase reporter and are representative of at least three independent experiments. (B) MCF-12A cells were cotransfected with WT TBX3 −141 base pair promoter luciferase reporter or a reporter in which the indicated AP-1 (AP1mt) or Smad-binding-element (SBEmt) was mutated and JunB or Smad3/4 expression constructs and luciferase activity analyzed. (C) MCF-12A cells were treated with 5 ng/ml TGF-β1 for 3 h and chromatin immunoprecipitation assays performed with antibodies against JunB, Smad4, or IgG (negative control). Immunoprecipitated DNA was assayed by qRT-PCR with primers against the TBX3 promoter. (D) For EMSA, biotin-labeled, double-stranded oligonucleotide probes containing the homologous WT SBE −67/−58 site were incubated with nuclear extracts from MCF-12A cells (lanes 2–6). Observed complexes are indicated by arrows on the left. Unspecific complexes are indicated by *NS on the right. Competition analyses were carried out in the presence of 5× (lanes 3 and 4) or 25× (lanes 5 and 6) molar excess of unlabeled homologous probes. The complex bands observed for the SBE site are indicated by the two lowest arrows. Right, biotinylated DNA probes of the TBX3 promoter containing the homologous WT or MT SBE were immobilized on streptavidin beads and incubated with nuclear extracts from MCF-12A cells treated with or without 5 ng/ml TGF-β1 for 3 h. The DNA-bound protein complexes were isolated and analyzed by Western blotting using antibodies to JunB or Smad4. Bars, SD. *p < 0.05.
TGF-β1 treatment enhances binding of JunB and Smad4 to the TBX3 promoter
To confirm that JunB and the Smad proteins can bind to the proximal region of TBX3 promoter in vivo, we performed a chromatin immunoprecipitation (ChIP) assay. The results obtained show that compared with the immunoglobulin G (IgG) control, JunB and Smad4 were able to bind to the TBX3 promoter in untreated cells and that in the presence of TGF-β1 this binding was enhanced by 2.5-fold (JunB) and 5.5-fold (Smad4) increase in signal, respectively (Figure 5C). These data suggest that JunB and Smad4 directly bind the proximal TBX3 promoter during basal regulation and that this mechanism is significantly enhanced by TGF-β1 treatment.
To demonstrate that JunB and/or Smad4 specifically bind the SBE −67 site in the TBX3 promoter, we first used nuclear extract isolated from TGF-β1–treated MCF-12A cells and a biotin-labeled probe containing WT SBE at −67 in an electromobility-shift assay (EMSA; Figure 5D, left). Four complexes were observed (lane 2; see arrows), and the lowest two were competed efficiently by non–biotin-labeled homologous WT competitor oligonucleotides (lanes 3 and 4, two lower arrows) but not by the oligonucleotide in which the SBE −67 was mutated (MT; lanes 5 and 6). The topmost complex was not competed by either WT or MT competitors, suggesting that it may be a nonspecific (NS) band or represent a complex that binds biotin. Furthermore, the fourth complex was competed by both WT and MT competitors and may thus represent a nuclear factor bound to the probe at a site adjacent to the SBE. To confirm that it was indeed JunB and Smad4 in the nuclear extract used in Figure 5D that bound the SBE at −67 in the TBX3 promoter, we performed a DNA affinity immunoblot assay. To this end, the same nuclear extracts used for EMSAs were incubated with biotinylated DNA probes containing either the WT or MT SBE-67 oligos. The results show that in the absence of TGF-β1 stimuli, both JunB and Smad4 but not pSmad3 bound the probes carrying the WT −67. However, when TGF-β1 was present, all three proteins could bind the WT −67, and their binding affinity was greatly decreased when this SBE was mutated (Figure 5D, right). Taken together, these results suggest that in response to TGF-β1, JunB and Smad4 form protein complexes at SBE-67 to directly activate TBX3 expression.
The antiproliferative and promigratory roles of TGF-β1 are mediated in part by TBX3
TGF-β1 has an antiproliferative and a promigratory role, and recent findings from our laboratory reported similar roles for TBX3 (Peres et al., 2010; Mowla et al., 2011). We therefore speculated that these biological effects of TGF-β1 are mediated in part by TBX3. To test this, we generated an MCF-12A cell line (lenti-MCF-12A) that contains a doxycycline (Dox)-inducible siRNA that specifically targets and knocks down TBX3. The efficacy of the system was confirmed by Western blotting (Figure 6A). When control cells were treated with TGF-β1, TBX3 protein levels increased, which corresponded with a decrease in proliferation as measured by BrdU incorporation (Figure 6, A and B). However, when TBX3 expression was knocked down there was no statistical difference in the proliferative ability of control and TGF-β1–treated cells, suggesting that TBX3 was required for the inhibitory effect of TGF-β1 on cell proliferation. Furthermore, scratch motility assays show that TBX3 also plays an important role in mediating the promigratory effect of TGF-β1 on these cells (Figure 6C). These results were reproducible when TBX3 was knocked down in HaCaT cells using an siTBX3 approach (Figure 6, D–F).
FIGURE 6:

TBX3 mediates TGF-β1–regulated cell proliferation and migration. Western blot analyses show lentiviral shRNA-mediated knockdown of TBX3 in MCF-12A cells (A) or siTBX3-mediated knockdown in HaCaT cells (D) in the presence or absence of 3 h TGF-β1 treatment. (B) MCF-12A cell lines were incubated with BrdU for 3 h, and BrdU-positive nuclei were visualized by fluorescence microscopy to measure cell proliferation. (E) Net cell growth of HaCaT cells as assessed using the MTT assay. MCF-12A (C) and HaCaT cells (F) were subjected to cell scratch migration assay. Bars, SD. *p < 0.05, **P < 0.001.
DISCUSSION
T-box transcription factors play important roles in embryonic development, and several lines of evidence suggest that some members have a role in cell cycle regulation and cancer progression. In particular, haploinsufficiency of the human TBX3 causes the ulnar–mammary syndrome, which is characterized in part by mammary gland hypoplasia or aplasia, and its overexpression has been associated with breast cancer. Furthermore, we and others have reported that this overexpression contributes directly to the oncogenic process, because when TBX3 levels are depleted in breast cancer and melanoma cells, proliferation is enhanced but tumor formation and invasion are inhibited (Renard et al., 2007; Peres et al., 2010; Mowla et al., 2011). There is limited information, however, on the mechanisms that up-regulate TBX3 and in particular those involved in regulating its oncogenic activity. Here we show, consistent with data obtained for TBX3-knockdown studies, that overexpression of TBX3 inhibits cell proliferation but promotes migration of normal epithelial cells. Of importance, we provide novel data to show that the stimulation of the TGF-β1 signaling pathway results in up-regulation of TBX3 expression and that TBX3 is a key mediator of its antiproliferative and promigratory roles in epithelial cells.
TGF-β1 is an intercellular ligand that has a well-established antiproliferative and promigratory role in epithelial cells (Massagué, 2012). On the basis of our data that TBX3 similarly promotes migration and invasion while having a negative effect on cell proliferation, we speculated that TBX3 may be downstream of TGF-β1 (Peres et al., 2010). Here we show that TGF-β1 transcriptionally activates TBX3, leading to an increase in TBX3 mRNA and protein expression in normal human breast epithelial cells and keratinocytes. Indeed, we provide in vitro and in vivo data showing that this transcriptional activation is mediated by cooperation between Smad3/4 and JunB. This is consistent with previous reports that JunB and Smad3 can form a complex in vitro (Verrecchia et al., 2001) and that the transcriptional activity of Smads in the canonical TGF-β1 signaling pathway frequently relies on partner transcription factors, such as JunB, Sp1, Egr1, ATF3, and forkhead family members (Chen et al., 1997; Feng et al., 2000; Verrecchia et al., 2001; Kang et al., 2003; Seoane et al., 2004; Fortin and Bernard, 2010). For example, Sp1 physically interacts with a complex of Smads to mediate TGF-β1–induced p15Ink4B, an important Cdk inhibitor responsible for the antiproliferative role of TGF-β1 (Feng et al., 2000), and Smad3 cooperatively interacts with either c-Jun or JunB to up-regulate expression of the COL7A1 promoter (Verrecchia et al., 2001).
This study also demonstrates that TBX3 is a pivotal player in TGF-β1–mediated antiproliferation and promigration. Although we did not explore the TBX3 target genes downstream of the TGF-β1 signaling pathway, it is tempting to speculate that one of them would be E-cadherin, a key epithelial cell adhesion molecule. TBX3 has been shown to promote melanoma cell migration by directly repressing E-cadherin (Rodriguez et al., 2008; Boyd et al., 2013), and a study by Vincent et al. (2009) showed that TGF-β1 signaling induces a SNAIL-Smad3/4 complex that negatively regulates E-cadherin in breast epithelial. Of interest, they demonstrated that whereas knockdown of Smad3/4 significantly rescued the repression of E-cadherin by TGF-β1, knockdown of Snail had only a marginal effect, suggesting that other transcriptional repressors may also be required for this repression. Our study also shows that knocking down TBX3 does not completely abrogate TGF-β1–induced migration. It would therefore be worthwhile to examine the effect of knocking down both TBX3 and Snail on E-cadherin levels, as well as to determine its effect on TGF-β1–induced breast epithelial cell migration. It is important to note that TBX3 expression has been demonstrated to be up-regulated in SNAIL-expressing cells (Humtsoe et al., 2012), and our unpublished data show the expression of SNAIL is reduced in TBX3-knockdown cells. The foregoing observations suggest that TBX3 and SNAIL can positively regulate one another, and since they are both able to repress E-cadherin, there may be a complex interplay among these three factors.
MATERIALS AND METHODS
Cell lines and culture conditions
Normal human breast epithelial MCF-12A cells were maintained in complete media consisting of DMEM/Ham's F12 supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 0.1 μg/ml cholera toxin (Sigma, St. Louis, MO), 0.5 μg/ml hydrocortisone (Calbiochem, Billerica, MA), 10 μg/ml insulin (Novorapid; Novo Nordisk, Copenhagen, Denmark), 20 ng/ml epidermal growth factor (Gibco, Life Technologies, Carlsbad, CA), and 5% horse serum (Highveld Biological, Lyndhurst, South Africa). Human keratinocyte HaCaT cells were maintained in DMEM (Highveld Biological, Lyndhurst, UK) supplemented with 10% (vol/vol) FBS, 200 U/ml penicillin, and 100 μg/ml streptomycin. All cell lines were maintained in a 5% CO2 humidified incubator at 37°C. For TGF-β1 treatment, we used 5 ng/ml rhTGF-β1 (R&D Systems, Minneapolis, MN) diluted in 4 mM HCl and 1 mg/ml bovine serum albumin. For transcription inhibitor experiments, cells were pretreated with 5 μg/ml actinomycin D (Sigma-Aldrich) for 1 h and then treated with TGF-β1 for 3 h. For inhibition of de novo protein synthesis experiments, cells were pretreated with 30 μg/ml cycloheximide (Sigma-Aldrich) for 1 h and then treated with TGF-β1 for 3 h.
Plasmid constructs
The human TBX3 promoter luciferase reporter constructs were described previously (Mowla et al., 2011). The pCMV-JunB expression vector was generously provided by Michael Birrer (National Cancer Institute, National Institutes of Health, Bethesda, MD), and the pCMV-Smad3 and pCMV-Smad4 expression vectors were provided by Rik Derynck (University of California at San Francisco, San Francisco, CA).
Western blot analysis
Cells were harvested and protein prepared as described previously (Davis et al., 2008). Primary antibodies used were as follows: rabbit monoclonal anti–phospho-Smad3 (Ser-423/425) antibody (C25A9; Cell Signaling, Beverly, MA), rabbit polyclonal anti-JunB (sc-73), goat polyclonal anti-Smad2/3 (sc-6202), rabbit polyclonal anti-Smad4 (sc-7154; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-TBX3 (42-4800; Zymed, Invitrogen, San Francisco, CA), and rabbit polyclonal anti-p38 (M0800; Sigma-Aldrich).
Quantitative real-time PCR
Total RNA was extracted from cells using the High Pure RNA Isolation Kit (Roche, Basle, Switzerland)). Reverse transcription of RNA was performed according to the manufacturer's instructions using the ImProm-II reverse transcription system (A3800; Promega, Madison, WI). PCR was conducted with the SensiMix Lite Kit (QT 405-05; Quantace, Bioline, Taunton, MA) according to the manufacturer's protocol. Real-time PCR was performed on a LightCycler Version 4 (Roche) using the following parameters: denaturation, 15 min at 95°C; annealing and amplification at 35 cycles, 15 s at 94°C, 20 s at 55°C, and 20 s at 72°C; melting temperature, 15 s at 65°C; and a cooling step, 30 s at 40°C. Each DNA sample was quantified in duplicate, and a negative control without cDNA template was run with every assay to assess the overall specificity. Melting curve analyses were carried out to ensure product specificity, and data were analyzed using the 2−ΔΔCt method. Relative mRNA expression levels were normalized to glucuronidase β (GUSB) for each reaction, with PCR efficiency correction calculated using the formula ratio = EtargetCPtarget(control – sample)/ErefCPref(control – sample), where E is the real-time PCR efficiency and CP is the crossing point. Primers used to amplify the human TBX3 (QT00022484) and GUSB (QT00046046) were purchased from Qiagen (Germantown, MD).
Generation of MCF-12A cell line overexpressing TBX3
The MCF-12A cells were transiently transduced using adenovirus with human TBX3 cDNA cloned into the vector component of an Adeno-X Tet-off expression system 1 (Clontech, Mountain View, CA). The virus was purified using the Adeno-X Maxi purification kit (Clontech). All steps were performed according to the manufacturer's instructions.
Transfection and luciferase assays
Transient transfections were performed using FuGENE HD (Roche) according to the manufacturer's instructions. MCF-12A cells were plated at 1 × 105 cells/well of a 12-well plate, 24 h before transfection. Cells were cotransfected with 400 ng of a TBX3-luciferase reporter plasmid plus 40 ng of the JunB or 30 ng each of the Smad3 and Smad4 expression plasmids or corresponding amounts of an empty-vector plasmid. Luciferase activities were measured using the Luminoskan Ascent luminometer (Thermo Labsystems, Franklin, MA). All transfections were performed in duplicate, and at least three independent experiments were done to confirm reproducibility. Firefly luciferase values were expressed relative to empty-vector control.
Site-directed mutagenesis of the TBX3 promoter
Site-directed mutagenesis was performed on the −141/+38–base pair TBX3 promoter-luciferase reporter as a template using PFU polymerase reagents (Promega). Mutations were introduced as follows (only sense strand is presented, and mutations are indicated with lowercase letters): AP1 (−86/−69), 5′-GGTCCGAAAGgGTagAAGAGCCtcTagAGAGGCCTCCGGC-3′; SBE (−67/−58), 5′-GCCAATCAAGAaGCtTtCGGCTCCCCGC-3′. The integrity of each mutant construct was verified using agarose gel electrophoresis and sequencing.
Generation of a lentiviral-inducible MCF-12A shTBX3 cell line
The MCF-12A cells were stably transduced using a tetracycline-inducible third-generation lentiviral system in which a short hairpin TBX3 RNA (shTBX3) sequence was cloned into the pHIV7-TetRIRESeGFP lentiviral vector (pTIG-shTBX3; Aagaard et al., 2007). Lentiviral infectious particles were produced by transfecting HEK293T cells using the calcium phosphate method. Virus-containing medium was collected after 24 h, and virus was concentrated by ultracentrifugation (OptimaTML-80 XP, Beckman Coulter, Brea, CA). The lenti-MCF-12A cells were produced by transducing the parental MCF-12A cell line using virus concentrate along with 8 μg/ml Polybrene (hexadimethrine bromide; Sigma-Aldrich) at 37°C overnight. Successful transduction was monitored by cells expressing green fluorescent protein (GFP) 48–96 h after infection, using an Axiovert fluorescent microscope (Carl Zeiss, Jena, Germany), and a pure population was obtained by fluorescence-activated cell sorting for GFP expression using a FACSVantage SE cell sorter (Becton Dickinson, Franklin Lakes, NJ). Induction of knockdown transcripts was achieved by treating the cells with 1 μg/ml doxycycline (Sigma-Aldrich), and effective knockdown was assessed by Western blot analysis with appropriate antibodies.
siRNA sequences and transfection
The anti-Smad4 siRNA (SI00076041), anti-TBX3 siRNA (SI00083503), and a control (nonsilencing) siRNA were purchased from Qiagen. Anti-JunB siRNA (sc-35726) was purchased from Santa Cruz Biotechnology. The cells were transfected with siRNAs using HiPerFect (Qiagen) according to the manufacturer's instructions.
Chromatin immunoprecipitation assays
ChIP assays were carried out as previously described (Prince et al., 2004). Briefly, MCF-12A cells treated with or without TGF-β1 for 3 h were fixed in 1% formaldehyde and the chromatin extracted, sonicated, and immunoprecipitated using antibodies against JunB (sc-73), Smad4 (sc-7154), or IgG (negative control; Santa Cruz Biotechnology). DNA precipitated was analyzed by qRT-PCR using human TBX3–specific primer pairs or a nonspecific promoter region (GUSB; Qiagen). Crossing values (Ct) of JunB or Smad4 precipitated DNA were normalized against the Ct values of IgG. Fold enrichment was determined using the ∆∆Ct method: fold enrichment = 2−(∆Ct1 − ∆Ct2), where ∆Ct1 is the ChIP of interest and ∆Ct2 is the IgG.
Immunofluorescence
MCF-12A or HaCaT cells grown on coverslips 3 or 12 h after TGF-β1 treatment were fixed with 4% paraformaldehyde for 10 min at room temperature before permeabilization in 0.2% Triton X-100 for 10 min. Slides were incubated overnight at 4°C with a rabbit anti-TBX3 polyclonal antibody (42-4800; Zymed, Invitrogen, San Francisco, CA) and then with the appropriate secondary antibody coupled to Cy3 (Jackson ImmunoResearch Laboratories, West Grove, PA) and visualized by fluorescence microscopy (Axiovert). All images were processed by Photoshop CS4 Extended Version 11.0.1 (Adobe, San Jose, CA).
Electromobility-shift assay
Nuclear extracts from 3-h TGF-β1–treated MCF-12A cells were prepared as previously described (Smith et al., 2011). A final concentration of 100 nM oligo was labeled with biotin, using the DNA 3′-end Biotinylation Kit (Pierce, Rockford, IL), according to the manufacturer's instructions and annealed. Protein-oligo binding reactions were prepared with nuclear extract, 5× incubation buffer (100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.9, 250 mM KCl, 2.5 mM dithiothreitol [DTT], 10 mM EDTA, 5 mM MgCl2, 20% Ficoll 400), poly(dI/dC) (1 μg/μl), DTT (10 mM), and unlabeled wild type (5′-GCCAATCAAGAGGCCTCCGGCTCCCCGC-3′) or mutant competitor (5′-GCCAATCAAGAAGCTTTCGGCTCCCCGC-3′). Proteins bound to the biotinylated probes were analyzed by PAGE and transferred to nylon membrane. DNA was cross-linked to the membrane using a UV Stratalinker 1800 apparatus (Stratagene, Santa Clara, CA) and processed for chemiluminescence detection using the LightShift Chemiluminescent EMSA Kit (Pierce) according to the manufacturer's instructions. Signals were visualized with a UVP Biospectrum imaging system (VisionWorks LS software; UVP, Upland, CA).
DNA affinity immunoblot assay
Biotinylated DNA oligos and nuclear extract were the same as for EMSA. For each DNA-binding reaction, nuclear extract was incubated with biotinylated DNA probe in binding buffer (20 mM Tris-HCl, pH 7.6, 50 mM NaCl, 1 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 5% glycerol, and 10 ng/μl poly(dI-dC)). The beads were washed with binding buffer and boiled in protein loading buffer (125 mM Tris-HCl, pH 6.5, 0.4% SDS, 10% β-mercaptoethanol, and 20% glycerol). Proteins bound to the biotinylated probes were analyzed by SDS–PAGE, followed by immunoblotting using rabbit monoclonal anti–phospho-Smad3 (Ser-423/425; C25A9; Cell Signaling), rabbit polyclonal anti-JunB (sc-73), or anti-Smad4 (C-19) antibodies (Santa Cruz Biotechnology).
BrdU incorporation assay
BrdU incorporation assays were performed as described previously (Davis et al., 2008) using 10 μM BrdU and a mouse monoclonal anti-BrdU antibody (6 μg/ml; Roche), followed by a secondary IgG coupled to Alexa 488 (1:1000; Molecular Probes, Carlsbad, CA).
MTT assay
Net cell growth was monitored using the MTT assay, according to the manufacturer's instructions (Roche).
In vitro cell migration assay
Cell migration in culture was measured using a two-dimensional in vitro scratch motility assay as previously described (Peres et al., 2010). Briefly, MCF-12A cells were transduced with or without Adeno-TBX3 and treated with or without Dox for 48 h before being grown to 100% confluency. The wound areas were measured over a period of 8 h, and the area of the wound was measured using ImageJ software (National Institutes of Health, Bethesda, MD). The same method was applied to HaCaT siControl and siTBX3 cells.
Statistical analysis
Statistical analysis was performed by using the two-sample t test (Excel; Microsoft, Redmond, WA).
Acknowledgments
This work was supported by grants from the South African Medical Research Council, the National Research Foundation, the Cancer Association of South Africa, the Cancer Research Initiative of South Africa, and the University of Cape Town. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Abbreviations used:
- AD
actinomycin D
- BrdU
5-bromo-2-deoxyuridine
- ChIP
chromatin immunoprecipitation
- Dox
doxycycline
- EMSA
electromobility-shift assay
- IgG
immunoglobulin G
- MT
mutant
- NS
nonspecific
- qRT-PCR
quantitative real-time PCR
- R-Smad
receptor Smad
- SBE
Smad-binding element
- siRNA
small interfering RNA
- TGF-β1
transforming growth factor β1
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-05-0273) on September 11, 2013.
The authors declare no conflict of interest.
REFERENCES
- Aagaard L, Amarzguioui M, Sun G, Santos LC, Ehsani A, Prydz H, Rossi JJ. A facile lentiviral vector system for expression of doxycycline-inducible shRNAs: knockdown of the pre-miRNA processing enzyme Drosha. Mol Ther. 2007;15:938–945. doi: 10.1038/sj.mt.6300118. [DOI] [PubMed] [Google Scholar]
- Bamshad M, et al. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet. 1997;16:311–315. doi: 10.1038/ng0797-311. [DOI] [PubMed] [Google Scholar]
- Boyd SC, et al. Oncogenic B-RAF(V600E) signaling induces the T-Box3 transcriptional repressor to repress E-cadherin and enhance melanoma cell invasion. J Invest Dermatol 133, 1269–1277. 2013 doi: 10.1038/jid.2012.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Kortlever RM, Lingbeek M, Trettel F, MacDonald ME, van Lohuizen M, Bernards R. TBX-3, the gene mutated in ulnar-mammary syndrome, is a negative regulator of p19ARF and inhibits senescence. J Biol Chem. 2002;277:6567–6572. doi: 10.1074/jbc.M110492200. [DOI] [PubMed] [Google Scholar]
- Carlson H, Ota S, Campbell CE, Hurlin PJ. A dominant repression domain in Tbx3 mediates transcriptional repression and cell immortalization: relevance to mutations in Tbx3 that cause ulnar-mammary syndrome. Hum Mol Genet. 2001;10:2403–2413. doi: 10.1093/hmg/10.21.2403. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Davis E, Teng H, Bilican B, Parker MI, Liu B, Carriera S, Goding CR, Prince S. Ectopic Tbx2 expression results in polyploidy and cisplatin resistance. Oncogene. 2008;27:976–984. doi: 10.1038/sj.onc.1210701. [DOI] [PubMed] [Google Scholar]
- Fan W, Huang X, Chen C, Gray J, Huang T. TBX3 and its isoform TBX3+2a are functionally distinctive in inhibition of senescence and are overexpressed in a subset of breast cancer cell lines. Cancer Res. 2004;64:5132–5139. doi: 10.1158/0008-5472.CAN-04-0615. [DOI] [PubMed] [Google Scholar]
- Feng XH, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Feng XH, Lin X, Derynck R. Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15(Ink4B) transcription in response to TGF-beta. EMBO J. 2000;19:5178–5193. doi: 10.1093/emboj/19.19.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, Kuperwasser C. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci USA. 2010;107:21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J, Bernard DJ. SMAD3 and EGR1 physically and functionally interact in promoter-specific fashion. Cell Signal. 2010;22:936–943. doi: 10.1016/j.cellsig.2010.01.019. [DOI] [PubMed] [Google Scholar]
- Hoogaars WMH, Barnett P, Rodriguez M, Clout DE, Moorman AFM, Goding CR, Christoffels VM. TBX3 and its splice variant TBX3 + exon 2a are functionally similar. Pigment Cell Melanoma Res. 2008;21:379–387. doi: 10.1111/j.1755-148X.2008.00461.x. [DOI] [PubMed] [Google Scholar]
- Humtsoe JO, Koya E, Pham E, Aramoto T, Zuo J, Ishikawa T, Kramer RH. Transcriptional profiling identifies upregulated genes following induction of epithelial-mesenchymal transition in squamous carcinoma cells. Exp Cell Res. 2012;318:379–390. doi: 10.1016/j.yexcr.2011.11.011. [DOI] [PubMed] [Google Scholar]
- Ito A, Asamoto M, Hokaiwado N, Takahashi S, Shirai T. Tbx3 expression is related to apoptosis and cell proliferation in rat bladder both hyperplastic epithelial cells and carcinoma cells. Cancer Lett. 2005;219:105–112. doi: 10.1016/j.canlet.2004.07.051. [DOI] [PubMed] [Google Scholar]
- Kang Y, Chen C-R, Massague J, Massagué J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11:915–926. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Lomnytska M, Dubrovska A, Hellman U, Volodko N, Souchelnytskyi S. Increased expression of cSHMT, Tbx3 and utrophin in plasma of ovarian and breast cancer patients. Int J Cancer. 2006;118:412–421. doi: 10.1002/ijc.21332. [DOI] [PubMed] [Google Scholar]
- Lyng H, Brøvig RS, Svendsrud DH, Holm R, Kaalhus O, Knutstad K, Oksefjell H, Sundfør K, Kristensen GB, Stokke T. Gene expressions and copy numbers associated with metastatic phenotypes of uterine cervical cancer. BMC Genom. 2006;7:268. doi: 10.1186/1471-2164-7-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. TGFβ signalling in context. Nat Rev. Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Moses H, Barcellos-Hoff MH. TGF-beta biology in mammary development and breast cancer. Cold Spring Harbor Perspect Biol. 2011;3:a003277. doi: 10.1101/cshperspect.a003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowla S, Pinnock R, Leaner VD, Goding CR, Prince S. PMA-induced up-regulation of TBX3 is mediated by AP-1 and contributes to breast cancer cell migration. Biochem J. 2011;433:145–153. doi: 10.1042/BJ20100886. [DOI] [PubMed] [Google Scholar]
- Peres J, Davis E, Mowla S, Bennett DC, Li JA, Wansleben S, Prince S. The highly homologous T-box transcription factors, TBX2 and TBX3, have distinct roles in the oncogenic process. Genes Cancer. 2010;1:272–282. doi: 10.1177/1947601910365160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince S, Carreira S, Vance KW, Abrahams A, Goding CR. Tbx2 directly represses the expression of the p21(WAF1) cyclin-dependent kinase inhibitor. Cancer Res. 2004;64:1669–1674. doi: 10.1158/0008-5472.can-03-3286. [DOI] [PubMed] [Google Scholar]
- Renard C-A, et al. Tbx3 is a downstream target of the Wnt/beta-catenin pathway and a critical mediator of beta-catenin survival functions in liver cancer. Cancer Res. 2007;67:901–910. doi: 10.1158/0008-5472.CAN-06-2344. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Aladowicz E, Lanfrancone L, Goding CR. Tbx3 represses E-cadherin expression and enhances melanoma invasiveness. Cancer Res. 2008;68:7872–7881. doi: 10.1158/0008-5472.CAN-08-0301. [DOI] [PubMed] [Google Scholar]
- Rowley M, Grothey E, Couch FJ. The role of Tbx2 and Tbx3 in mammary development and tumorigenesis. J Mamm Gland Biol Neoplasia. 2004;9:109–118. doi: 10.1023/B:JOMG.0000037156.64331.3f. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le H-V, Shen L, Anderson SA, Massagué J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Smith J, Mowla S, Prince S. Basal transcription of the human TBX3 gene, a key developmental regulator which is overexpressed in several cancers, requires functional NF-Y and Sp1 sites. Gene. 2011;486:41–46. doi: 10.1016/j.gene.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Tian F, DaCosta Byfield S, Parks WT, Yoo S, Felici A, Tang B, Piek E, Wakefield LM, Roberts AB, Byfield SD. Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res. 2003;63:8284–8292. [PubMed] [Google Scholar]
- Verrecchia F, Vindevoghel L, Lechleider RJ, Uitto J, Roberts AB, Mauviel A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene. 2001;20:3332–3340. doi: 10.1038/sj.onc.1204448. [DOI] [PubMed] [Google Scholar]
- Vincent T, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11:943–950. doi: 10.1038/ncb1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarosh W, Barrientos T, Esmailpour T, Lin L, Carpenter PM, Osann K, Anton-culver H, Huang T. TBX3 is overexpressed in breast cancer and represses p14 ARF by interacting with histone deacetylases. Cancer Res. 2008;68:693–699. doi: 10.1158/0008-5472.CAN-07-5012. [DOI] [PubMed] [Google Scholar]