

Abstract
The single-operation deracemization of 3H indolines and tetrahydroquinolines is described. An asymmetric redox approach was employed, in which a phosphoric acid catalyst, oxidant and reductant are present in the reaction mixture. The simultaneous presence of both oxidant and reductant was enabled by phase separation, and resulted in the isolation of highly enantioenriched starting materials in high yields.
Simple and kinetic resolutions, in which one enantiomer of a racemic mixture is selectively removed from the other, remain important means for the isolation of enantioenriched materials in an industrial setting.1 While attractive in its simplicity, since one enantiomer is being removed, the theoretical yield of the desired optically active material is limited to 50% (eq 1). This restriction has been addressed through dynamic kinetic processes.2 In such a process, both enantiomers of a racemate proceed through a shared common reactive species, which undergoes transformation to a new optically active product (eq 2). However, if optically active starting material is desired, (i.e. a resolution rather than a transformation), this strategy requires additional steps to convert the product back to starting material.3,4
| (1) |
 |
(2) |
 |
(3) |
In order to resolve a racemate in a dynamic sense, one enantiomer of the starting material must be converted to the other. In the case of the direct interconversion of enantiomers proceeding through a single shared reaction pathway, equilibrium is reached between the thermodynamically equivalent starting material and product. The principle of microscopic reversibility dictates that under such circumstances, the racemate must be returned.5 Instead, to realize such a deracemization,6 the forward and reverse reactions must follow distinct mechanistic pathways; in other words, two sets of reagents to drive opposing reactions are necessary. In analogy to a dynamic kinetic transformation, deracemization can be accomplished by proceeding through a shared, achiral intermediate and reforming the stereocenter in an enantioselective manner (eq 3).
One archetypal approach to destroying and generating stereocenters is through oxidation and reduction chemistry. In order to test a redox method for deracemization, a reaction platform must be identified in which a substrate reacts with an oxidant, the subsequent oxidized intermediate reacts with a reductant, and at least one of those steps is rendered enantioselective by a chiral catalyst. As there exists an abundance of effective catalytic methods for enantioselective oxidation and enantioselective reduction of a wide variety of substrates, many such combinations can be identified.
Despite viable candidates for reaction development, accomplishment of a redox-driven deracemization as a single operation is largely precluded by issues of chemoselectivity. A single-operation procedure requires the simultaneous presence of both an oxidant and reductant in the reaction medium, and their direct reaction is likely to be the most kinetically favorable process. Previously reported deracemization reactions have avoided this reactivity issue either through separation into two distinct operations,7 or through the use of enzymes with highly specific reaction profiles to avoid reagent quenching.8 To the best of our knowledge, nonenzymatic single-operation chemically induced deracemization has not been reported.9
We considered that a single-operation deracemization might be accomplished by nonenzymatic means if the relative concentrations of both oxidant and reductant in bulk solution were kept low enough compared to the substrate. This could minimize their direct reaction, and still allow for each reagent’s reaction with the substrate. We have recently reported on the use of chiral phosphate anions for phase-transfer catalysis, in which undesired background reaction is minimized through the use of poorly soluble cationic reagents.10 We hypothesized, in the case of a redox-driven deracemization, that the undesired reaction between oxidant and reductant might similarly be suppressed through phase separation. Specifically, we envisioned that if both an oxidant and a reductant with poor solubility in nonpolar organic solvents were employed, their direct reaction could be disfavored relative to reaction with the substrate.
This reaction scheme would constitute a triphasic system,11 in which the substrate is dissolved in organic reaction solvent, and the oxidant and the reductant exist mainly as solids out of solution (solid-organic-solid phase-separation). Alternatively, if either of the reagents has high water solubility (e.g. if it is a salt), it could be partitioned into an aqueous phase, representing an aqueous-organic-solid phase-separation. By limiting the majority of reagent interaction to the phase interfaces, direct quenching of oxidant and reductant could be minimized while still allowing for their individual reactions with the solubilized substrate, thus permitting the possibility of deracemization.
Chiral phosphate anions have been shown to promote the reaction of insoluble cationic electrophiles with soluble substrates in nonpolar solvents,10 and their conjugate acids are well-known catalysts for the enantioselective reduction of mines through activation toward hydride delivery.12,13 By selecting appropriate reagents (a cationic oxidant and a reductant with minimal solubility in reaction solvent), we initially hoped to show that a single chiral catalyst could mediate not just distinct, but actually opposing processes (Figure 1).
Figure 1.

Schematic representation of a phase-separation enabled deracemization.
3H-indoline 1a was identified as a test substrate, as it met the aforementioned criteria for deracemization: it is readily oxidized to 3H indole 2a by oxopiperidinium salt 314 in toluene under anionic phase-transfer conditions (addition of the chiral phosphoric acid TCYP, 5a, and inorganic base to generate the anionic phosphate); and the enantioselective reduction of 2a by a hydride source, Hantzsch ester 4a in the presence of chiral phosphoric acid catalysts is precedented.15 We were able to reproduce these results using the conjugate acid of the phase-transfer catalyst, 5a.
Though the enantioselectivity of the oxidation step was low under the listed conditions, (s < 1.1) the high selectivity of the reductive second step still permitted the possibility of an effective deracemization. While this feature would allow for this methodology to be performed as two distinct operations, it was our goal to show that by choosing appropriate conditions, an equally effective single-operation deracemization could be developed. The observation that both oxopiperidinium 3 and Hantzsch ester 4a have low solubility in toluene allowed us to evaluate our hypothesis (7.8 mM and 2.0 mM respectively) (Scheme 1).
Scheme 1.

Identification of a reaction system.
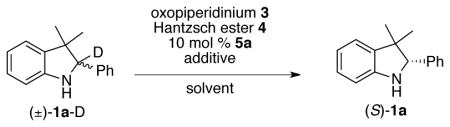
In order to interpret the one-pot deracemization reaction outcome in a more useful manner, deuterated 3H indoline 1a-D, was used, and deuterium erosion (which occurs by oxidation and subsequent hydride delivery from the Hantzsch ester) was monitored as an indicator of conversion. Given the high enantioselectivity of the reduction, we were confident that conversion of the deuterated substrate and enantiomeric excess would track closely. To evaluate the feasibility of solid-organic-solid phase-separation, two equivalents of reductant and oxidant were added to substrate in the presence of 10 mol % 5a in toluene under phase-transfer (basic) conditions. The indoline was reisolated as the race-mate with no detectable deuterium erosion (Table 1, entry 1). The consumption of oxidant, which could be followed qualititatively by the dissipation of its bright yellow color, appeared faster than in the presence of substrate alone, and it is most likely that the oxidant was consumed completely by reaction with reductant rather than substrate. Since basic conditions may impede the enantioselective reduction of any oxidized intermediate, we also ran the reaction under nonbasic conditions (entry 2), though again no deuterium erosion was observed.
Table 1.
Development of reaction conditions for the single-operation deracemization of 3H indolines.
| |||||
|---|---|---|---|---|---|
| entry | Hantzsh ester (equiv.)a | solvent | additive (equiv.) | %D erosion | %ee |
| 1 | 4a (2.0) | PhCH3 | Na2CO3 (2.0) | 0 | 0 |
| 2 | 4a (2.0) | PhCH3 | -- | 0 | 0 |
| 3 | 4a (2.0) | 1:1 PhCH3/H2O | -- | 5 | 4 |
| 4 | 4a (2.0) | 9:1:10 Hex/Et2O/H2O | -- | 68 | 67 |
| 5 | 4a (2.0) | 9:1:10 Hex/Et2O/H2O | Na2CO3 (2.0) | 57 | 42 |
| 6 | 4a (2.0) | 9:1:10 Hex/Et2O/H2O | HCl (1.0) | 84 | 81 |
| 7 | 4b (2.0) | 9:1:10 Hex/Et2O/H2O | HCl (1.0) | 56 | 56 |
| 8 | 4c (2.0) | 9:1:10 Hex/Et2O/H2O | HCl (1.0) | 97 | 92 |
| 9 | 4c (1.5) | 9:1:10 Hex/Et2O/H2O | HCl (1.0) | 94 | 91 |
| 10 | 4d (1.5) | 9:1:10 Hex/Et2O/H2O | HCl (1.0) | 96 | 92 |
| 11b | 4d (1.5) | 9:1:10 Hex/Et2O/H2O | HCl (1.0) | 98 | 94 |
| 12b,c | 4d (1.5) | 9:1:10 Hex/Et2O/H2O | HCl (1.0) | 97 | 94 |
Equimolar amount of oxopiperidinium used;
(S)-TRIP, 5b, used as catalyst instead of (S)-TCYP;
Reaction run in stepwise fashion;
Given the high water solubility of the oxammonium salt (~70 mM),14a we rationalized that aqueous-organic-solid phase-separation might more effectively partition oxidant and reductant. Indeed, when the oxidant was added as an aqueous solution,16 5% deuterium erosion was observed, along with 4% ee (entry 3), indicating that a small fraction of the substrate had been oxidized and subsequently reduced enantioselectively. Switching the organic phase to a 9:1 hexanes/diethyl ether mixture resulted in a significant improvement in both conversion and enantioenrichment (entry 4). This result observation is in line with our phase-separation hypothesis, as both oxopiperidinium 3 (0.07 mM) and Hantzsch ester 4a (0.4 mM) have lower solubility in the less polar organic solvent mixture. Under the analogous basic conditions, though comparable conversion was observed, the indoline was isolated in a significantly deteriorated enantioenrichment (entry 5). Conversely, we found that addition of one equivalent of strong acid led to improved results in the presence of an aqueous phase (entry 6).17,18
By altering the solubility properties of the Hantzsch ester reductant, we hoped to further eliminate reagent quenching and thus improve enantioenrichment of the isolated product. While benzyl Hantzsch ester 4b gave worse results compared to the ethyl analog (entry 7), use of 4-Cl-Bn Hantzsch ester 4c improved conversion of deuterated substrate, also reflected in the product enantioenrichment (entries 8). Furthermore, oxidant and reductant stoichiometry could be lowered from 2.0 to 1.5 equivalents without compromising enantioselectivity (entry 9). 2,6-Cl2-Bn Hantzsch ester 4d (which has a measured solubility of 0.08 mM in 9:1 hexanes/diethyl ether) gave slightly improved conversion (entry 10), and its highly crystalline nature makes it easier to handle than 4c. Use of the chiral phosphoric acid TRIP (5b) in place of TCYP as catalyst led to a further increase in enantioselectivity (entry 11). Under these conditions, the enantioenrichment matched that of a stepwise oxidation-reduction sequence (entry 12), indicating that the starting racemate had been fully converted under reaction conditions. Absolute stereochemistry of the product was confirmed through X-ray crystallography of the hydrochloride salt, and is in agreement with the literature assignment.15
The isotopologue 1a was deracemized to the same degree as 1a-D (Table 2, 2a), and 3H indolines of various substitution patterns were then subjected to the same single-operation deracemization conditions (a slight excess of reductant was used to ensure full reduction), and compared to their stepwise oxidation-reduction to determine efficacy. Spirocyclic products were obtained in good yields and high enantiomeric excesses (1b and 1c). Substitution around the indoline arene was well tolerated, as in the cases of benzoindoline 1d and 5-bromoindoline 1e. Substrates with 2-aryl functionality were subjected to reaction conditions and isolated in good to high enantiomeric excess, including para-(1f, 1j, 1k), meta- (1g, 1h, 1i), and ortho- (1h) substituted 2-arylindolines. Electron rich arenes (1f, 1g) were suitable substrates, though in the case of 1f, the enantio selectivity of the reduction (rather than degree of deracemization) was slightly diminished. Electron poor substrates (1e, 1h, 1i, 1j, 1k) were also isolated in high enantiomeric excess, however in these cases a higher loading of oxidant (1.75 equivalents) and reductant (1.80 equivalents) was needed for full deracemization.19
Table 2.
Substrate scope for substituted 3H indolines.
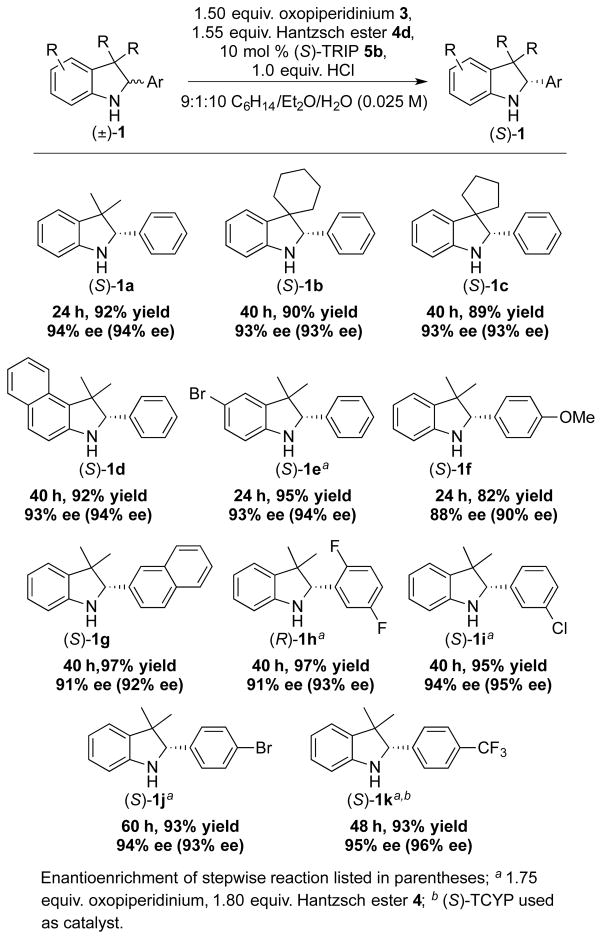
|
Enantioenrichment of stepwise reaction listed in parentheses;
1.75 equiv. oxopiperidinium, 1.80 equiv. Hantzsch ester 4;
(S)-TCYP used as catalyst.
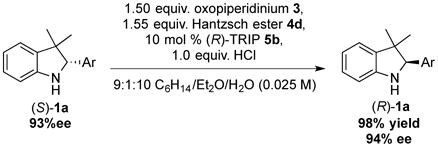
Under similar reaction conditions, substituted tetrahydroquinoline substrates were similarly effectively deracemized (Scheme 2).15b In these instances, three or more equivalents of oxidant and reductant were required since a second oxidation of the intermediate dihydroquinoline to quinoline occurred rapidly. Additionally, highly enantioenriched starting material was converted to the opposite enantiomer under the same reaction conditions as were used for racemic starting material. When (S)-1a (93% ee) was submitted to reaction conditions, the antipode (R)-1a was isolated in excellent yield and enantiomeric excess (eq 4).
Scheme 2.

Substrate scope for substituted tetrahydroquinoilnes.
 |
(4) |
The development of a single-operation deracemization enabled by a three-phase system indicates the ability of phase-separation to suppress undesired reactivity: it has allowed us to develop a class of transformation for which there was no purely chemical protocol. That it has been used to enable what would be a minor or fully inaccessible reaction pathway under more typical reaction conditions (cf Table 1, entries 1, 2), demonstrates its efficacy. Future studies aimed at applying phase separation to develop reaction sequences of incompatible transformations are ongoing.
Supplementary Material
Acknowledgments
We gratefully acknowledge NIHGMS (RO1 GM104534) for financial support. A.D.L. was supported by a Dauben Fellowship from UCB Department of Chemistry. We would like to thank Dr. Gregory L. Hamilton for helpful discussions, William J. Wolf for obtaining crystallographic data, and Dr. Joerg P. Hehn and Ji Lee for experimental assistance.
Footnotes
ASSOCIATED CONTENT
Procedures, spectral data, x-ray crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Pellissier H. Adv Synth Catal. 2011;353:1613. [Google Scholar]; (a) Fogassy E, Nógrádi M, Kozma D, Egri G, Pálovics E, Kiss V. Org Biol Chem. 2006;4:3011–3030. doi: 10.1039/b603058k. [DOI] [PubMed] [Google Scholar]; (b) Keith JM, Larrow JF, Jacobsen EN. Adv Synth Catal. 2001;343:5–26. [Google Scholar]
- 2.(a) Wolf C. Dynamic Stereochemistry of Chiral Compounds: Principles and Application. RSC; 2007. [Google Scholar]; (b) Faber K. Chem Eur J. 2001;7:5004–5010. doi: 10.1002/1521-3765(20011203)7:23<5004::aid-chem5004>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]; (c) Steinreiber J, Faber K, Griengl H. Chem Eur J. 2008;14:8060–8072. doi: 10.1002/chem.200701643. [DOI] [PubMed] [Google Scholar]
- 3.Dynamic kinetic transformations of secondary alcohols to their ester-protected forms have been reported chemo-enzymatically: Persson BA, Larsson ALE, Le Ray M, Bäckvall JE. J Am Chem Soc. 1999;121:1645–1650.Pamies O, Bäckvall JE. Trends Biotechnol. 2004;22:130–135. doi: 10.1016/j.tibtech.2004.01.005.Martín-Matute B, Bäckvall J-E. In: In Organic Synthesis with Enzymes in Non-Aqueous Media. Carrea G, Riva S, editors. Wiley-VCH; Weinheim: 2008. pp. 113–144.More recently by a pure chemical variant was reported: Lee SY, Murphy JM, Ukai A, Fu GC. J Am Chem Soc. 2012;134:15149–15153. doi: 10.1021/ja307425g.
- 4.For examples of chemoenzymatic dynamic kinetic transformations of amines see: Reetz MT, Schimossek K. Chimia. 1996;50:668–669.Paetzold J, Bäckvall JE. J Am Chem Soc. 2005;127:17620–17621. doi: 10.1021/ja056306t.For a review see: Kim Y, Park J, Kim MJ. Chem Cat Chem. 2011;3:271–277.
- 5.(a) Blackmond DG, Matar OK. J Phys Chem B. 2008;112:5098–5104. doi: 10.1021/jp7118586. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG. Angew Chem Int Ed. 2009;48:2648–2654. doi: 10.1002/anie.200804566. [DOI] [PubMed] [Google Scholar]
- 6.While other definitions exist (for example, see ref. 2b–c), deracemization is used in this context in its strict sense, indicating that the obtained product differs from the starting material only in that it is enantioenriched. For a relevant discussions see: Turner NJ. Curr Opin Chem Biol. 2010;2:115–121. doi: 10.1016/j.cbpa.2009.11.027.Gruber CC, Lavandera I, Faber K, Kroutil W. Adv Synth Catal. 2006;348:1789.
- 7.(a) Shimada Y, Miyake Y, Matsuzawa H, Nishibayashi Y. Chem Asian J. 2007;2:393–396. doi: 10.1002/asia.200600354. [DOI] [PubMed] [Google Scholar]; (b) Adair GRA, Williams JMJ. Chem Commun. 2007:2608–2609. doi: 10.1039/b704956k. [DOI] [PubMed] [Google Scholar]
- 8.For selected examples see: Soda K, Oikawa T, Yokoigawa K. J Mol Catal B. 2001;11:149–153.Carr R, Alexeeva M, Enright E, Eve TSC, Dawson MJ, Turner NJ. Angew Chem Int. 2003;42:4807–4810. doi: 10.1002/anie.200352100.Dunsmore CJ, Carr R, Fleming T, Turner NJ. J Am Chem Soc. 2006;128:2224–2225. doi: 10.1021/ja058536d.Carr R, Alexeeva M, Dawson MJ, Gotor-Fernández V, Humphrey CE, Turner NJ. Chem Bio Chem. 2005;6:637–639. doi: 10.1002/cbic.200400329.Voss CV, Gruber CC, Faber K, Knaus T, Macheroux P, Kroutil W. J Am Chem Soc. 2008;130:13969–13972. doi: 10.1021/ja804816a.For a review see: Hall M, Bommarius AS. Chem Rev. 2011;111:4088–4110. doi: 10.1021/cr200013n.
- 9.Enantioenrichment of a racemate based on the physical process of selective crystallization from solution is known for compounds which crystallize as enantiopure conglomerates: Küenburg B, Czollner L, Frohlich J, Jordis U. Org Process Res Dev. 1999;3:425–431.Yagishita F, Ishikawa H, Onuki T, Hachiya S, Mino T, Sakamoto M. Ang Chem Int Ed. 2012;51:13023–13025. doi: 10.1002/anie.201205097.
- 10.Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Science. 2011;334:1681–1684. doi: 10.1126/science.1213918.Phipps RJ, Hiramatsu K, Toste FD. J Am Chem Soc. 2012;134:8376–8379. doi: 10.1021/ja303959p.Wang YM, Wu J, Hoong C, Rauniyar V, Toste FD. J Am Chem Soc. 2012;134:12928–12931. doi: 10.1021/ja305795x.Honjo T, Phipps RJ, Rauniyar V, Toste FD. Angew Chem Int Ed. 2012;51:9684–9688. doi: 10.1002/anie.201205383.Phipps RJ, Toste FD. J Am Chem Soc. 2013;135:1268–1271. doi: 10.1021/ja311798q.Shunatona HP, Früh N, Wang Y-M, Rauniyar V, Toste FD. Angew Chem Int Ed. 2013;52:7724–7727. doi: 10.1002/anie.201302002.For reviews see: Phipps RJ, Hamilton GL, Toste FD. Nature Chem. 2012;4:603–614. doi: 10.1038/nchem.1405.Brak K, Jacobsen EN. Angew Chem, Int Ed. 2013;52:534–561. doi: 10.1002/anie.201205449.Mahlau M, List B. Angew Chem, Int Ed. 2013;52:518–533. doi: 10.1002/anie.201205343.
- 11.(a) Cohen BJ, Kraus MA, Patchornik A. J Am Chem Soc. 1977;99:4165–4167. [Google Scholar]; (b) Davies IW, Matty L, Hughes DL, Reider PJ. J Am Chem Soc. 2001;123:10139–10140. doi: 10.1021/ja016877v. [DOI] [PubMed] [Google Scholar]
- 12.Hoffmann S, Seayad AM, List B. Angew Chem Int Ed. 2005;44:7424–7427. doi: 10.1002/anie.200503062.Storer RI, Carrera DE, Ni Y, MacMillan DWC. J Am Chem Soc. 2006;128:84–86. doi: 10.1021/ja057222n.Li G, Liang Y, Antilla JC. J Am Chem Soc. 2007;129:5830–5831. doi: 10.1021/ja070519w.For some relevant recent reviews see: Rueping M, Sugiono E, Schoepke FR. Synlett. 2010:852–865.Kampen D, Reisinger CM, List B. Top Curr Chem. 2010;291:395–456. doi: 10.1007/978-3-642-02815-1_1.Wang Z, Jiang Z. Asian J Chem. 2010;22:4141–4149.
- 13.During the preparation of this manuscript, a report using a chiral phosphoric acid catalyst and racemic indoline as a reductant led to highly enantioselective reduction of N-arylimines and concomitant oxidative kinetic resolution of the indoline: Saito K, Shibata Y, Yamanaka M, Akiyama T. J Am Chem Soc. 2013;135:11740–11743. doi: 10.1021/ja406004q.
- 14.(a) Bobbitt JM. J Org Chem. 1998;63:9367–9374. [Google Scholar]; (b) Qiu JC, Pradhan PP, Blanck NB, Bobbitt JM, Bailey WF. Org Lett. 2012;14:350–353. doi: 10.1021/ol203096f. [DOI] [PubMed] [Google Scholar]
- 15.(a) Rueping M, Brinkmann C, Antonchick AP, Atodiresei I. Org Lett. 2010;12:4604–4607. doi: 10.1021/ol1019234. [DOI] [PubMed] [Google Scholar]; (b) Rueping M, Antonchick AP, Thiessmann T. Angew Chem Int Ed. 2006;45:3683–3686. doi: 10.1002/anie.200600191. [DOI] [PubMed] [Google Scholar]
- 16.(a) Rueping M, Thiessmann T. Chem Sci. 2010;1:473–476. [Google Scholar]; (b) Scroggins ST, Chi Y, Fréchet JMJ. Angew Chem Int Ed. 2010;49:2393–2396. doi: 10.1002/anie.200902945. [DOI] [PubMed] [Google Scholar]
- 17.(a) Li G, Antilla JC. Org Lett. 2009;11:1075–1078. doi: 10.1021/ol802860u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rueping M, Azap C. Angew Chem Int Ed. 2006;45:7832–7835. doi: 10.1002/anie.200603199. [DOI] [PubMed] [Google Scholar]
- 18.The observation that the reaction is faster under acidic conditions implies that the catalyst may not act as an anionic phase-transfer catalyst. Preliminary investigations indicate that the oxidation does proceed more rapidly in the presence of the phosphoric acid, and further investigations on mechanism are ongoing. For reviews of phosphoric acid catalysis see: Akiyama T. Chem Rev. 2007;107:5744–5758. doi: 10.1021/cr068374j.Terada M. Synthesis. 2010:1929–1982.
- 19.See ref. 14b for a discussion of relative rate of oxidation using oxopiperidinium salts.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.