

Abstract
There have been significant advances with regard to BRAF-targeted therapies against metastatic melanoma. However, the majority of patients receiving BRAF inhibitors (BRAFi) manifest disease progression within a year. We have recently shown that melanoma patients treated with BRAFi exhibit an increase in melanoma-associated antigens and in CD8+ tumor-infiltrating lymphocytes in response to therapy. To characterize such a T-cell infiltrate, we analyzed the complementarity-determining region 3 (CDR3) of rearranged T-cell receptor (TCR) β chain-coding genes in tumor biopsies obtained before the initiation of BRAFi and 10–14 d later. We observed an increase in the clonality of tumor-infiltrating lymphocytes in 7 of 8 patients receiving BRAFi, with a statistically significant 21% aggregate increase in clonality. Over 80% of individual T-cell clones detected after initiation of BRAFi treatment were new clones. Interestingly, the comparison of tumor infiltrates with clinical responses revealed that patients who had a high proportion of pre-existing dominant clones after the administration of BRAFi responded better to therapy than patients who had a low proportion of such pre-existing dominant clones following BRAFi. These data suggest that although the inhibition of BRAF in melanoma patients results in tumor infiltration by new lymphocytes, the response to treatment appears to be related to the presence of a pre-existing population of tumor-infiltrating T-cell clones.
Keywords: BRAF, T cells, TILs, melanoma, vemurafenib
Introduction
Within the past several years, there have been major advances in the treatment of metastatic melanoma, including the use of therapeutics that specifically target oncogenic mutations such as alterations in BRAF. Mutations in BRAF indeed occur in near half of melanoma cases.1,2 The treatment of patients with melanoma harboring BRAF mutations results in a large proportion of objective responses, and several agents targeting mutant BRAF are now approved for use in individuals with stage IV disease.3,4 However, despite dramatic initial responses, the majority of melanoma patients do not achieve a durable response upon treatment with BRAF inhibitors (BRAFi), with disease progression occurring within several months after the initiation of therapy (generally, 6 mo for BRAFi monotherapy, 10 mo when BRAFi are combined with MEK inhibitors).4,5 Alternative therapeutic strategies to achieve long-term clinical responses are therefore urgently needed.
Compelling evidence indicates that oncogenic BRAF mutations contribute to the immune escape of malignant cells and that targeting this pathway may increase the immunogenicity of melanoma. The initial evidence in support of this notion came from in vitro studies demonstrating that the administration of a BRAFi is associated with an augmentation in melanoma-associated antigens as well as with an increased reactivity of antigen-specific T cells.6 More recently, these findings were corroborated in melanoma patients treated with BRAFi, who exhibited not only an increase in melanoma-associated antigens but also a rather therapy-friendly tumor microenvironment, containing decreased levels of immunosuppressive cytokines and vascular endothelial growth factor (VEGF).7-9 Importantly, we and others have reported a significant increase in tumor-infiltrating CD8+ T cells within 10–14 d of the administration of a BRAF inhibitor.7,10 Taken together, these observations suggest a potential synergy between BRAF-targeted agents and immunotherapeutic strategies against melanoma, though several important questions remain unanswered.
Indeed, it is still unclear whether the BRAFi-dependent increase in tumor infiltration originates from the necrotic demise of malignant cells or rather reflects the elicitation of a primary immune response involving antigen-specific T cells. Based on the observations reported above, we hypothesized that the immune infiltrate associated with BRAFi would constitute a primary response, and that the administration of a BRAFi would increase the clonality of tumor-infiltrating lymphocytes (TILs). However, the analysis of TILs by techniques such as flow cytometry in our patient population was prevented by the limited availability of tumor biopsies, especially after the initiation of BRAF-targeted therapy. To circumvent this limitation, we utilized a multiplex PCR strategy to amplify the CDR3 region of the T-cell receptor (TCR) β chain-coding gene, spanning the variable region formed by the junction of the V, D, and J segments and their associated non-templated insertions.11 The resulting 60-bp nucleotide sequence could be used as an identifier or “tag” for a particular clone across different samples. In 8 metastatic melanoma patients harboring BRAF mutations, we sequenced the CDR3 region in tumor biopsies obtained before treatment (day 0) and 10–14 d after the initiation of BRAF-targeted therapy). The goal of the present study was to better define the T-cell infiltrate elicited by BRAFi and hence allow for the development of therapeutic strategies that specifically harness BRAFi-induced tumor infiltration.
Results
We (and others) have previously shown that BRAF inhibition is associated with a significant increase in tumor-infiltrating CD8+ T cells within 10–14 d of treatment.7,10 In order to investigate the clonality of BRAFi-elicited TILs and gain insights into the antigen specificity of such a T-cell response, we sequenced the variable CDR3 region in serial biopsies collected before treatment and 10–14 d after the initiation of BRAF-targeted therapy from 8 metastatic melanoma patients harboring BRAF mutations. The age of these individuals ranged from 25 to 72 y and patients had multiple sites of disease (Table 1). All patients manifested a decrease in the size of targeted lesions after the administration of BRAF inhibitors, as assessed by radiographical methods. In particular, 5 patients achieved a partial response, 1 manifested a complete response and 2 exhibited stable disease (Fig. 1 and Table 1).
Table 1. Patient characteristics.
| ID# | Age | Gender | Site | Treatment | Response | PFS (months) |
|---|---|---|---|---|---|---|
| 7 |
56 |
M |
n, sc |
BRAFi + MEKi |
CR (100%) |
18 |
| 9 |
36 |
M |
br, n |
BRAFi + MEKi |
PR (-45%) |
7 |
| 10 |
37 |
F |
li, n, sc |
BRAFi + MEKi |
SD (-13%) |
3 |
| 11 |
72 |
M |
br, sc |
BRAFi + MEKi |
PR (-80%) |
10 |
| 14 |
25 |
M |
b, n, sc, lu |
BRAFi + MEKi |
PR (-64.9%) |
8 |
| 16 |
43 |
M |
n, sc |
BRAFi + MEKi |
SD (-19.5%) |
11 |
| 19 |
61 |
M |
lu, n, sc |
BRAFi + MEKi |
PR (-48.7%) |
13, ongoing |
| 24 | 70 | F | br, li, lu, n, sc | BRAFi | PR (-53%) | 2 |
Patients with metastatic melanoma harboring BRAFV600E (confirmed by genotyping) were enrolled in clinical trials and treated with a BRAF inhibitor (BRAFi) alone or combined with a MEK inhibitor (BRAFi + MEKi). Abbreviations: B, bone; br, brain; CR, complete response; li, liver; lu, lung; n, nodal; PFS, progression-free survival; PR, partial response; sc, subcutaneous; SD, stable disease.
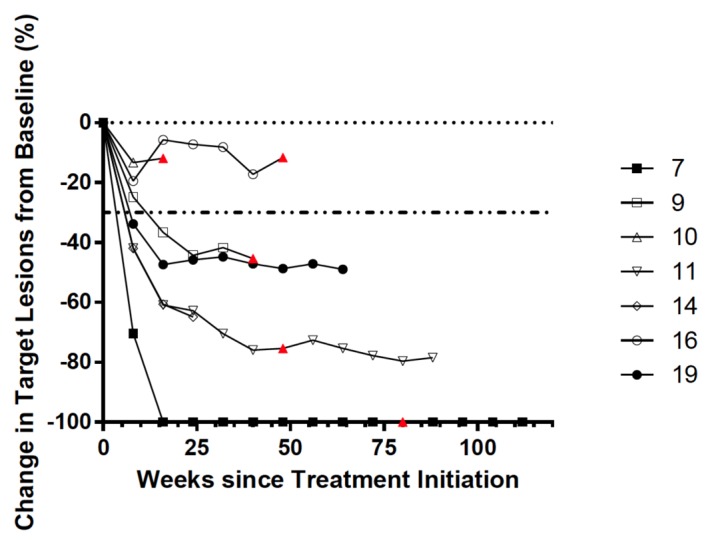
Figure 1. Tumor burden in melanoma patients treated with BRAF inhibitors. The size of neoplastic lesions (assessed the longest linear dimension) in melanoma patients receiving BRAF inhibitors (BRAFi) is plotted against time. The horizontal dashed line indicates a decrease of 30% in target lesion size, which demarks an “objective response” according to the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1.32 Solid data points refer to patients who are currently on trial, clear data points to individual that dropped out of the study. Red triangles mark time-to-progression. Patient 24 was on commercial drugs so official assessment of tumor burden were not performed.
Treatment with BRAFi increases TIL clonality but does not alter the overall proportion of dominant clones
The TIL clonality, a term used to quantify the diversity of clones and frequency of any given clone, was determined using a software provided by Adaptive Biotechnologies. The equation to calculate clonality is 1-(entropy)/log2(# of productive unique reads), where the entropy term takes into account the variation of clone frequencies. In this setting, a maximally diverse cell population in which every sequence is represented one time would be associated with a clonality score of 0, and a perfectly monoclonal population (a theoretical situation approached in some patients with chronic lymphocytic leukemia) would obtain a clonality score of 1. Using the technique described above, we analyzed the clonality of our samples and observed a significant increase in TIL clonality upon BRAFi-based therapy in 7 out of 8 patients (Fig. 2A). Importantly, we wanted to determine if the increase in clonality correlated with response to treatment. To do this, we first took into consideration the dominant clones, i.e., the clones that make up the largest percentage of the total T-cell population, in patients under treatment with BRAFi. We sorted these clones by frequency, each clone being listed once, focused on the top 5% of them, and asked what percentage of the overall T-cell population these clones represented. We determined that the top 5% of clones represented between 40–60% of the overall T-cell population, but no correlation existed between these values and the response of melanoma patients to therapy. Similarly, we observed no correlation with disease outcome when we performed this analysis for the top 2.5%, 1%, or 0.5% of clones (Fig. 2B).

Figure 2. BRAF inhibitors increase the clonality of tumor-infiltrating lymphocytes. (A) Changes in clonality of tumor-infiltrating lymphocytes (TILs) 10–14 d after the initiation of BRAF inhibitors are shown as fold change as compared with pre-treatment values, as a box and whiskers plot (n = 8 patients). The bottom and top of the box represent the 25th and 75th percentile, respectively, while the bar indicates the median value. (B) Percentage of the total TIL population occupied by the 5%, 2.5%, 1%, and 0.5% dominant TIL clones. Patient samples are sorted along the X-axis based on their maximal response to treatment. CR, complete response; PR, partial response; SD, stable disease.
BRAFi-based therapy results in new TIL clones in patients with metastatic melanoma
Next, we wanted to see if the increase in clonality elicited by BRAFi in melanoma patients was associated with the infiltration of neoplastic lesion by novel TIL clones. To this aim, we analyzed the biopsies obtained from patients on BRAF inhibitors for the presence of clones that were absent from pre-treatment biopsies. Interestingly, ~80% of the individual clones detected in the bioptic samples from treated patients were shown to be new clones, suggesting that there is a considerable influx of T cells within the neoplastic lesions of melanoma patients treated with BRAFi (Fig. 3A).

Figure 3. A high proportion of pre-existing clones within the dominant T-cell population after the administration of BRAF inhibitors correlates with improve response to treatment. (A) The percentage of new T-cell clones infiltrating neoplastic lesions after the administration of BRAF inhibitors (BRAFi) was determined by comparing the presence of a tumor-infiltrating lymphocyte (TIL) in biopsies taken before treatment and 10–14 d after the of therapy. Individual patient data are presented with the mean ± SEM for the entire patient cohort (n = 8). (B) Percentage of the top 5%, 2.5%, 1%, and 0.5% dominant TIL clones that were present before the inhibition of therapy. Patient samples are sorted along the X-axis based on their maximal response to treatment. CR, complete response; PR, partial response; SD, stable disease.
The clinical response to BRAFi is associated with a pre-existing population of TIL clones
To further understand the role of TIL clones in therapeutic efficacy of BRAFi, we focused on the top 5%, 2.5%, 1%, or 0.5% dominant clones found in the biopsies of treated patients and examined whether these clones were present within neoplastic lesions before the initiation of therapy. We then correlated the percentage of pre-existing dominant clones with the clinical response to BRAFi. Two groups clearly emerged: patients with a high proportion of pre-existing dominant clones after the administration of BRAFi exhibited a good response, whereas patients who had a low proportion of such clones on treatment had poor disease outcome (Fig. 3B). Specifically, the 3 patients with the lowest proportion of pre-existing dominant clones exhibited the poorest responses of the whole cohort at all time points measured (Fig. 1 and 3B). Of note, there was no clear correlation between the proportion of pre-existing dominant clones upon BRAFi-based therapy and progression-free survival, though the number of patients analyzed was admittedly low. Overall, these data suggest that the response to BRAFi may correlate with the presence of pre-existing dominant T-cell clones, and perhaps also that pre-existing TIL populations may be more important than new TILs in the elicitation of therapeutically-relevant immune responses by BRAF-targeting therapies.
Discussion
The profound clinical response of metastatic melanoma patients to BRAFi demonstrates a significant advance in cancer therapeutics. Although transient, such a response is indeed associated with an increase in melanoma-associate antigens and TILs as well as with tumor microenvironment that supports the effects of therapy.7-10 Interestingly, 95.3% of patients manifest a decrease in disease burden upon treatment with BRAFi, but only 43% display an objective response.3 Although numerous mechanisms of resistance to BRAFi have been described (including BRAF mutations, splice isoforms, copy number alterations, fusions, and allele specific expression),12-23 differences in the immune response elicited by BRAF inhibition have not been well studied. A better understanding of the immunological mechanisms triggered by BRAFi could help us to explain the diversity in both maximal response to therapy and progression free survival of melanoma patients subjected to BRAF-targeting therapies, and may have significant translational implications.
Here, we analyzed the clonality of TILs in melanoma patients on BRAFi. Importantly, the patients included in this study include objective responders and non-responders, both groups demonstrating an increase in T-cell infiltrate. However, differences were seen between these groups of patients when we looked deeply into the origin of the T-cell clones that infiltrated neoplastic lesions in response to BRAFi.
T-cell clonality can be studied by observing changes in the variable V, D, J region of the CDR3-coding sequence within tumor biopsies. This variable region plays a critical role in determining antigen specificity.24,25 Our results confirms previous findings from Yazdi et al., who used laser-capture micro-dissection to isolate different TIL clusters and demonstrated that many T-cell clones with different TCR rearrangements may be detected within one primary malignant melanoma.26 The significant increase in aggregate clonality upon the administration of BRAFi suggests that other factors are involved in response to treatment. The kinetics of such an increase in clonality (after 10–14 d) is not surprising, as Buchholz et al. have shown that a spectrum of T-cell expansion kinetics exists, ranging from slow-dividing, long-lived T cells to fast-proliferating, short-lived cell subsets in which a single T lymphocyte can lead to 70,000 descendants in infection models.27
In our small cohort of patients, there was a striking difference in the relationship between the maximal response to therapy and the presence of new dominant clones. This suggests that the response to BRAFi may rely on pre-existing TILs rather than on the infiltration of neoplastic lesions by new T-cell clones. The specificity of this clonal response requires further investigation. The increase in clonality observed herein may be suggestive of an antigen-specific response, though there are several obstacles to answering this question definitively. First, the amount of bioptic material (especially in the case of treated patients) is quite limited, implying that conventional methods to assess an antigen-specific response such as tetramer staining could not be performed. Moreover, use of technical platforms such as ELISPOT requires a step of in vitro stimulation, which may significantly alter the clonal composition of TILs. Finally, in line with the novelty of the TCR sequencing approach used in this manuscript, we could not rely on a data set describing the antigen specificity of each particular TCR. Such data sets, allowing for the determination of antigen specificity, may become available in the future, along with the diffusion of this technology.
The influence of the anatomical site of disease on TIL clonality is not well understood, and our data do not completely address this question. The majority of biopsies taken in the context of this study were from cutaneous or subcutaneous lesions, and only a few of them from other sites (e.g., lymph nodes). The precise characterization of TCR clonality in cutaneous vs. visceral sites of disease may provide further insights to this issue. Another interesting question is whether the diversity of TIL clonality is expected between distinct biopsy sites or whether it is due to BRAFi. Our data suggest that this diversity is due to BRAF inhibition, although to definitively answer this question one would need to obtain serial biopsies of the same and different lesions from the same patient (both before treatment and in the course of BRAF-targeting therapy), which would be challenging.
Many other questions arise with our data. What is the pre-existing TIL population and why is it present in some patients and not in others? Is it an expansion of such a pre-existing population or an activation of an already dominant yet exhausted population of T cells that mediate therapeutic responses to BRAFi? Is the presence of pre-existing TILs in the lesions of treated patients paralleled by a circulating biomarker that may represent an early indicator of response to therapy?
Of note, we have previously observed a deleterious effect of MEK inhibition on T lymphocytes in vitro, raising the concern that MEK inhibitors may alter T-cell function in patients.6 However, we observed no significant difference in the absolute number of CD8+ TILs between patients receiving BRAFi alone or a BRAFi plus a MEK inhibitor,7 although functional studies on these cells were not performed. These preliminary data suggest that MEK inhibition does not significantly impact T-cell function in patients. In line with our findings, Shindo et al. demonstrated that MEK inhibitors selectively suppress the alloreactivity of naïve T cells in the course of graft-vs.-host-disease without a significant effect on the memory T-cell population.28 This suggests that MEK inhibitors may have a less pronounced effect on memory T cells than on their naïve counterparts. CD8+ TILs are expected to comprise a significant fraction of antigen-experienced memory cells, and may therefore be rather insensitive to MEK inhibition. To date, this hypothesis has not been tested.
Additional translational implications of our findings relate to recent advances in the treatment of melanoma patients with immune checkpoint inhibitors, such as monoclonal antibodies specific for cytotoxic T lymphocyte-associated protein 4 (CTLA4), programmed cell death 1 (PDCD1, best known as PD-1) and CD274 (best known as PD-L1).29-31 The characterization of TIL populations along the lines of our study may provide insights into the propensity of patients to respond to this form of immunotherapy. In addition, this approach may provide a rationale for combination immune checkpoint blockers and BRAFi.
This is the first report to study the clonality of the T-cell infiltrate in melanoma patients treated with BRAFi and its correlation with disease outcome. Given the small number of patients involved in this study, we look forward to other groups testing our conclusions in independent patient cohorts, a work that is currently underway.
Materials and Methods
Patient samples
Metastatic melanoma patients harboring the BRAFV600E mutation (confirmed by genotyping) were enrolled in a clinical trial at Massachusetts General Hospital and treated with a BRAFi alone (vemurafenib) or combined with a MEK inhibitor (dabrafenib + trametinib) (Table 1). Tumor biopsies were obtained before treatment (day 0) and 10–14 d after treatment initiation. Formalin-fixed tissues were analyzed to confirm the presence of viable tumor cells upon hematoxylin and eosin (H&E) staining. Tissues were immediately processed for genomic DNA extraction. Explicit consent on the procedure was obtained as per the institution review board-approved protocol.
Preparation of genomic DNA
Clinical samples were received in the form of tumor tissue. For DNA purification, tissue was digested overnight at 55°C in extraction buffer (10 mM Tris pH 8.0, 25 mM EDTA, 0.5% sodium dodecyl sulfate, 100 mM NaCl) supplemented with 20 mg/mL proteinase K. Residual debris were removed by the addition of 80 μL of Protein Precipitation Solution (Promega #A795A) and centrifugation. Genomic DNA was precipitated by adding an equal volume of isopropanol. The DNA pellet was washed with 70% ethanol, air-dried and re-dissolved in sterile water.
CDR3 sequencing and clonality
CDR3 regions were sequenced on an Illumina HiSeq sequencer with at least 5-fold coverage by ImmunoSEQ™ sequencing (Adaptive Biotechnologies). The sequencing assay relies on a multiplex PCR reaction with forward primers annealing to each V segment and reverse primers annealing to each J segment, resulting in the selective amplification of rearranged VDJ segments from each cell in a format compatible with sequencing.11 Clonality scores were calculated for our patient samples using a software by Adaptive Technologies, clonality being defined as 1-(entropy)/log2(# of productive unique sequences), where the entropy term takes into account of the varying clone frequency. Clonality values are given through the ImmunoSeq Analyzer software, according to which a maximally diverse population is associated with a clonality score of 0 and a perfectly monoclonal population with a clonality score of 1. Analyzing the clones as a percent of total was performed to control for the number of unique reads in each specific patient sample.
Statistics
Statistical analyses were performed using the GraphPad Prism software or the R statistical package.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Citation: Cooper ZA, Frederick DT, Juneja VR, Sullivan RJ, Lawrence DP, Piris A, Sharpe AH, Fisher DA, Flaherty KT, Wargo JA. BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. OncoImmunology 2013; 2:e26615; 10.4161/onci.26615
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/26615
References
- 1.Flaherty KT, Hodi FS, Bastian BC. Mutation-driven drug development in melanoma. Curr Opin Oncol. 2010;22:178–83. doi: 10.1097/CCO.0b013e32833888ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. BRIM-3 Study Group Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. Combined BRAF; MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 7.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khalili JS, Liu S, Rodríguez-Cruz TG, Whittington M, Wardell S, Liu C, Zhang M, Cooper ZA, Frederick DT, Li Y, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18:5329–40. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, Chen JQ, Li HS, Watowich SS, Yang Y, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19:393–403. doi: 10.1158/1078-0432.CCR-12-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, Scolyer RA. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–94. doi: 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- 11.Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR, Carlson CS, Warren EH. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med. 2010;2:47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. 2011;364:772–4. doi: 10.1056/NEJMcibr1013704. [DOI] [PubMed] [Google Scholar]
- 16.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–5. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, Kellet M, Storm PB, Resnick AC. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci USA. 2013;110:5957–62. doi: 10.1073/pnas.1219232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–61. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009;106:20411–6. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov 2013; 3:338-49;; 10.1158/2159-8290.CD-12-0313 [DOI] [PMC free article] [PubMed]
- 23.Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE, Garraway LA. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov 2013; 3:350-62;; 10.1158/2159-8290.CD-12-0470 [DOI] [PMC free article] [PubMed]
- 24.Katayama CD, Eidelman FJ, Duncan A, Hooshmand F, Hedrick SM. Predicted complementarity determining regions of the T cell antigen receptor determine antigen specificity. EMBO J. 1995;14:927–38. doi: 10.1002/j.1460-2075.1995.tb07074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goyarts EC, Vegh Z, Kalergis AM, Hörig H, Papadopoulos NJ, Young AC, Thomson CT, Chang HC, Joyce S, Nathenson SG. Point mutations in the beta chain CDR3 can alter the T cell receptor recognition pattern on an MHC class I/peptide complex over a broad interface area. Mol Immunol. 1998;35:593–607. doi: 10.1016/S0161-5890(98)00056-X. [DOI] [PubMed] [Google Scholar]
- 26.Yazdi AS, Morstedt K, Puchta U, Ghoreschi K, Flaig MJ, Rocken M, Sander CA. Heterogeneity of T-cell clones infiltrating primary malignant melanomas. J Invest Dermatol. 2006;126:393–8. doi: 10.1038/sj.jid.5700082. [DOI] [PubMed] [Google Scholar]
- 27.Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Gräf P, Verschoor A, Schiemann M, Höfer T, Busch DH. Disparate individual fates compose robust CD8+ T cell immunity. Science. 2013;340:630–5. doi: 10.1126/science.1235454. [DOI] [PubMed] [Google Scholar]
- 28.Shindo T, Kim TK, Benjamin CL, Wieder ED, Levy RB, Komanduri KV. MEK inhibitors selectively suppress alloreactivity and graft-versus-host disease in a memory stage-dependent manner. Blood. 2013;121:4617–26. doi: 10.1182/blood-2012-12-476218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]