

Abstract
Atoh1 is a transcription factor that regulates neural development in multiple tissues and is conserved among species. Prior mouse models of Atoh1, though effective and important in the evolution of our understanding of the gene, have been limited by perinatal lethality. Here we describe a novel point mutation of Atoh1 (designated Atoh1trhl) underlying a phenotype of trembling gait and hearing loss. Histology revealed inner ear hair cell loss and cerebellar atrophy. Auditory Brainstem Response (ABR) and Distortion Product Otoacoustic Emission (DPOAE) showed functional abnormalities in the ear. Normal lifespan and fecundity of Atoh1trhlmice provide a complementary model to facilitate elucidation of ATOH1 function in hearing,central nervous system and cancer biology.
Introduction
Atoh1 (Math1), a basic helix-loop-helix transcription factor homolog of the Drosophilaatonal gene is involved in a wide range of developmental changes in mice and humans.A critical role in neurogenesis has been shown for Atoh1 in proper hindbrain development [1]. Unconscious proprioception is dysfunctional in mice lacking Atoh1, with cerebella missing the external granule layer and bereft of the granule cells essential for proper functioning [2]. Atoh1 has also been implicated in the creation and interplay of complex neural circuits that link hearing, proprioception and arousal with tasks like respiration, likely through a pathway of glutamatergic neurons [1]. Secretory cells in the gut, such as goblet, enteroendocrine and paneth cells, arise from a secretory cell lineage through aAtoh1-dependent pathway [3]. Atoh1 behavesas anoncogene in medullablastomas[4] but acts as a tumor suppressorgene in adenomatous polyposiscarcinoma[5]. Apparently, the Atoh1 gene plays many significant roles in the development of several organs, systems and diseases.
Previous studies regarding the function of the Atoh1 gene are mostly on mutants generated from a gene-driven approach (manipulating known sequences through gene-gene targeting). It is shown that Atoh1 is a positive regulator of hair-cell differentiation during cochlear development[6], [7].Atoh1-homozygous-null mice lack hair cellsin the vestibular organs and cochleae and was the first model demonstrating the phenotype associated with Atoh1 loss of function [7].Adenovirus-mediated overexpression of Atoh1 has been reported to produce a large number of hair cells in cultured adult utricular maculae from rats[8] and in cochleaefrom mature guinea pigs[9]. Earlier studies regarding Atoh1 mutant mice have been limited to the prenatal stage because the targeted null mutants die shortly after birth due to respiratory failure [2]. A few studies circumvented lethality in Atoh1 deficiency using a conditional knockout strategy [10], [11], but the authors of the Pan paper reported possible leakiness in expression resulting from incomplete recombination and a lifespan of a month or less. The conditional knockout strategy has been invaluable in understanding in vivo function of Atoh1 and has played a significant role in helping the scientific community elucidate the primary role of Atoh1 in development. However, some disadvantages do exist with this approach, such as the efficiency of gene deletion varying depending on the gene locus position and the Cre activity. It is also time consuming and expensive, requiring two transgenic lines (Flox and Cre mice), to generate conditional knockout mouse models [12].
In contrast to a gene-driven approach, a phenotype-driven approach to study gene function is an alternate method with a different set of advantages and drawbacks. This tactic overcomes the lethality early in life and is a similar way in which human mutation is generated. The phenotype-driven approach includes spontaneous and induced mutations such as those that are chemically induced. Here, we report anethylmethanesulfonate (EMS) induced hypomorphicmutant Atoh1trhlmouse model with a normal life span and cerebellar atrophy accompanied by a failure to form a morphologically and functionally normal inner ear. This mouse mutant could be useful model to explore human disease associated with the Atoh1 gene due to a natural development process and having postnatal vitality. It is important to understand the expression and regulation of Atoh1, not just for the generation of hair cells in the inner ear, but also for the possible contribution of this gene to diseases such as medulloblastoma in which the Atoh1 gene is thought to be improperly activated [11]. We can take advantage of this new AtohItrhl mutant mouse model to investigate many aspects of physiology, molecular biology, proteomics and the development of a variety of hearing and balance pathologies.
Materials and Methods
Mice
The mutant strain was first established at The Jackson Laboratory (JAX, Bar Harbor, Maine) from a mutagens program as previously described [13]. In brief, culture media contains embryonic stem (ES)cells of 129/Sv ×C57BL/6Jwere treated with the point mutagen ethylmethanesulfonate (EMS). Then the treated ES cells were injected into C57BL/6J blasto-cysts by standard methods. Chimeric mice generated from EMS-treated ES cells were mated with C57BL/6J partners to recover the recessive mutations. The trhl phenotype was first identified from the third generation mice with a ‘click box’, where mice showed a reduced Preyer reflex (pinna twitch) and then were confirmed by ABR testto have defective hearing. Progeny of later generations were noticed to have leg tremors. The Atoh1trhl/trhlmutation was moved to the C57BL/6J mouse background by backcrossing 10 generations.
Later, this mouse strain was imported to Case Western Reserve University. Mice were housed in ventilated racks at 21°C in a 12 h light–dark cycle with food and water given ad libitum. The mice used as control groups were either C57BL/6J wild type or Atoh1trhl/+, depending on the stage of the experiment, and were all managed in the same fashion as the Atoh1trhl/trhlmice. All animal protocolswere approved by the Institutional Animal Care and Use Committee of Case Western Reserve University (R01DC007392).
Genotyping Assay
A DNA pooling method was used as previously described [14]. Genomic DNA was prepared from tail tips of Atoh1trhl/trhl,Atoh1trhl/+and C57BL/6J (B6)wild-type mice. Briefly, 2-mm tail tips were each digested with 0.3 ml of 50 mMNaOH in a 0.5 ml Eppendorf tube at 95°C for 10 min. A total of 26 µl of 1 M Tris-HCl was then added to each tube. The mixtures were centrifuged at 12,000 x g for 5 min and the DNA concentration in supernatants was measured using a BioPhotometer (Eppendorf AG, Hamburg, Germany). Genomic DNA screening to identify the alteration in the Atoh1 gene was performed using PCR with primers designed to detect the Sau96 I site present in wild-type and absent in mutant mice: (Sau494 5′-GAAGGTGATGGTGGTCATTTTT-3′and Sau756 5′-ACAGGTGAATGGGGTACAGAAG-3′). Also,thesingle exon of the Atoh1 gene was sequenced for detailed comparison between mutants and controls using Sequencher 4.0. (http://genecodes.com; Gene Codes Corporation, Ann Arbor, MI, USA). To confirm the mutation, reverse transcription PCR was run with initial RNA from Atoh1 mutant and wild-type control mice. Mice around 2 months of age were sacrificed and RNA (DNA-free) was collected from the inner ears and temporal bones. cDNA was synthesized using the SuperScriptTM First-Strand Synthesis System (Invitrogen). The same gene-specific primers for the single exon of Atoh1 were used in RT-PCR as described above for genotyping. Ten microliters of each PCR product were analyzed by agarose gel electrophoresis after exposure to the restriction enzyme Sau96 I.
cDNA preparation, RT-PCR, and quantitative RT-PCR
Atoh1trhl/trh1, Atoh1trhl/+and B6 wild-type mice were sacrificed under Avertin anesthesia conditions at 1 day or 3 days of age. The inner ears (50 mg) were quickly isolated for total RNA and cDNA preparation as described in the previously [4], [15]. One µg of total RNA from each sample was used as template for cDNA synthesis. The 20 µl reaction mixture contained 50 mMKCl, 10 mMTris-HCl, pH 9.0 (at 25°C), 0.01% Triton X-100, 2 mM MgCl2, 250 nM of each primer (forward and reverse), 200 µM dNTP, 1 µl of cDNA and 0.5 U of Taq DNA polymerase (New England Biolabs, Inc., Ipswich, MA, USA). Quantitative-RT-PCR using SYBR green PCR mix (Applied Biosystems) in an Applied Biosystems 7300 Real-Time PCR system) was performed using the following primers: GAPDH (5′-AAC TTT GGC ATT GTG GAA GG-3′ and 5′-GTC TTC TGG GTG GCA GTG AT-3′), Atoh1 (5′-GTA AGG AGA AGC GGC TGT G-3′ and 5′-AGC CAA GCT CGT CCA CTA-3′), Rab15 (5′-GGC TTG GGC TGT GTC ATT G-3′ and 5′-GGC AGA CAG GCC AGG AAA-3′), Selm (5′-TCG TGC TGT TAA GCC GAA ATT-3′ and 5′-CCG GGT CAT TTG GCT GAG T-3′), and Barhl1 (5′-GAG CGG CAG AAA TAC CTG AG-3′ and 5′-GGT CCA GAT TGG AAA CCA GA-3′).
Generation of Cytocochleograms
Cytocochleograms were obtained bythe following method described previously[15]. Briefly, the organ of Corti was carefully microdissected out and mounted in glycerin on glass slides. The surface preparations were stained with Ehrlich's hematoxylin solution and examined under a light microscope.Hair cells were counted as present if the cell body intact. Inner and outer hair cell counts were made over 0.12-0.24-mm intervals of the organ of Corti, beginning at the apex and continuing toward the base. Individual cochleograms were constructed to show the percentage of hair cells missing as a function of distance from the apex. Composite cochleograms were generated by calculating the means at each distance point for each genotype, averaging (n = 5) mice of each genotype.
Histology and SEM
Mice were subjected to an overdose of carbon dioxide, decapitated, and the inner ears quickly removed. The tympanic bullae were opened and a small hole was made in the round window of each exposed cochleae. 4% paraformaldehyde fixative was gently perfused through the round window, followed by immersion of cochleae in the same fixative. The cochleae were decalcified when needed for up to 6 h with Cal-Ex solution. The organ of Corti was carefully isolated by microdissection and mounted in glycerin on glass slides. The surface preparations were stained with Ehrlich's hematoxylin solution. Over 0.12–0.24 mm intervals under light microscopy (Leica DFC500, Leica Microsystems, Wetzlar, Germany) hair cells were counted as present if the cell body and cuticular plate were intact. This procedure was performed to generate the cytocochleograms both to determine where hair cells were missing along the length of the cochlea and also to examine hair cell loss over a time course.
For the silver nitrate staining, mice from each group were sacrificed by CO2. Organs of the cochlea and vestibular system were dissected, and the round window and oval window were pierced. Another small hole was drilled on the apical surface of the cochlea. 1.0% silver nitrate solution was gently perfused through the cochlea for 1 minute and the procedure was repeated three times. Specimens were fixed in 10% formalin for 2 h and exposed to sunlight for approximately 1 h to finish all procedures of this staining. Stained tissue was then examined under the microscope for hair bundle orientation.
Mice cerebella were dissected and embedded in paraffin wax after following the appropriate dehydration and embedding protocol. Tissue was then sectioned and stained with hematoxylin and counterstained with eosin after deparaffinizing and rehydrating sections. Tissue was also stained with cresyl violet to better view neurons [16]. Sections were then analyzed and photographed under the microscope at increasing magnifications.
For the SEM images,inner ears from Atoh1trhl/trhl and wild-type mice weredissected after transcardial perfusion with 4% paraformaldehyde (PFA) and then immersed in 2.5% glutaraldehyde in 0.1 M phosphate buffered saline (PBS, pH = 7.2) for 4 hours at 4°C. Dissection was performed to expose the middle ear cavities and inner ear basilar membrane. After post-fixation in 1% OsO4 in 0.1 M PBS (pH = 7.2, 1 hour), samples were washed in 0.1 M PBS (pH = 7.2), dehydrated in increasing concentrations of ethanol, dried in a BAL-TEC CPD 030 critical point dryer (BAL-TEC GmbH, Witten, Germany) according to manufacturer's instructions, and analyzed on a Hitachi S-4500 scanning electron microscope (Hitachi, Tokyo, Japan) at 5 kV.
Evaluation of auditory function in mice
The ABR methodology has been previously described and includes criteria for evaluating hearing loss in response to various stimuli [17]. ABR thresholds were obtained for each stimulus (clicks, and 8, 16, and 32 kHz tone-bursts) by reducing the sound pressure level (SPL) at 10 dB steps and finally at 5 dB steps up and down to identify the lowest level at which an ABR pattern could be recognized. ABR was measured at various intervals for Atoh1 mutant and control mice (at ages ≥14 days) up until 8 months of age (n = 5 per group).
To test the function of outer hair cells of different mice at different time points, we used DPOAE measurement, which was conducted for pure tones from 2 to 36 kHz. Frequencies were acquired with an F2-F1 ratio of 1∶22. Five stimulation levels ranging from 65 to 25 dB SPL in 10 dB steps were used to assay DPOAE at multiple ages (n = 5 per group).
Results and Discussion
Gross phenotype, genetic mapping and identification of the Atoh1trhl mutation define the first viable normal-life-span mutant without conditional gene targeting
The trhlmodel is the result of anEMS induced hypomorphic allele of the Atoh1 geneon the C57BL/6J (B6) mouse strain[13]. The phenotype of these hypomorphic mutant mice includes a trembling gait and hearing loss (symbol: trhl). Homozygous mutant mice exhibit a shaky gait at weaning age and a progressive hearing loss starting at 3 weeks of age (as we tested) or earlier and culminating in near-total deafness by 8 months, with a lifespan that is comparable to wild-type mice. This lifespan is in comparison to two previously described targeted mutations of Atoh1which affected inner-ear hair cells and the cerebellum as well as the arousal system, with mice homozygous for the null mutations dying of respiratory failure shortly after birth[2], [7].
To genetically map trhl, we generated 230 F2 progeny mice from a cross of B6-trhl x CAST/Ei. We used a pooled DNA strategy[1], [14], [18]to localize the mutation to chromosome 6. We then refined the map position of the mutation to 29 cM from the centromere and narrowed the candidate gene interval to a 6 cM region between D6Mit364 and D6Mit71 where there are three genes that related to cerebellum like ataxia. These three genes are glutamate receptor ionotropic delta 2 (Grid2), catenin (cadherin associated protein) alpha 2 (Ctnna2) and Atoh1. We tested thethree genes as a candidate gene for thetrhlmutation by sequence comparison.
We first found no differences for all exons of Grid2 and Ctnna2as well as 500 bp of the upstream promoter regions for both genes. Thenwe identified a G-Atransition mutation at the 600th base pair (bp) position of the Atoh1 gene (Fig. 1A) in DNA from mutant mice.The mutation resulted in an amino acid substitution ofmethionine (M) to isoleucine (I) at the 200th amino acid residue in the helix-loop-helix (HLH) DNA binding domain, which is highly conserved between species (Fig. 1D). ASau96 I restriction site was lost secondary to the mutation; accordingly, we developed a PCR assay to distinguish trhl/+ (H), trhl/trhl(M)and +/+ (W) genotypes. Primers Sau494 and Sau756 were used for PCR identification of wild-type and mutant genotypes and Sau96 I-digested products were run on an agarose gel (Fig. 1B). Two products were seen in the +/+ wild-type samples, which had the restriction site present, while only a single band was produced in the mutant trhl/trhlsamples. The heterozygous mouse yields three PCR fragments, having one allele of each type. The trhlmutation might cause slight changes in final protein configuration and/or 3D structure of the protein with the mutation from methionine to isoleucine, whichmay allow partial development of hair cells and the central nervous system to produce a distinctive phenotype without decreasing the viability of the animal.
Figure 1. Identifying the mutation in Atoh1trhl/trhl mice.
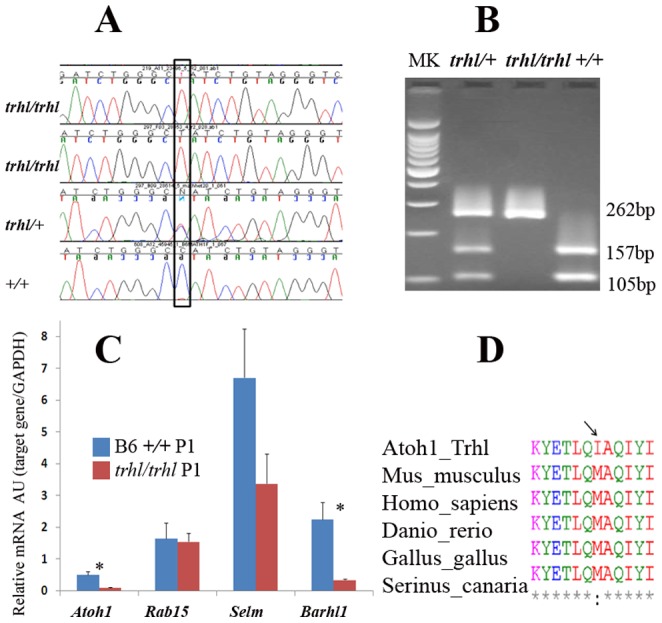
A)Sequencing comparison over the candidate gene interval reveals a G-A transition at nucleotide 600 in the 6 cM region of chromosome 6 that includes the Atoh1 gene. The black box indicates the mutated nucleotide. B) The Sau96 I restriction site (GGNCC; from bp 596 – 600) was altered in the mutant, facilitating genotyping. Lane MK, 100 bp ladder. PCR of a 262 bp fragment in the mutation-containing region of the Atoh1 gene, followed by Sau96 I digestion of the PCR product, revealed digestion of product into two smaller bands of 157 bp and 105 bp in the wild-type mice (+/+) and one intact 262 bp fragment in the homozygous mutant mice (trhl/trhl) due to the absence of the restriction site. The heterozygous mutant (trhl/+) displayed three bands with one mutated allele lacking the restriction site and one wild-type allele with the restriction site present. C) Gene expression levels were measured by quantitative RT-PCR. All samples were run in triplicate and normalized to the GAPDH relative expression level. The 2−ΔΔCT method was used for statistical analysis. AU, arbitrary units. mRNA levels were significant lower for mutant (trhl/trhl) mice than wildtype at P1 for Ahoh1 and Barhl1 genes at P21 (*P<0.05). Error bars were generated by calculating the SEM, (n = 3 mice, 6 ears, at postnatal 1 day). D) Analysis of the conservation of methionine (M) across species and the change to isoleucine (I) in Atoh1 mutants. M to I substitution is shown by the arrow at the 200th amino acid residue in the helix-loop-helix (HLH) DNA-binding domain of the ATOH1protein in the trhlmutant mouse. M in the ATOH1 protein is conserved at position 200 across species.
mRNA expression of several genes specific to Atoh1 targets was down-regulated in the inner ear tissue from Atoh1trhlmice
To determine theexpression of the genes known to be direct Atoh1 targets, we examined the mRNA levels of Atoh1, Rab15, Selm and Barhl1 in inner ear tissue from thehomozygous mutant mice (trhl/trhl P1) (n = 3 mice, 6 ears, at 1 day) compared tothe wild-type mice (B6 +/+ P1)(n = 3 mice, 6 ears, at 1 day). The mRNA of Atoh1, and Barhl1 were significantly down-regulated in thehomozygous mutant mice compared to wild-type mice at 1 day (shown in Fig. 1C). However, there was no significant difference in mRNA levels of Rab15 in inner ear tissue between mutant mice and wild-type mice. It is widely acceptedthat Barhl1 is one direct target of Atoh1 in the developing neural tube or cerebellum[5], [12], [19], which is sufficient for the initial generation of those hair cells even in the absence of Barhl1expression. The expression of Selm was down-regulated in trhl/trhl samples, though not significant, which were in accordance with previously report[13], indicating a G-Atransition mutation affects the regulating target genes of Atoh1.
Histologic and scanning electron microscope examinationof hair cells
Surface preparations from mouse cochleae were stained with Ehrlich's hematoxylin solution and examined under a light microscope.Individual cytocochleograms were constructed for each mouse to show the percentage of hair cells missing as a function of distance from the apex, relative to age-matched heterozygous mice. The study revealed that in homozygous Atoh1trhlmice, outer hair cells (OHC) and inner hair cells (IHC) exhibited losses throughout the cochleae that were already present at the initial 3weeks time point, and exhibitedsimilar, but progressive, losses at 7 weeks of age as compared to heterozygote controls, shown in the composite figure using the means (Fig. 2).Hair cell loss was greatest at the mid-basal (greater than 40% distance from apex) turn of the cochleae in both three-week-old (Fig. 2A) and seven-week-old homozygous mutants (Fig. 2C). Cochlear surface preparations stained with silver nitrate (AgNO3) were used to examine loss of stereociliary bundles in OHC and IHC.Figure 3A shows a representative cochleae stained with silver nitrate to examine orientation and structure along the full length of the organ, with 3B showing a magnified view. In 3A, the arrows and arrowheads indicate sites of missing cells, while the arrows in 3B show direction of orientation of hair cells. There are more cells missing in the trhlmutant than in controls and the hair cells present are oriented haphazardly. Hair cells in the organ of Corti were viewed under SEM (Fig. 3C) to examine structure more closely. SEM views show the classic disorientation of hair bundles and missing cells in homozygous mutants as compared to the wild-type controls, confirming the inner ear morphological dysfunction of the model.
Figure 2. Cytochochleograms show hair cell loss in mutants.

Cytocochleograms indicate percentage of outer (OHC) and inner (IHC) hair-cell loss as a function of percent distance from the cochlear apex, where 0% distance describes the most apical region of the cochlea and 100% distance describes the most basal region of the cochlea, excluding the cochlear hook. Hair-cell loss is shown for three-week-old trhl homozygotes (n = 5) (A) and heterozygotes (n = 5) (B) and for seven-week-old trhl homozygotes (n = 5) (C) and heterozygotes (n = 5) (D). Data points represent the mean percentage, with error bars indicating standard deviation. There is clear evidence of hair-cell loss to a much greater degree in the homozygous mutants as compared to the heterozygous controls.
Figure 3. Stained cochlear sections and SEM reveal hair cell loss and altered morphology.

Representative cochlear sections fromAtoh1trhl/trhland control Atoh1trhl/+ mice were stained with AgNO3to examineOHC and IHC morphology.(A) At P21, sections were compared from all three turns of the cochleae in heterozygous (n = 3, representative view of Atoh1trhl/+, upper panels) and homozygous (n = 3, representative view of Atoh1trhl/trhl, lower panels) mutant mice. Arrows indicate sites of OHC absence; arrowhead/triangles indicate sites of IHC absence. (B) At P21, representative comparative high magnification views of sections from mutant mice and heterozygous controls are shown from the mid-turns of the cochleae, which showed the greatest degree of hair-cell loss. Arrows indicate orientation of the stereociliary bundles. Thedisorientation is clear in the Atoh1trhl/trhlmice with arrows aligned much less uniformly than in the control. (C) Cochleae were analyzed under the scanning electron microscopeto reveal a more detailed view of stereociliary bundle orientation. The hair-cell bundle appears as a light-colored, V-shaped (normal morphology, seen in the top panel) band within each hair cell. The lower panel shows the disorientation of bundles and missing hair cells in the homozygous mutant mice in contrast to the consistent structure of bundles in heterozygous mutant mice.
Structural alterations in cerebella of Atoh1trhlmice
The structure and histology of the cerebella of homozygous Atoh1trhl mice were abnormal compared to normal littermates. Atoh1trhl/trhl mice lacked robust external granular cell layers (EGL), as shown by H&E staining and cresyl violet staining. The homozygous mutant cerebella were reduced in size and lacked surface foliation (Fig. 4). Upon visual inspection of the samples (n = 3 per group), in contrast to the relative lack of granule neurons, relatively normal numbers of Purkinje cells and neurons of the deep cerebellar nuclei were present in Atoh1trhl/trhl mice. However, the Purkinje cells, which normally localize beneath the EGL, did not form a distinct, organized layer as seen in 4E.Some Purkinje cells localized improperly at the periphery of the cerebellum while a significant subpopulation even failed to migrate from their initial central area as is evident in the representative section in Figure 4F. Staining with glial fibrillary acidic protein[20] and nestin[21], both of which label glial cells, demonstrated properly localized radial glial cells and no excessive gliosis throughout the cerebella (data not shown). These results suggest that Atoh1is essential for formation of the EGL, likely through regulation of downstream effectors. The disorganization of the Purkinje cells in particular may be a function of either the Atoh1 gene's effect on cell migration or due to the direct disturbance of the EGL. In Atoh1trhl/trhl mice, absence of the EGL led to a lack of foliation of the cerebella, which was typically observable in stained sections from normal embryos (n = 3 per group) starting from day 18 of the embryonic period (E18). The observations in this mutant mouse are thus consistent with those of Ben-Arie and colleagues in the Atoh1-null mutant mouse with concern to the cerebellum [2] and the model can be further used to investigate postnatal function and development because of vitality beyond birth.
Figure 4. Atoh1trhl/trhlmutant compared to wild-typecontrol cerebellum morphology.

Cerebellar abnormalities in H&E-stainedand cresyl violet-counterstained sections through the cerebellum of a representative wild-type control (left panels) and a Atoh1trhl/trhl(right panels) mouse shown under increasing magnification from 5X to 63X (n = 3 per group).Cerebella from each genotype were examined. The cerebellum in wild type is well developed and foliated(A), but it is smooth(B) in Atoh1trhl/trhlmice.The external granule layer (EGL)is indicated by the arrow in both middle panels. In (D) the EGL of theAtoh1trhl/trhlmice is significantly underdeveloped as compared to (C).Under high magnification, the Purkinje cell layer (PCL), indicated by the star in both (E) and (F), appears disorganized in the mutant, with some Purkinje cells (asterisk in F) located deep in the cerebellum,where they are not normally found.
Progressive hearing loss indicated by ABR and DPOAE
To test hearing and to detect any progressive changes, we measured auditory brainstem response (ABR) thresholds over time, beginning at 21 days postnatum (P21) and at a number of time points up to 420 days. Measurement of hearing at P21 indicated a higher threshold and lower amplitude with more prominent central peaks in the Atoh1trhl/trhl strain, in comparison to normal and heterozygous mice (n = 5 per group; Fig. 5A). It is important to note that the background strain of mice C57BL/6J has an underlying hearing loss mutation of Cdh23753Athat causes progressive hearing loss and deafness starting at 10 months of age. Our data isconsistent with hair cell loss and possible auditory neuron loss in Atoh1trhl/trhl mice that is starting much earlier as evidenced by issues with hearing at the first P21 time point and most likely related to the induced hypomorphic Atoh1 mutation.We found the sensorineural hearing loss to be slowly progressive, with a phase of accelerated loss at approximately 3 months postnatal (Fig. 5B). The sensorineural hearing loss and its progression were more prominent at higher frequencies, corresponding to hair cell loss in the basal cochlear region.Distortion product otoacoustic emission (DPOAE) recordings (Fig. 5C) showed a comparative decrease in the homozygous Atoh1 vs. heterozygous Atoh1 mutant vs. the B6 +/+ control (n = 5 per group). The reduced DPOAE is indicative of the expected reduced function of the inner ear and its hair cells. This decline corresponded to the histological observations about the loss of hair cells, more in the Atoh1 homozygous mutants than heterozygous (Fig. 2) as well as ABR recordings.
Figure 5. Hearing tests indicate progressive hearing loss in mutants.
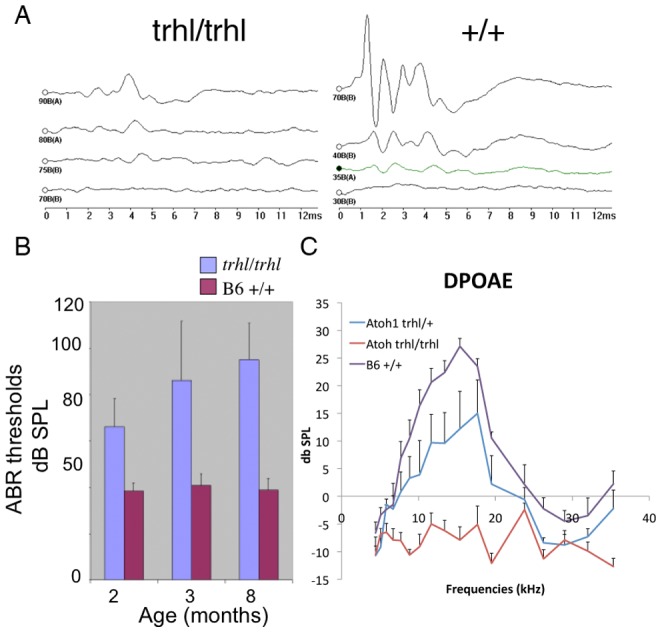
(A)Representative ABR thresholds (with click) from wild-type B6 (right panel) and mutant Atoh1trhl/trhl(left panel) mice recorded at 21 days of age (P21); n = 5 were tested for each genotype. Note the differences in threshold, waveform morphology and amplitude of the peaks. The X-axis represents time in milliseconds and Y-axis represents amplitude of the action potentials in microvolts. Each waveform, at a different, declining dB level to examine response, is stacked onto the same graph. The right panel displays a much stronger response to the stimulus and clear waveforms at 70 dB but the left panel does not show a very strong response even at 90 dB. (B)Average of ABR thresholds evoked by 16 kHz pure tone recorded from wild-type and Atoh1trhl/trhlmice at ages 2, 3, and 8 months (n = 5 for each grouping). ABR threshold was higher for mutant strains and progressively worsened over the measured time period, in comparison to littermates. Error bars represent standard deviation. The graph in (C) showsDPOAE amplitudes in mice of three different genotypes at P80 (n = 10 for each genotype). DPOAE amplitudes of the homozygous mutant mice were undetectable at all frequencies while B6 +/+ mice had normal DPOAE amplitudes. The heterozygous had minor reduction of DPOAE amplitudes at 15-20 kHz. Error bars indicate standard error at each time point, for each mouse group.
Biological Significance, Clinical Significance, and Future Prospects
Since this model displays hair cell loss and neuronal alteration, it is important to understand the molecular mechanisms that underlie specification and differentiation of immature and/or proliferating epithelial cells into mature hair cellstogether withcomponents of relevant neuronal pathways. This knowledge may allow elucidation of the requirements for triggering cell repair and/or regeneration after injury. Identification and manipulation of candidate genes that control and regulate hair cell differentiation would facilitate restoration of hair cells in a damaged organ of Corti. The homozygous Atoh1trhlmouse thus provides a valuable model to study the effects of the Atoh1 gene on postnatal ear anatomy and function. It also provides a good model for studying gene therapy, asAtoh1 gene transfer could be a potential treatment to promote hair cell regeneration[8], [9], [22]. The role of Atoh1 was explored byBerminghamand colleagues[7] who demonstrated that at embryonic day 18.5, Atoh1 expression is restricted to the hair cells of the developing sensory epithelia. However, using quantitative real time-polymerase chain reaction (RT-PCR) analysis, Zheng[23]showed that Atoh1 is expressed in adults, albeit at lower levelsthan during development. The progressive hearing dysfunction and loss of hair cellsintrhl/trhl mice suggests that Atoh1 is required not only for formation but also for maintenance of hair cells in adult mice. The mutated Atoh1 gene might cause long-term or permanent expression changes of downstream genes such as LIM homeobox protein 2 (Lh2a), LIM homeobox protein 9 (Lhx9), BarH-like 1 (Barhl1)and BarH-like 2 (Barhl2) [24], [25], which can be investigated in future studies. Barhl1has been extensively studied and appears to play an essential role in the migration and survival of cerebellar and pre-cerebellar neurons in mice as well as a rolein normal hair cell function where it is expressed in inner ear hair cells, but most abundantly in cochlear outer hair cells[20], [21], [26], [27].Progressive loss of cochlear hair cells in Barhl1-null mice indicates a crucial role for Barhl1in the maintenance of these sensory cells. The onset of hair cell defects in the Barhl1 –/–resulting in early low frequency loss and progressive loss around 3 months [20]suggests that Barhl1 is likely to act downstream of Atoh1-dependentinner ear development which is sufficient for the initial generation of those hair cells even in the absence of Barhl1expression. Similarly, analysis of Barhl2-flanking sequences identified an enhancer that can be activated by the basic helix-loop-helix factor Atoh1 and can drive spinal cord-specific gene expression [25].Atoh1 must be expressed in sufficient quantity to appropriately act on downstream genes such as Barhl1 and Barhl2, but Atoh1 over-expression has been associated with increased granule neuron precursor proliferation and also with medulloblastoma, one of the most common pediatric nervous system tumors [11]. Additional research is needed to explore these associations to reach a better understanding of the interplay between these factors to better direct possible routes of future therapies. Based on a exclusive search in Pubmed, OMIN (http://www.ncbi.nlm.nih.gov/omim?term=atoh1), http://www.hgvs.org/dblist/dblist.html and genecards.org search, there is no survival human mutation although many polymorphism in human genome (including 32 SNPs- Single Nucleotide Polymorphismand one SSLP-Single Sequence Length Polymorphisms) in ATOH1.
According to informatics.jax.org (http://www.informatics.jax.org/marker/MGI:104654),all 9 alleles of Ahoh1 aretargeted alleles, and they either die early in life or need complicated breeding/gene manipulation to order show desired phenotype. This makes the Atoh1trhlmousewith normal lifespan an uniquely valuable model to study the effects of the Atoh1 gene on postnatal ear anatomy and function in hearing, central nerves system and cancer biology.
Conclusion
Since the trhl mutant mice have a normal life span, unlike previous Atoh1-nullmutations, which are perinatally lethal, we can take advantage of this newmodel to investigate many aspects of downstream hearing and balance pathophysiology, molecular biology, proteomics and cancer biology. We feel that this unique model can offer an interesting alternate pathway to examine Atoh1 function to complement other models and not necessarily replace them. It is important to note that this model is a hypomorphic mutation and that Atoh1 has more of an effect developmentally than on the neurogenesis and inner ear hair cell formation [1], [3]. The effect of this mutation on other Atoh1affected tissues is currently unknown but is an important avenue to investigate in the future. Inaddition to the normal life span, the Atoh1mutant mice display unique features as a model for progressive age related hearing loss. These characteristics may reflect a crucial requirement for Atoh1in the lifelong maintenance of cochlear hair cells. Conceivably, an understanding of this requirement at the molecular level will provide significant insights into the pathology of aging-related deafness, creating further demand for this model as a significant tool to explore human diseases including lung cancers[19], intestinal tumorigenesis, [5] and neuroblastomaformation [19], [28].
Acknowledgments
We thank Dr. Cindy Benedict-Alderfer for editorial assistance with this manuscript. We thank John C. Schimenti for donating the initial breeding pair of the trhl mice, Drs. Ken Johnson and Susan L. Ackerman for helpful initial discussions on candidate gene identification.
Funding Statement
This project was supported by the National Institutes of Health, National Institute on Deafness and Other Communication Disorders (R01DC009246, R21DC005846, R01DC009246 and R01DC007392), Foundation of Taishan Scholar (tshw20110515 and ZR2012HZ004) and National Natural Science Foundation of China (81271085). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Rose MF, Ren J, Ahmad KA, Chao HT, Klisch TJ, et al. (2009) Math1 is essential for the development of hindbrain neurons critical for perinatal breathing. Neuron 64: 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, et al. (1997) Math1 is essential for genesis of cerebellar granule neurons. Nature 390: 169–172. [DOI] [PubMed] [Google Scholar]
- 3. Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY (2001) Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294: 2155–2158. [DOI] [PubMed] [Google Scholar]
- 4. Tian C, Liu XZ, Han F, Yu H, Longo-Guess C, et al. (2010) Ush1c gene expression levels in the ear and eye suggest different roles for Ush1c in neurosensory organs in a new Ush1c knockout mouse. Brain Res 1328: 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saba R, Johnson JE, Saito T (2005) Commissural neuron identity is specified by a homeodomain protein, Mbh1, that is directly downstream of Math1. Development 132: 2147–2155. [DOI] [PubMed] [Google Scholar]
- 6. Murre C, McCaw PS, Baltimore D (1989) A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell 56: 777–783. [DOI] [PubMed] [Google Scholar]
- 7. Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, et al. (1999) Math1: an essential gene for the generation of inner ear hair cells. Science 284: 1837–1841. [DOI] [PubMed] [Google Scholar]
- 8. Shou J, Zheng JL, Gao WQ (2003) Robust generation of new hair cells in the mature mammalian inner ear by adenoviral expression of Hath1. Mol Cell Neurosci 23: 169–179. [DOI] [PubMed] [Google Scholar]
- 9. Kawamoto K, Ishimoto S, Minoda R, Brough DE, Raphael Y (2003) Math1 gene transfer generates new cochlear hair cells in mature guinea pigs in vivo. J Neurosci 23: 4395–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pan N, Jahan I, Kersigo J, Kopecky B, Santi P, et al. (2011) Conditional deletion of Atoh1 using Pax2-Cre results in viable mice without differentiated cochlear hair cells that have lost most of the organ of Corti. Hear Res 275: 66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Flora A, Klisch TJ, Schuster G, Zoghbi HY (2009) Deletion of Atoh1 disrupts Sonic Hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science 326: 1424–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawauchi D, Saito T (2008) Transcriptional cascade from Math1 to Mbh1 and Mbh2 is required for cerebellar granule cell differentiation. Dev Biol 322: 345–354. [DOI] [PubMed] [Google Scholar]
- 13. Lai HC, Klisch TJ, Roberts R, Zoghbi HY, Johnson JE (2011) In vivo neuronal subtype-specific targets of Atoh1 (Math1) in dorsal spinal cord. J Neurosci 31: 10859–10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taylor BA, Navin A, Phillips SJ (1994) PCR-amplification of simple sequence repeat variants from pooled DNA samples for rapidly mapping new mutations of the mouse. Genomics 21: 626–632. [DOI] [PubMed] [Google Scholar]
- 15. Han F, Yu H, Tian C, Chen HE, Benedict-Alderfer C, et al. (2012) A new mouse mutant of the Cdh23 gene with early-onset hearing loss facilitates evaluation of otoprotection drugs. Pharmacogenomics J 12: 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim D, Ackerman SL (2011) The UNC5C netrin receptor regulates dorsal guidance of mouse hindbrain axons. J Neurosci 31: 2167–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng QY, Johnson KR, Erway LC (1999) Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res 130: 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, et al. (2003) Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet 12: 3075–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pham TV, Hartomo TB, Lee MJ, Hasegawa D, Ishida T, et al. (2012) Rab15 alternative splicing is altered in spheres of neuroblastoma cells. Oncol Rep 27: 2045–2049. [DOI] [PubMed] [Google Scholar]
- 20. Li S, Price SM, Cahill H, Ryugo DK, Shen MM, et al. (2002) Hearing loss caused by progressive degeneration of cochlear hair cells in mice deficient for the Barhl1 homeobox gene. Development 129: 3523–3532. [DOI] [PubMed] [Google Scholar]
- 21. Lim J, Choi KW (2003) Bar homeodomain proteins are anti-proneural in the Drosophila eye: transcriptional repression of atonal by Bar prevents ectopic retinal neurogenesis. Development 130: 5965–5974. [DOI] [PubMed] [Google Scholar]
- 22. Zheng JL, Gao WQ (2000) Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat Neurosci 3: 580–586. [DOI] [PubMed] [Google Scholar]
- 23. Zheng JL, Shou J, Guillemot F, Kageyama R, Gao WQ (2000) Hes1 is a negative regulator of inner ear hair cell differentiation. Development 127: 4551–4560. [DOI] [PubMed] [Google Scholar]
- 24. Bermingham NA, Hassan BA, Wang VY, Fernandez M, Banfi S, et al. (2001) Proprioceptor pathway development is dependent on Math1. Neuron 30: 411–422. [DOI] [PubMed] [Google Scholar]
- 25. Chellappa R, Li S, Pauley S, Jahan I, Jin K, et al. (2008) Barhl1 regulatory sequences required for cell-specific gene expression and autoregulation in the inner ear and central nervous system. Mol Cell Biol 28: 1905–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li S, Qiu F, Xu A, Price SM, Xiang M (2004) Barhl1 regulates migration and survival of cerebellar granule cells by controlling expression of the neurotrophin-3 gene. J Neurosci 24: 3104–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McEvilly RJ, Erkman L, Luo L, Sawchenko PE, Ryan AF, et al. (1996) Requirement for Brn-3.0 in differentiation and survival of sensory and motor neurons. Nature 384: 574–577. [DOI] [PubMed] [Google Scholar]
- 28. Klisch TJ, Xi Y, Flora A, Wang L, Li W, et al. (2011) In vivo Atoh1 targetome reveals how a proneural transcription factor regulates cerebellar development. Proc Natl Acad Sci U S A 108: 3288–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]