

Abstract
HNP1 is a human alpha defensin that forms dimers and multimers governed by hydrophobic residues, including Tyr16, Ile20, Leu25, and Phe28. Previously, alanine scanning mutagenesis identified each of these residues and other hydrophobic residues as important for function. Here we report further structural and functional studies of residues shown to interact with one another across oligomeric interfaces: I20A-HNP1 and L25A-HNP1, plus the double alanine mutants I20A/L25A-HNP1 and Y16A/F28A-HNP1, and the quadruple alanine mutant Y16A/I20A/L25A/F28A-HNP1. We tested binding to HIV-1 gp120 and HNP1 by surface plasmon resonance, binding to HIV-1 gp41 and HNP1 by fluorescence polarization, inhibition of anthrax lethal factor, and antibacterial activity using the virtual colony count assay. Similar to the previously described single mutant W26A-HNP1, the quadruple mutant displayed the least activity in all functional assays, followed by the double mutant Y16A/F28A-HNP1. The effects of the L25A and I20A single mutations were milder than the double mutant I20A/L25A-HNP1. Crystallographic studies confirmed the correct folding and disulfide pairing, and depicted an array of dimeric and tetrameric structures. These results indicate that side chain hydrophobicity is the critical factor that determines activity at these positions.
Introduction
Defensins are a family of antimicrobial peptides of innate immunity broadly active against bacteria, viruses and toxins [1], [2], [3], [4], [5], [6]. They are small (2–5 kDa), stabilized structurally by three disulfide bonds, form oligomers, and are divided into α [6], β [7] and θ [8] structural classes. There are six human α-defensins: human neutrophil peptides (HNP) 1–4 [9], [10], [11], [12], [13] and human defensins (HD) 5–6 [14], [15]. The structural basis for defensin antimicrobial activity against bacteria and viruses is only partially understood, but it is clear that defensins possess two fundamental characteristics: cationicity and hydrophobicity. α-Defensins are positively charged, and their activity against Gram negative bacteria such as Escherichia coli is thought to be mediated by the electrostatic attraction between cationic arginine residues and the anionic phospholipids of bacterial membranes [16]. By contrast, the human α-defensins are more selective for Gram positive strains, and specific, chiral interactions with lipid II molecules have been proposed to be primarily responsible for activity against Gram positive bacterium, Staphylococcus aureus [17], as is the case with several other defensins [18], [19], [20], [21]. Defensins provide two types of protection against bacilli: they are active against Bacillus cereus directly with a high degree of potency [22], [23], and they bind to and inhibit bacterial toxins such as anthrax lethal factor (LF) [24], [25], [26], [27], [28]. A picture of the myriad functions of HNP1 and HD5 is emerging that identifies hydrophobicity as more critical than cationicity, which can be explained partially by the hydrophobic effect on the α-defensin “canonical” dimer. For HNP1, the canonical dimer interface is formed by the antiparallel extensions of the β2 strands stabilized by the reciprocal main-chain hydrogen bonds contributed by Thr18 and Ile20, and the hydrophobic packing of the side chains of Tyr16, Tyr21, Phe28 and the Cys2–Cys30 disulfide [27], [29]. Underlying these hydrophobic contacts are the aromatic rings of Trp26 [28], constituting the bulk of the hydrophobic core of the monomer.
Alanine scanning mutagenesis is a powerful tool for exploring the contributions of individual side chains to aspects of protein and peptide function such as binding [30], [31], [32], stability [33], [34], [35], and catalysis [36], [37]. Alanine scanning mutageneses of both HNP1 [28] and HD5 [26] identified hydrophobic residues as the most important for activity, especially HNP1 Trp26 and HD5 Leu29. In the alanine scan of HNP1, the hydrophobic residues of Tyr16, Ile20, Leu25 and Phe28 were also shown to be significant, since the Y16A, I20A, L25A and F28A mutants all had decreased bactericidal activity against S. aureus [28]. Surface plasmon resonance (SPR) studies supported the antibacterial assays, identifying F28A-HNP1 as the second lowest self-association on the HNP1 surface of all of the alanine scanning mutants, which corresponded to its second worst antibacterial activity, after W26A. However, in spite of the power of alanine scanning, it can generate misleading results when at least two side chains interact, which can cause non-additivity [38], [39]. In order to circumvent this potential shortcoming and shed light upon the cumulative effects of side chain interactions, if any, multiple mutants can be constructed. Crystallography identified two pairs of side chains that would be particularly insightful: Tyr16/Phe28 and Ile20/Leu25. Tyr16 and Phe28 mapped to the dimer interface, making side chain contacts with the opposing monomer. The canonical dimer interface of HNP1 is formed by the antiparallel extensions of the β2 strands stabilized by the reciprocal main-chain hydrogen bonds contributed by Thr18 and Ile20, and the hydrophobic packing of the side chains of Tyr16, Tyr21, Trp26, Phe28 and the Cys2–Cys30 and Cys4–Cys19 disulfides (Fig. 1A) [27], [28]. Further analysis of probable quaternary structures by the Protein Interfaces, Surfaces and Assemblies (PISA) software indicates that the HNP1 dimer forms a tetramer or dimer of dimers in a crystal. The dimer-dimer interface consists of the Ile20 and Leu25 side chains and the assembly is maintained exclusively through van der Waals interactions (Fig. 1B).
Figure 1. Quaternary structure of wild type HNP1.
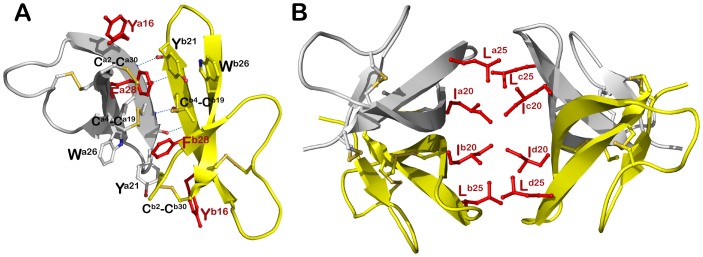
(A) Dimeric and (B) tetrameric assembly of HNP1 in a crystal (PDB:3GNY, [27]). Disulfide bonds and residues involved in oligomerization are shown as balls and sticks and reciprocal main chain hydrogen bonds are shown as blue dashes. Residues subjected to Ala-substitutions in presented studies are highlighted in red.
Here we report the x-ray crystal structures of I20A-HNP1, I20A/I25A-HNP1, Y16A/F28A-HNP1 and Y16A/I20A/L25A/F28A-HNP1. The functional consequences of each mutation are also investigated using SPR, fluorescence polarization, an enzyme kinetic assay to quantify LF inhibition, and the virtual colony count assay to quantify antibacterial activity.
Materials and Methods
Synthesis and Folding of Defensins
All five HNP1 mutants (Table 1) were synthesized on an ABI 433A synthesizer using an optimized HBTU activation/DIEA in situ neutralization protocol developed by Kent and coworkers for Boc chemistry solid phase peptide synthesis (SPPS) [40]. All peptides were purified by C18 reversed phase high performance liquid chromatography (RP-HPLC), and their molecular masses ascertained by electrospray ionization mass spectrometry (ESI-MS) (data not shown). Correct folding of the Y16A/F28A, I20A/L25A, I20A, and L25A mutants was achieved at 0.25 mg/mL in 25% N,N-dimethylformamide, 2 M urea, 50 mM Tris/HCl, 3 mM reduced and 0.3 mM oxidized glutathione (pH 8.3) overnight at room temperature [41]. Y16A/I20A/L25A/F28A-HNP1, with the side chains of Cys9 and Cys29 acetamidomethyl (Acm) protected, was first oxidized at 0.25 mg/mL in 50 mM Tris/HCl buffer (pH 8.3) by stirring in the open air overnight to form two disulfide bridges, Cys2–Cys30 and Cys4–Cys19. The desired folding intermediate, confirmed by disulfide mapping through digesting with 0.1 mg/mL chymotrypsin for 1.5 hr at room temperature in 50 mM Tris/HCl, 20 mM CaCl2, 0.005% Triton X-100 (pH 8.3), was then treated by 0.5 mM iodine for 45 min at 0.5 mg/mL in an acidic solution containing 0.1 M citric acid, 0.2 M HCl and 20% methanol to deprotect Acm and form the third disulfide bond. The reaction was quenched by 0.2 M ascorbic acid, and the fully folded peptide was purified to homogeneity by RP-HPLC and its molecular mass verified by ESI-MS. Defensin stock solutions prepared with water were quantified spectroscopically at 280 nm using molar extinction coefficients calculated according to the algorithm of Pace et al [42].
Table 1. The amino acid sequences of wild type HNP1 and HNP1 analogs.
| HNP1 | ACYCRIPACI10 AGERRYGTCI20 YQGRLWAFCC30 |
| Y16A/F28A | ACYCRIPACI10 AGERRAGTCI20 YQGRLWAACC30 |
| Y16A/I20A/L25A/F28A | ACYCRIPACI10 AGERRAGTCA 20 YQGRAWAACC30 |
| I20A/L25A | ACYCRIPACI10 AGERRYGTCA 20 YQGRAWAFCC30 |
| I20A | ACYCRIPACI10 AGERRYGTCA 20 YQGRLWAFCC30 |
| L25A | ACYCRIPACI10 AGERRYGTCI20 YQGRAWAFCC30 |
Crystallization and Data Collection
Lyophilized HNP1 mutant proteins were dissolved in water (20 mg/mL), mixed in a 1∶1 ratio (1 µl total) with appropriate precipitant solutions and left to equilibrate in a hanging drop at room temperature. Crystals were flash frozen in liquid nitrogen after briefly soaking in the crystallization condition plus 15–25% glycerol prior to data collection.
For the I20A-HNP1 and I20A/L25A-HNP1 mutants, data were collected using a rotating anode x-ray generator Rigaku-MSC Micromax 7 and a Raxis-4++ image plate detector (at the X-ray crystallography core facility, University of Maryland, Baltimore). Diffraction data for HNP1 Y16A/F28A and Y16A/I20A/L25A/F28A mutants were collected at the Stanford Synchrotron Radiation Light Source (SSRL) BL7-1 beamline on an ADSC Quantum 315 area detector. All data were processed and reduced with HKL2000 [43]. Structures were solved by molecular replacement with Phaser [44] from the CCP4 suite based on the coordinates of the HNP1 monomer (PDB: 3GNY). Refinement was carried out with Refmac [45] and/or Phenix [46] and model building was done with COOT [47]. Data collection and refinement statistics are shown in Table 2. Ramachandran statistics were calculated with Molprobity [48] and illustrations were prepared with Pymol molecular graphics (http://pymol.org) or Molscript [49].
Table 2. Data collection and refinement statistics.
| Crystals of HNP1 mutants | ||||||||
| I20A | I20A/L25A | Y16A/F28A | Y16A/I20A/L25A/F28A | |||||
| Data collection | ||||||||
| Wavelength, Å | 1.54 | 1.54 | 0.98 | 1.00 | ||||
| Space group | P41 | C2 | H3 | H3 | ||||
| Cell parameters | ||||||||
| a, b, c, Å | 37.7, 37.7, 40.5 | 75.0, 62.6, 42.5 | 88.6, 88.6, 54.1 | 83.8, 83.8, 51.3 | ||||
| α, β, γ, ° | 90, 90, 90 | 90, 99.7, 90 | 90, 90, 120 | 90, 90, 120 | ||||
| Molecules/a.u. | 2 | 8 | 4 | 4 | ||||
| Resolution, (Å)a | 50-1.7 (1.73-1.7) | 50-1.66 (1.69-1.66) | 50-2.0 (2.03-2.00) | 50.0-1.9 (1.93-1.90) | ||||
| # of reflections | ||||||||
| Total | 17,297 | 73,805 | 58,691 | 60,651 | ||||
| Unique | 8,953 | 22,465 | 10,671 | 10,457 | ||||
| Rmerg b, % | 13.9 (67.4) | 8.1 (44.8) | 10.3 (75.9) | 3.6 (96.0) | ||||
| I/σ | 12.7 (2.0) | 16.5 (1.6) | 21.6 (1.3) | 56 (2.2) | ||||
| Completeness, % | 100 (99.0) | 98.3 (84.1) | 99.5 (98.5) | 99.1 (100) | ||||
| Redundancy | 4.8 (3.8) | 2.3 (3.3) | 5.5 (4.2) | 5.8 (5.8) | ||||
| Refinement Statistics | ||||||||
| Resolution, Å | 27.6-1.72 | 20-1.70 | 18.0-2.00 | 24.2-1.90 | ||||
| Rc, % | 20.1 | 18.5 | 19.0 | 19.9 | ||||
| Rfree d, % | 22.9 | 23.1 | 22.8 | 23.6 | ||||
| # of atoms | ||||||||
| Protein | 470 | 1,815 | 900 | 876 | ||||
| Water | 26 | 209 | 8 | 8 | ||||
| Ligand/Ion | 6 | 6 | 0 | 0 | ||||
| Overall B value (Å)2 | ||||||||
| Protein | 38.2 | 28.6 | 64.8 | 72.7 | ||||
| Water | 46.3 | 37.8 | 60.8 | 67.0 | ||||
| Ligand/Ion | 67.6 | 20.9 | – | – | ||||
| Root mean square deviation | ||||||||
| Bond lengths, Å | 0.015 | 0.024 | 0.018 | 0.007 | ||||
| Bond angles,° | 1.6 | 1.9 | 1.9 | 1.33 | ||||
| Ramachandrane | ||||||||
| favored, % | 94.6 | 96.8 | 96.4 | 94.4 | ||||
| allowed, % | 5.4 | 3.2 | 3.6 | 3.6 | ||||
| outliers, % | 0.0 | 0.0 | 0.0 | 0.0 | ||||
| PDB code | 4LBB | 4LBF | 4LB1 | 4LB7 | ||||
Values in parentheses are for highest-resolution shell.
R merge = ∑|I - <I>|/∑I, where I is the observed intensity and <I> is the average intensity obtained from multiple observations of symmetry-related reflections after rejections.
R = ∑||Fo|- | Fc||/∑|Fo |, where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree = defined by by Brünger [68].
Calculated with MolProbity [48].
SPR Based wt-HNP1 and gp120 Binding
Experiments were performed at 25°C on a BIAcore T100 System (BIAcore, Inc., Piscataway, NY). The assay running buffer was 10 mM HEPES, 150 mM NaCl, 0.05% surfactant P20, pH 7.4 (±3 mM EDTA). 233 response units (RU) of HNP1 or 2770 RU of gp120 were immobilized on CM5 sensor chips using the amine-coupling chemistry. Analytes were introduced into the flow-cells at 30 µl/min in the running buffer. Association and dissociation were assessed for 5 and 10 min, respectively. After each analysis, the sensor chip surfaces were regenerated with 30 mM HCl for HNP1, 10 mM glycine solution (pH 2.0) and 10 mM NaOH for gp120, and equilibrated with the running buffer before the next injection. Binding isotherms were analyzed with BIAevaluation software.
Fluorescence Polarization-based Defensin and N36-gp41 Binding
An N-terminally acetylated N36 (HIV-1 gp160 546–581) peptide derived from the N-terminus of gp41 was synthesized by Boc-chemistry SPPS and purified to homogeneity by preparative C18 RP-HPLC. Succinimidyl ester-activated carboxyfluorescein (FAM-NHS) was covalently conjugated to N-acetylated N36 peptide via its Lys574 (HIV gp160 numbering) side chain in DMF, and the resultant product N-acetyl-N36-FAM was HPLC-purified and lyophilized. The defensin-N36 binding experiments were performed in 384-well plates on a Tecan Infinite M1000 multimode plate reader. 2-fold serially diluted defensins were prepared in PBS and incubated with 50 nM FAM-labeled N36 in a total volume of 100 µl per well. After a 30 min incubation at room temperature, fluorescence polarization values were measured at exciting and emitted wavelengths of 470 nm and 530 nm, respectively.
LF inhibition kinetics
A 2-fold dilution series of defensin, ranging from 1024 to 1 nM in 20 mM HEPES buffer containing 1 mM CaCl2 and 0.5% Nonidet P-40 (pH 7.2), was incubated at 37°C for 30 min with 1 µg/mL (∼10 nM) of LF. Then, 20 µl of 1 mM LF substrate was added into each well to a final concentration of 100 µM in a total volume of 200 µl. Kinetic measurements of LF enzymatic activity were monitored at 405 nm over 30 min at 37°C on a Tecan Infinite M1000 microplate reader. Data were presented in a plot showing percent inhibition versus defensin concentration, from which IC50 values (the concentration of defensin that reduced the enzymatic activity of LF by 50%) were derived by a non-linear regression analysis [27], [28].
Virtual Colony Count
Antibacterial assays against Escherichia coli strain American Type Culture Collection (ATCC) 25922, Staphylococcus aureus ATCC 29213, and Bacillus cereus ATCC 10876 were conducted using the Virtual Colony Count 96-well kinetic turbidimetric method [22]. Strains were obtained from ATCC (Manassas, VA). A 2-fold dilution series of defensin, ranging from 256 to 1 µg/mL, was incubated with ∼5×105 virtual colony forming units (CFUv)/mL bacteria at 37°C for 2 h in 10 mM sodium phosphate buffer, pH 7.4, 1% tryptic soy broth (TSB), followed by addition of twice-concentrated Mueller-Hinton broth and kinetic measurements of bacterial growth at 650 nm every 5 minutes over 12 h using a Tecan Infinite M1000 plate reader set to shake 3s orbitally before each read. B. cereus was also assayed using a 2-fold dilution series of defensin ranging from 4-0.016 µg/mL. The 10 mM sodium phosphate incubation buffer included 1% TSB to increase the sensitivity of bacteria to defensin activity [9]. The zero time point was discarded and the optical density at the 5-minute time point was subtracted from subsequent growth kinetic optical density readings, as previously described [23]. Sextuplicate calibration curves were measured at a threshold change in optical density at 650 nm of 0.05. The virtual LD50 (vLD50), vLD90, vLD99, and vLD99.9 were reported as the defensin concentration that resulted in survival rates of 0.5, 0.1, 0.01, and 0.001, respectively. Measurements were done in triplicate on three separate days, except the assay of Y16A/I20A/L25A/F28A-HNP1 against B. cereus from 256 to 1 µg/mL, which was done in triplicate on the same 96-well plate. Statistical p-values were calculated as the output of the paired two-tailed Microsoft Excel T.TEST function.
Results
Total Chemical Synthesis and Oxidative Folding of HNP1 Analogs
Crude HNP1 mutants, after HF cleavage and ether precipitation, gave rise to molecular masses in agreement with the expected values calculated on the basis of the average isotopic compositions of reduced defensins. Using the efficient protocol established for folding of HNP1, the reduced analogs Y16A/F28A-HNP1, I20A/L25A-HNP1, I20A-HNP1, and L25A-HNP1 productively folded in the presence of 2 M urea and 25% DMF, giving the three disulfide bridges of Cys2–Cys30, Cys4–Cys19 and Cys9–Cys29. For Y16A/I20A/L25A/F28A-HNP1, Cys9 and Cys29 were strategically selected to be protected by Acm during peptide synthesis. The correctly folded product could then be produced in high yield through a 2-step folding procedure. Two folding species were produced after the first air-oxidation reaction, one of which generated three desired major segments after chymotrypsin digestion: [AC2Y][AAC29(Acm)C30] (found 791.3 Da, calculated 791.9 Da), [QGRAW] (found 617.3 Da, calculated 616.7 Da) and [C4RIPAC9(Acm)IAGERRAGTC19AY] (found 1979.6 Da, calculated 1980.3 Da), confirming the presence of disulfide bridges between Cys2–Cys30 and Cys4–Cys19. This folding intermediate was then iodine-oxidized to form the third disulfide bond between Cys9 and Cys29.
Crystallographic Monomers of HNP1 Analogs are Similar to the Previously Determined Structure of the HNP1 Monomer
The crystal structures of I20A-HNP1, I20A/L25A-HNP1, Y16A/F28A-HNP1 and Y16A/I20A/L25A/F28A-HNP1 were solved by molecular replacement at resolutions between 1.66–2.0 Å. In each case, multiple copies of defensin molecules were present in the asymmetric unit of the crystal (Table 2). Structural alignment of these crystallographically independent monomers reveals no changes to the core structure, which is the same as in wild type HNP1 (Fig. S1). When superimposed, the root-mean-square (RMS) deviations between 30 equivalent Cα-atoms of mutants and wild-type HNP1 are in the range of 0.51–0.78 Å. There are no changes to the network of three disulfide bridges. Both the disulfide bond connectivity and stereochemistry are identical to wild-type HNP1. In addition to the flexible termini, the major difference is observed for the loop region connecting the second and third β strands. Since the β2–β3 loop of HNP1 is involved in intensive crystal contacts, these differences are attributed to the effect of crystal packing rather than determined by the intrinsic properties of the backbone.
Mutants Exhibit a Variety of Oligomerization Structures
Analysis of HNP1 quaternary structures by PISA indicates that Tyr16 and Phe28 are essential for HNP1 dimerization contributing 16 and 11%, respectively, of the solvent-accessible surface buried by each monomer at the dimer interface. Recently we have shown that single point mutations of Y16A or F28A had no effect on the ability of HNP1 to form stable dimers in crystals [28]. To further test the role of Tyr16 and Phe28 in mediating the HNP1 dimer, we prepared the double mutant Y16A/F28A-HNP1. Analysis of intermolecular contacts within the Y16A/F28A-HNP1 crystal indicates that mutant monomers assemble into compact dimers reassembling canonical dimers of wild-type HNP1 (Fig. 2). The replacement of the bulky side chains of Tyr16 and Phe28 by linear Ala side chains allows the dimer to pack more tightly and form an additional set of H-bonds on the opposite face of the dimer. The new set of four H-bonds pins the short β1 strand of one monomer to the equivalent strand of the other monomer to form a two-stranded antiparallel β-sheet (Fig. 2A). These include the reciprocal main-chain H-bonds contributed by the carbonyl oxygen of Cys2 and the nitrogen of Cys4 and main chain nitrogen of Ala1 of monomer a and the hydroxyl of Tyr3 of monomer b. Formation of this dimer results in the average burial of more than 495 Å2 of molecular surface per each monomer, which compares to an average of 370 Å2 for the wild-type HNP1 dimer (PDB:3GNY, [27]). The significant increase in the average value of the molecular surface buried within inter-monomer interactions of Y16A/F28A-HNP1 as compared to the wild-type dimer is attributed to the presence of additional interactions mediated by the β1 strands. Similarly, a more compact dimer was formed by the replacement of the single Phe28 at the dimer interface [28]. Superimposition analysis (Fig. S2) of the Y16A/F28A-HNP1 and F28A-HNP1 dimers reveals very close similarity as shown by an average RMDS value of 0.6 Å for 60 aligned Cα atoms and an almost identical molecular surface buried within the dimer (495 and 500 Å2 per monomer for the Y16A/F28A-HNP1 and F28A-HNP1 dimer, respectively). Introduction of mutations at the dimer interface have no effect on the ability of Y16A/F28A-HNP1 to form a stable tetramer, which closely resembles the wild type HNP1 tetramer (Fig. S2).
Figure 2. Dimerization of Y16A/F28A-HNP1.

(A) Ribbon diagram of the Y16A/F28A HNP1 dimer with hydrophilic (left panel) and hydrophobic (right panel) residues stabilizing the dimer shown as balls and sticks. Mutated residues are shown in red and H-bonds are displayed as blue dashes. In addition to the main chain hydrogen bonds formed between Thr18 and Ile20, three new hydrogen bonds, between Cys2 and Cys4 and between the hydroxyl of Tyr3 and Ala1 help stabilize the opposite face of the dimer. (B) Superposition of Y16A/F28A and wild type HNP1 dimers. Y16A/F28A is in cyan and green and wild type HNP1 in grey and yellow.
Surprisingly, although the asymmetric unit of the I20A-HNP1 crystal contained two monomers they were not arranged into a dimer. Analysis of intermolecular contacts within the crystal unambiguously rules out the formation of any quaternary structure for the I20A-HNP1 mutant.
We failed to obtain diffracting crystals of the L25A-HNP1 mutant. I20A/L25A-HNP1 crystallizes as a tetramer with two independent tetramers in the asymmetric unit of the crystal (Fig. 3 and Fig. 4). The RMSD between tetramers is 0.744 Å. Unlike wild type HNP1 and most other HNP1 mutants, the tetramer I20A/L25A-HNP1 is not formed as a dimer of dimers. Each monomer contributes equally to tetramer stability with a combined buried surface area of approximately 2760 Å2 (as compared to 2280 Å2 of the wild type HNP1 tetramer). Tyr16 and Phe28 make up the bulk of the hydrophobic core with contributions from Tyr21 and the adjacent disulfide bonds of Cys4–Cys19 and Cys2–Cys30. In addition, there are two core sets of hydrogen bonds in the tetramer center that help in its stability, utilizing main chain atoms from Ala1, Cys2, and Cys30 and the hydroxyl of Tyr16 (Fig. 3). The tetramer is further stabilized by a hydrogen bond between Arg14 and Tyr21 of one pair of subunits.
Figure 3. I20A/L25A-HNP1 mutant tetramer.

The tetramer is stabilized by intensive hydrophobic interactions contributed mainly by the side chain atoms of Phe28 and Tyr21 and Cys4–Cys19 and Cys2–Cys30 The close-up view shows one of two hydrogen bond networks formed at the tetramer interface. The residues contributing to the tetramer formation are shown as balls and sticks. H-bond distances are given in Angstroms.
Figure 4. I20A/L25A-HNP1 tetramers.

(A) Ribbon and (B) and α-carbon traces of the two independent tetramers of I20A/L25A-HNP1 present in the asymmetric unit of the crystal. Tetramers could be superimposed with the RMSD of 0.744 Å.
Y16A/I20A/L25A/F28A-HNP1 forms a dimer which is very similar to the Y16A/F28A-HNP1 dimer (the average RMSDs between 60 equivalent Cα-atoms of 0.4 Å, Fig. 5). Solvent-accessible surface buried due to Y16A/I20A/L25A/F28A-HNP1 and Y16A/F28A-HNP1 dimer formation corresponds to 490 and 495 Å2 per monomer, respectively, indicating that these dimers are energetically indistinguishable. Surprisingly, although Y16A/I20A/L25A/F28A-HNP1 has mutated Ile20 and Leu25 that were previously identified as essential for wild type tetramer formation, analysis of intermolecular contacts within the crystal indicates that Y16A/I20A/L25A/F28A-HNP1 dimers arrange into a tetramer resembling closely the wild-type tetramer architecture (Fig. 6). Replacement of Ile20 and Leu25 side chains at the tetramer interface by less ‘bulky’ side chains of Ala allows Y16A/I20A/L25A/F28A-HNP1 dimers to pack more tightly together (Fig. 5B). The Y16A/I20A/L25A/F28A-HNP1 tetramer is stabilized through hydrophobic interactions involving Ala20 and a network of direct and water-mediated H-bonds formed between Thr18 and four water molecules that were ‘trapped’ at the tetramer interface (Fig. 5A). The buried interface area for Y16A/I20A/L25A/F28A-HNP1 tetramer formation is 1450 Å2 per dimer as compared to 1140 Å2 buried at the wild type HNP1 tetramer interface.
Figure 5. Y16A/F28A and Y16A/I20A/L25A/F28A-HNP1 dimers.

(A) Ribbon and (B) α-carbon traces of all Y16A/F28A and Y16A/I20A/L25A/F28A HNP1 dimers present in the asymmetric unit of the crystal. Y16A/F28A dimers are in green and Y16A/I20A/L25A/F28A dimers in red. Disulfide bonds are shown in yellow. The RMSD is 0.216 Å between Y16A/F28A dimers and 0.093 Å between Y16A/I20A/L25A/F28A dimers.
Figure 6. Quaternary structure of Y16A/I20A/L25A/F28A-HNP1.

(A) Putative tetrameric assembly of Y16A/I20A/L25A/F28A-HNP1 in the crystal. Residues involved in tetramer formation are shown as balls and sticks with the mutated residues colored in red. The network of direct and water-mediated H-bonds formed at the tetramer interface is shown as blue dashes. (B) Comparison of Y16A/I20A/L25A/F28A-HNP1 and wild type HNP1 tetramers. Tetramers were aligned based on AB dimers and colored cyan and orange (Y16A/I20A/L25A/F28A-HNP1) and yellow and grey (HNP1, PDB:3GNY, [27] ). The red arrow indicates the shift of the CD dimer of Y16A/I20A/L25A/F28A-HNP1 relative to the CD dimer of wild-type HNP1.
Binding to HNP1 and gp120 as Determined by Surface plasmon Resonance
We tested the ability of HNP1 and its mutants to bind to immobilized HNP1 or to the HIV-1 protein gp120 by surface plasmon resonance. (Fig. 7) Against both the HNP1 and gp120 surfaces, response units were ranked HNP1> L25A>I20A>I20A/L25A>Y16A/F28A>Y16A/I20A/L25A/F28A. HNP1 bound to immobilized HNP1 with a 20-fold greater response at 8000 nM than Y16A/I20A/L25A/F28A-HNP1 bound to HNP1, and HNP1 bound to immobilized gp120 with an 8-fold greater response at 8000 nM than Y16A/I20A/L25A/F28A-HNP1 bound to gp120.
Figure 7. Surface plasmon resonance binding curves.

Binding of HNP1 and mutants to 233 RU of HNP1 (left) and binding of HNP1 and mutants to 2770 RU of gp120 (right). The curves are plots of RU values at 300 s of association versus different defensin concentrations (from 0 nm to 8 µm). The RU values are the average readings from three measurements.
Interactions of HNP1 Analogs with N36 Peptide
HNP1 inhibits HIV-1 infectivity via multiple mechanisms [50], [51]. Recently, HNP1 has been shown to be able to interact with the heptad repeat domains of of HIV-1 gp41 comprising both N36 and C34 peptides, and contribute to the inhibition of fusion by interfering with the formation of the fusogenic gp41 structure [52]. An N-acetyl-N36 peptide labeled with 6-carboxyfluorescein (FAM) at Lys574 (HIV gp160 numbering) at 50 nM in PBS was incubated at room temperature for 30 min with a 2-fold dilution series of HNP1 mutants (0.024–50 µM) before fluorescence polarization was measured in a triplicate assay on a Tecan Infinite M1000 multimode plate reader. All HNP1 analogs except Y16A/I20A/L25A/F28A-HNP1 bound N36 in a dose-dependent fashion, reflected by increasing fluorescence polarization values with the increase in defensin concentration. The binding of HNP1 analogs to N36 was also structure–dependent, since the polarization values were in the order HNP1> L25A>I20A>I20A/L25A>Y16A/F28A>Y16A/I20A/L25A/F28A at almost all of the concentration series. (Fig. 8).
Figure 8. Defensin binding to N36 peptide as determined by fluorescence polarization.

An increase in fluorescence polarization is indicative of defensin binding to fluorecently labeled N36 peptide. The curves are obtained from the averages of three measurements.
Inhibition of Lethal Factor by HNP1 Analogs
The inhibition of lethal factor by various defensins was quantified at 37°C using an enzyme kinetic assay [27]. As shown as inhibition curves and IC50 values in Fig. 9, replacement of Ile20 and/or Leu25 with Ala slightly weakened inhibition of LF relative to wild type HNP1. However, the inhibitory activity of Y16A/F28A and Y16A/I20A/L25A/F28A in particular was greatly suppressed with the latter yielding an IC50 95-fold higher than that of wild type HNP1.
Figure 9. Inhibition of LF activity by different concentrations of HNP1 and mutant analogs.

Each inhibition curve is the mean of three independent enzyme kinetic measurements except Y16AF28A-HNP1, which is the mean of duplicate measurements.
Antibacterial Activity of HNP1 Analogs
Shown in Fig. 10 are virtual survival curves of E. coli, S. aureus, and B. cereus exposed to each defensin, and in Table 3 are the corresponding virtual lethal dose (vLD) values. Against S. aureus, the activity of all five HNP1 mutants was significantly undermined, giving activity in the rank order of HNP1> L25A>I20A>I20A/L25A>Y16A/F28A>Y16A/I20A/L25A/F28A at all defensin concentrations tested. By contrast, only the Y16A/F28A and Y16A/I20A/L25A/F28A mutations significantly reduced activity against E. coli and B. cereus. Notably, Y16A/I20A/L25A/F28A-HNP1 barely showed any activity at the maximum tested concentration of 256 µg/mL against S. aureus, whereas its activity remained less than vLD99.9 against E. coli and its vLD99.9 against B. cereus was 11-fold higher than HNP1. S. aureus growth curves were not necessarily parallel to non-defensin controls, suggesting that defensins can affect growth kinetics and delay threshold times after the addition of twice-concentrated Mueller-Hinton broth during the 12 h outgrowth phase of the virtual colony count assay.
Figure 10. Antibacterial activity of defensins as determined by virtual colony count.

E. coli ATCC 25922, S. aureus ATCC 29213 and B. cereus ATCC 10876 were exposed to HNP1 (◊; dotted line), I20A-HNP1 (▪), L25A-HNP1 (•), I20A/L25A-HNP1 (□), Y16A/F28A-HNP1 (○), and Y16A/I20A/L25A/F28A-HNP1 (Δ). Strains were exposed a twofold dilution series of defensins at concentrations varying from 1 to 256 µg/mL, except B. cereus was exposed to all defensins at 0.016 to 4 µg/mL and only Y16A/I20A/L25A/F28A-HNP1 at 1 to 256 µg/mL. Points equivalent to zero survival cannot be plotted on a logarithmic scale, such that against S. aureus virtual survival was zero above 32 µg/mL for HNP1, above 64 µg/mL for L25A-HNP1, and at 256 µg/mL for I20A-HNP1; against B. cereus virtual survival was zero at 4 µg/mL for HNP1. Each point is the mean of triplicate measurements, except HNP1 at 32 µg/mL against S. aureus (mean of two measurements; the third replicate gave a virtual survival value of zero) and L25A-HNP1 at 4 µg/mL against B. cereus (single measurement; the other two replicates gave virtual survival values of zero). Although error cannot easily be read directly from this plot, error values are quantified in Table 3.
Table 3. Antibacterial activities of HNP1 and mutants.
| Test organism | vLD50 (µg/mL) | |||||
| Wild-type HNP1 | I20A | L25A | I20A/L25A | Y16A/F28A | I20A/L25A/Y16A/F28A | |
| E. coli | 4.23±0.80 | 3.22±0.50 | 2.78±0.19 | 2.89±0.10 | 8.65±0.24a | 20.68±1.93a |
| S. aureus | <1 | 1.36±0.02a | 1.13±0.06b | 1.58±0.05a | 5.48±0.26a | 135.00±11.41a |
| B. cereus | 0.78±0.08 | 1.03±0.03 | 0.77±0.12 | 1.04±0.05 | 2.09±0.01a | 8.40±0.04a |
| vLD90 (µg/mL) | ||||||
| Wild-type HNP1 | I20A | L25A | I20A/L25A | Y16A/F28A | I20A/L25A/Y16A/F28A | |
| E. coli | 8.06±0.75 | 5.81±0.24 | 5.20±0.27 | 5.57±0.15 | 18.15±0.65a | 64.92±8.68a |
| S. aureus | 2.0±0.10 | 4.23±0.15a | 2.54±0.05a | 8.05±0.24a | 41.32±0.96a | >256a |
| B. cereus | 1.12±0.02 | 1.19±0.04 | 1.05±0.04 | 1.25±0.07 | 2.32±0.02a | 10.06±0.16a |
| vLD99 (µg/mL) | ||||||
| Wild-type HNP1 | I20A | L25A | I20A/L25A | Y16A/F28A | I20A/L25A/Y16A/F28A | |
| E. coli | 13.92±1.41 | 13.85±2.58 | 13.05±1.81 | 11.94±2.07 | 33.41±0.45a | 183.68±27.52a |
| S. aureus | 3.25±0.03 | 8.60±0.45a | 5.59±0.12a | 19.72±0.51a | 80.56±1.44a | >256a |
| B. cereus | 1.40±0.03 | 1.44±0.08 | 1.21±0.06 | 1.60±0.14 | 2.69±0.04a | 13.04±0.41a |
| vLD99.9 (µg/mL) | ||||||
| Wild-type HNP1 | I20A | L25A | I20A/L25A | Y16A/F28A | I20A/L25A/Y16A/F28A | |
| E. coli | 19.70±1.16 | 19.52±1.26 | 18.75±0.86 | 17.92±1.15 | 48.92±7.98 | >256a |
| S. aureus | 8.13±0.10 | 23.93±1.33a | 18.37±0.69a | 56.70±1.12a | 115.40±5.43a | >256a |
| B. cereus | 1.75±0.05 | 1.76±0.13 | 1.39±0.09a | 1.95±0.20 | 3.12±0.06a | 17.23±0.95a |
Virtual lethal doses vLD50, vLD90, vLD99, and vLD99.9 are the concentrations that resulted in a survival of 0.5, 0.1, 0.01, and 0.001, respectively. Mean ± SEM is presented for triplicate measurements except where noted.
Significantly different than wild-type HNP1 (paired t test, p<0.05).
Average of two measurements; the third measurement was <1 µg/mL.
Discussion
Defensins form a wide array of oligomeric structures. There is considerable structural diversity among the defensins, and the α-defensins are not the only group that has been shown to dimerize and multimerize. The authors of a study of the plant defensin NaD1 mutated a lysine to alanine and observed reduced activity, which they attributed to impaired dimer formation [53]. The activity could also have decreased due to a decrease in cationicity. This type of ambiguity does not apply to our present study, since we only mutated hydrophobic residues to alanine. The crystal structure of human β-defensin 2 depicted dimers and octamers, although the authors commented that it is unclear whether the octamer is physiologically relevant given the lack of conservation of the residues at the various subunit interfaces [54]. By contrast, a study of human β-defensins (HBD) 1–3 depicted HBD1 and 2 as monomers, while HBD3 crystallized as a dimer [55]. The enhanced activity of HBD3 compared to HBD1 and HBD2 may be related to its ability to dimerize. The θ-defensin retrocyclin-2 trimerized in solution, according to analytical ultracentrifugation and nuclear magnetic resonance results [56].
Starting with the crystal structure of HNP-3 [29], and the nuclear magnetic resonance (NMR) structures of HNP1 [57], [58], wild-type human α-defensin structures have typically depicted dimers. However, monomeric mutant forms of HNP1 and HD5 have been generated by N-methylating an amide bond at the dimer interface, thus breaking a hydrogen bond and introducing steric hindrance to the canonical dimerization scheme [26], [59]. In both cases, the monomeric mutant forms were as active against E. coli as the respective wild-type defensins. Other mutant forms have emphasized the versatility of the defensin framework. The HD5 canonical dimer with antiparallel β-strands existed in the crystal structure of the L29Abu-HD5 mutant, while the L29Nle-HD5 mutant exhibited a novel dimeric form with β-strands stacked in parallel [26]. Interestingly, the non-canonical L29Nle-HD5 molecule was functional, exhibiting less than wild-type activity against S. aureus but greater than wild-type activity against E. coli [26]. In the present study, we also discovered novel quaternary structures in the crystal, including a form of I20A-HNP1 that appears to be unable to form dimers or tetramers and a novel tetrameric form of I20A/L25A-HNP1. In addition, the canonical dimer formed by the quadruple mutant displayed a larger interface than wild-type. The HNP1 and HD5 results indicate that the presence of a canonical form of dimerization in the crystal does not correlate with α-defensin activity. Structural diversity may be a consequence of the small size of the peptides, leading to agile dynamic movements, combined with the rigidity of the tridisulfide structure that prevents major conformational transitions within the monomer. Defensin structural dynamics has been studied in solution in the context of binding to membranes or micelles [56], [60], [61]. The structure of HNP1 investigated using solid-state NMR suggested that the loop region between the first and second beta-strands is flexible and may change conformation when exposed to membranes [58]. However, the effect of membranes on HNP1 quaternary structure is poorly understood.
The stability of the dimer apparently varies from one α-defensin to the next. The HNP4 dimer is weakened by smaller hydrophobic side chains than HNP1 [62]. The HD5 dimer is stabilized by an additional short two-stranded beta sheet and hydrogen bond at the interface, yet dimerization is less important for HD5 than for HNP1 as indicated by the results of studies of forced monomeric defensins [26], [59]. However, a study of HD5 activity against non-enveloped viruses demonstrated that dimerization and multimerization are important for antiviral activity [63]. There is also diversity in modes of tetramerization among the α-defensins. The Ile20/Leu25-mediated HNP1 tetramer does not have an equivalent form in the cases of HNP4 or HD5, although there is evidence that HD5 forms tetramers [64]. Even though it has almost no direct antibacterial activity [22], HD6 exhibits the ability to form long chains of monomers [6], [62] and nanonets that trap bacteria and protect transgenic mice expressing HD6 from Salmonella enterica serovar Typhimurium infection [65].
Hydrophobicity has repercussions beyond oligomerization. In the case of HNP1, the finding that mutating four hydrophobic residues depicted in the crystal as contacting one another to alanine interferes with HNP1 function indicates that hydrophobicity at these positions is potentially evolutionarily significant. However, these results with HNP1 do not apply to α-defensins unviersally. Hydrophobicity is not conserved at any of the four positions (16, 20, 25, or 28) in an alignment of mammalian α-defensins. Radically different examples exist, such as the more basic rabbit neutrophil peptide NP-3a (Swiss-Prot identifier DEF1_RABIT), which has Ser at position 16 and Arg at positions 20, 25 and 28 (HNP1 numbering) [66]. Cationicity could be more significant than hydrophobicity in these cases, whereas for HNP1 and HD5 alanine scanning mutagenesis showed that hydrophobicity is clearly more important than cationicity [26], [28]. For HD5, positions 16, 20, 25, and 28 (HNP1 numbering; to translate into HD5 numbering add 1) are occupied by Ser, Glu, Leu and Leu, respectively, and the latter Leu, Leu29, has been shown to be the most important residue for antibacterial and LF-inhibiting activity [26]. A defensin like NP-3a would presumably form much different quaternary structures than HNP1, if any, because the electrostatic repulsion of the arginine side chains would make dimer and tetramer structures such as those described in the crystallographic section of this work unlikely.
The cumulative functional effect of the quadruple alanine mutations was similar to the effect of the single mutation, W26A [28]. This result sheds new light upon the role of Trp26, since interfering with this residue forming the hydrophobic core probably changes the conformation of several different side chains, including Phe28. The same study also showed that F28A had the second least SPR self-association of all HNP1 alanine scanning mutants, and the second lowest activity against S. aureus [28]. Interestingly, the F28A mutation did not prevent HNP1 from inhibiting LF and binding gp120, indicating that self-association and S. aureus activity are more sensitive to perturbations of HNP1 structure at this position. Here, our studies using the double and quadruple mutants that include the F28A mutation extend the previous findings. Unlike the F28A mutation alone, the Y16A/F28A double mutant and the quadruple mutant LF inhibition IC50 values increased 6-fold and 95-fold, respectively. In the virtual colony count assay, the double mutant Y16A/F28A-HNP1 had a vLD99.9 value against S. aureus 7-fold higher than HNP1, and Y16A/I20A/L25A/F28A-HNP1 failed to exhibit activity equivalent to the vLD90 level against S. aureus. The Y16A/F28A and Y16A/I20A/L25A/F28A mutants were also significantly less potent than HNP1 when assayed against both E. coli and B. cereus.
The functional consequences of the I20A and L25A mutations, alone or in concert, were milder than the Y16A/F28A double mutant or the quadruple mutant. LF inhibition was less than 2-fold different than wild-type for the single mutants and about 2-fold different for the I20A/L25A double mutant, as measured by IC50 values. In the virtual colony count assay, whereas the I20A and L25A mutations each made a significant difference against S. aureus, they did not make a significant difference against E. coli or B. cereus even in the I20A/I25A double mutant. Since Ile20 and Leu25 function as an isologous binding site for the formation of a dimer of dimers, their effect or lack thereof on E. coli is not surprising, given a previous study of N-methylated Ile20 that showed that dimerization is dispensible for E. coli activity [59]. The fact that the I20A/L25A double mutations were irrelevant, but the Y16A/F28A double mutations were quite relevant, for E. coli activity is insightful, because it demonstrates that the hydrophobicity of Tyr16 and Phe28 has repercussions beyond dimerization. If providing hydrophobic contacts for dimerization were the sole function of Tyr16 and Phe28, we would expect the activity of Y16A/F28A-HNP1 to be the same as wild-type HNP1, just as there was no difference between HNP1 or MeIle20-HNP1 when assayed against E. coli [59].
Functional assays correlated with each other and with hydrophobicity. Here we observed the binding affinity of HNP1 and mutants to HNP1 and gp120, as measured by SPR, and the N-terminus of HIV-1 gp41, as measured by fluorescence polarization. In all cases, binding was in agreement with LF inhibition and activity against S. aureus, in the order HNP1> L25A-HNP1> I20A-HNP1> I20A/L25A-HNP1> Y16A/F28A-HNP1> Y16A/I20A/L25A/F28A-HNP1. Correlation between these quantities was expected, given the previously observed correlation for mutants of Trp26 [28]. Self-association on the HNP1 surface was lessened by the mutations, reinforcing that these four positions are important for dimerization and multimerization. Diminished association with the two HIV-1 proteins also suggests that dimerization and multimerization are important for antiviral function. However, the diversity of monomeric, dimeric and tetrameric crystallographic forms suggest that propensity to form certain quaternary structures in the crystal does not always inform or predict defensin function.
Although these crystallographic results do not support the premise that crystal structures are necessarily uniformly functionally insightful, the convergence of evidence from the wide variety of other methods employed has yielded the ability to rank the contribution of each of the side chains mutated to alanine. According to the Wimley-White experimentally determined hydrophobicity scales for proteins at membrane interfaces [67], the five most hydrophobic residues are Trp>Phe>Tyr>Leu>Ile. Our studies of HNP1 have determined the relative importance of residues to be the same, save for the transposition of Ile and Leu: Trp26>Phe28> Tyr16> Ile20> Leu25. The Wimley-White scale was determined using large unilamellar vesicles comprised of the zwitterionic phospholipid palmitoyloleoylphosphatidylcholine; the scale might have differed had they used more anionic membranes typical of bacteria. Nevertheless, the results of our alanine scanning mutagenesis studies of HNP1 with single, double, and quadruple alanine mutants emphasize the importance of hydrophobicity as the primary factor that determines activity, and indicate that the importance of a residue is roughly proportional to its hydrophobicity regardless of structural location.
Although these residues are shown to be at oligomeric interfaces in most crystal structures, the consequences of decreasing hydrophobicity at any of these positions on crystallographic results indicates that the canonical dimer is delicate. In the dynamic environment of the liquid phase as opposed to the solid crystal, a loose association between monomers may allow hydrophobic residues to exchange their affinities for one another for interactions with carbon atoms in diverse targets such as bacterial membranes, enzymes and proteins. Weak van der Waals interactions can be interchangeable and do not necessarily result in specificity. Therefore, while the significance of these four residues for function has been clearly established, the degree to which oligomers such as those implied by the canonical dimer contribute to defensin activity remains an open question worthy of further study.
Supporting Information
Stereo view of the backbone (A) and ribbon (B) traces of superimposed HNP1 mutant monomers with the monomers of wild type HNP1 (PDB:3GNY, [27] ). Wild type HNP1 is shown in green, I20A-HNP1 in turquoise, Y16A/F18A-HNP1 in gold, I20A/L25A-HNP1 in violet, and Y16A/I20A/L25A/F28A-HNP1 in coral. Disulfide bonds are shown in yellow, with sulfurs in yellow, nitrogens in blue, and oxygens in red. The crystal of the I20A-HNP1 mutant contained two defensin molecules in the asymmetric unit, and the I20A/L25A, Y16A/F28A and Y16A/I20A/L25A/F28A mutants crystallized with four defensin molecules in the asymmetric unit. Pairwise superposition of the crystallographically independent copies of I20A-HNP1, I20A/L25A-HNP1, Y16A/F28A-HNP1 and Y16A/I20A/L25A/F28A-HNP1 yielded average Cα RMDS values of 0.09, 0.66, 0.20, and 0.10 Å for 30 atoms, respectively.
(TIF)
Quaternary structure of Y16AF28A-HNP1. (A) Stereo view of the structural alignment of the Y16A/F28A-HNP1 and F28A-HNP1 dimers. Dimers were aligned based on monomer A and colored cyan and green (Y16A/F28A-HNP1) and pink and grey (F28A-HNP1, PDB:3LOE, [28]). Both dimers are stabilized by the same network of main chain H-bonds (shown as blue dashes). The H-bond formed between hydroxyl groups of Tyr3 in the F28A-HNP1 dimer (shown as magenta dashes) is replaced by the H-bond formed between the main chain nitrogen of Ala1 and the hydroxyl of Tyr3 in Y16A/F28A-HNP1 dimer (shown as light blue dashes). The molecular surface buried within the Y16A/F28A-HNP1 dimer is 494 Å2 per monomer, which compares to 500 Å2 for the F28A-HNP1 dimer. (B) Structural alignment of Y16A/F28A-HNP1 and wild type HNP1 tetramers. Tetramers were aligned based on the AB dimer and residues involved in tetramer formation are shown as balls and sticks. The Y16A/F28A-HNP1 dimers are colored as in (A) and the HNP1 dimers (PDB:3GNY, [27]) are colored red and blue. The molecular surface buried within the Y16A/F28A-HNP1 tetramer is 1420 Å2 per dimer, which compares to 1140 Å2 for the HNP1 tetramer.
(TIF)
Acknowledgments
L.Z. was a Guanghua Scholar supported by Xi’an Jiaotong University School of Medicine. We thank the X-ray Crystallography Core Facility of the University of Maryland at Baltimore for providing crystallographic equipment and resources. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and the National Institute of General Medical Sciences.
Funding Statement
This work was supported by the National Institutes of Health Grants AI072732 and AI061482 (to W.L.). L.Z. was a Guanghua Scholar supported by Xi’an Jiaotong University School of Medicine. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bevins CL (2006) Paneth cell defensins: key effector molecules of innate immunity. Biochem Soc Trans 34: 263–266. [DOI] [PubMed] [Google Scholar]
- 2. Ganz T (2003) Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 3: 710–720. [DOI] [PubMed] [Google Scholar]
- 3. Lehrer RI (2004) Primate defensins. Nat Rev Microbiol 2: 727–738. [DOI] [PubMed] [Google Scholar]
- 4. Selsted ME, Ouellette AJ (2005) Mammalian defensins in the antimicrobial immune response. Nat Immunol 6: 551–557. [DOI] [PubMed] [Google Scholar]
- 5. Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415: 389–395. [DOI] [PubMed] [Google Scholar]
- 6. Lehrer RI, Lu W (2012) alpha-Defensins in human innate immunity. Immunological reviews 245: 84–112. [DOI] [PubMed] [Google Scholar]
- 7. Pazgier M, Hoover DM, Yang D, Lu W, Lubkowski J (2006) Human beta-defensins. Cell Mol Life Sci 63: 1294–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lehrer RI, Cole AM, Selsted ME (2012) theta-Defensins: cyclic peptides with endless potential. The Journal of biological chemistry 287: 27014–27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ganz T, Selsted ME, Szklarek D, Harwig SS, Daher K, et al. (1985) Defensins. Natural peptide antibiotics of human neutrophils. J Clin Invest 76: 1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Selsted ME, Harwig SS, Ganz T, Schilling JW, Lehrer RI (1985) Primary structures of three human neutrophil defensins. J Clin Invest 76: 1436–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh A, Bateman A, Zhu QZ, Shimasaki S, Esch F, et al. (1988) Structure of a novel human granulocyte peptide with anti-ACTH activity. Biochem Biophys Res Commun 155: 524–529. [DOI] [PubMed] [Google Scholar]
- 12. Gabay JE, Scott RW, Campanelli D, Griffith J, Wilde C, et al. (1989) Antibiotic proteins of human polymorphonuclear leukocytes. Proc Natl Acad Sci U S A 86: 5610–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilde CG, Griffith JE, Marra MN, Snable JL, Scott RW (1989) Purification and characterization of human neutrophil peptide 4, a novel member of the defensin family. J Biol Chem 264: 11200–11203. [PubMed] [Google Scholar]
- 14. Jones DE, Bevins CL (1992) Paneth cells of the human small intestine express an antimicrobial peptide gene. The Journal of Biological Chemistry 267: 23216–23225. [PubMed] [Google Scholar]
- 15. Jones DE, Bevins CL (1993) Defensin-6 mRNA in human Paneth cells: implications for antimicrobial peptides in host defense of the human bowel. Febs Letters 315: 187–192. [DOI] [PubMed] [Google Scholar]
- 16. Lehrer RI, Barton A, Daher KA, Harwig SS, Ganz T, et al. (1989) Interaction of human defensins with Escherichia coli. Mechanism of bactericidal activity. J Clin Invest 84: 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Leeuw E, Li C, Zeng P, Diepeveen-de Buin M, Lu WY, et al. (2010) Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett 584: 1543–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oeemig JS, Lynggaard C, Knudsen DH, Hansen FT, Norgaard KD, et al. (2012) Eurocin, a new fungal defensin: structure, lipid binding, and its mode of action. The Journal of biological chemistry 287: 42361–42372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sass V, Schneider T, Wilmes M, Korner C, Tossi A, et al. (2010) Human beta-defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect Immun 78: 2793–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmitt P, Wilmes M, Pugniere M, Aumelas A, Bachere E, et al. (2010) Insight into invertebrate defensin mechanism of action: oyster defensins inhibit peptidoglycan biosynthesis by binding to lipid II. The Journal of biological chemistry 285: 29208–29216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, et al. (2010) Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 328: 1168–1172. [DOI] [PubMed] [Google Scholar]
- 22. Ericksen B, Wu Z, Lu W, Lehrer RI (2005) Antibacterial activity and specificity of the six human {alpha}-defensins. Antimicrob Agents Chemother 49: 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao L, Ericksen B, Wu X, Zhan C, Yuan W, et al. (2012) Invariant gly residue is important for alpha-defensin folding, dimerization, and function: a case study of the human neutrophil alpha-defensin HNP1. The Journal of biological chemistry 287: 18900–18912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim C, Gajendran N, Mittrucker HW, Weiwad M, Song YH, et al. (2005) Human alpha-defensins neutralize anthrax lethal toxin and protect against its fatal consequences. Proc Natl Acad Sci U S A 102: 4830–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mayer-Scholl A, Hurwitz R, Brinkmann V, Schmid M, Jungblut P, et al. (2005) Human neutrophils kill Bacillus anthracis. PLoS Pathog 1: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajabi M, Ericksen B, Wu X, de Leeuw E, Zhao L, et al. (2012) Functional determinants of human enteric alpha-defensin HD5: crucial role for hydrophobicity at dimer interface. The Journal of biological chemistry 287: 21615–21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wei G, de Leeuw E, Pazgier M, Yuan W, Zou G, et al. (2009) Through the looking glass, mechanistic insights from enantiomeric human defensins. J Biol Chem 284: 29180–29192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wei G, Pazgier M, de Leeuw E, Rajabi M, Li J, et al. (2010) Trp-26 imparts functional versatility to human alpha-defensin HNP1. J Biol Chem 285: 16275–16285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hill CP, Yee J, Selsted ME, Eisenberg D (1991) Crystal structure of defensin HNP-3, an amphiphilic dimer: mechanisms of membrane permeabilization. Science 251: 1481–1485. [DOI] [PubMed] [Google Scholar]
- 30. Ashkenazi A, Presta LG, Marsters SA, Camerato TR, Rosenthal KA, et al. (1990) Mapping the CD4 binding site for human immunodeficiency virus by alanine-scanning mutagenesis. Proceedings of the National Academy of Sciences of the United States of America 87: 7150–7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cunningham BC, Wells JA (1989) High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244: 1081–1085. [DOI] [PubMed] [Google Scholar]
- 32. Kristensen C, Kjeldsen T, Wiberg FC, Schaffer L, Hach M, et al. (1997) Alanine scanning mutagenesis of insulin. The Journal of biological chemistry 272: 12978–12983. [DOI] [PubMed] [Google Scholar]
- 33. Blaber M, Baase WA, Gassner N, Matthews BW (1995) Alanine scanning mutagenesis of the alpha-helix 115–123 of phage T4 lysozyme: effects on structure, stability and the binding of solvent. Journal of molecular biology 246: 317–330. [DOI] [PubMed] [Google Scholar]
- 34. Williams AD, Shivaprasad S, Wetzel R (2006) Alanine scanning mutagenesis of Abeta(1–40) amyloid fibril stability. Journal of molecular biology 357: 1283–1294. [DOI] [PubMed] [Google Scholar]
- 35. Yu MH, Weissman JS, Kim PS (1995) Contribution of individual side-chains to the stability of BPTI examined by alanine-scanning mutagenesis. Journal of molecular biology 249: 388–397. [DOI] [PubMed] [Google Scholar]
- 36. Gibbs CS, Zoller MJ (1991) Rational scanning mutagenesis of a protein kinase identifies functional regions involved in catalysis and substrate interactions. The Journal of biological chemistry 266: 8923–8931. [PubMed] [Google Scholar]
- 37. Morrison KL, Weiss GA (2001) Combinatorial alanine-scanning. Current opinion in chemical biology 5: 302–307. [DOI] [PubMed] [Google Scholar]
- 38. Dill KA (1997) Additivity principles in biochemistry. The Journal of biological chemistry 272: 701–704. [DOI] [PubMed] [Google Scholar]
- 39. Wells JA (1990) Additivity of mutational effects in proteins. Biochemistry 29: 8509–8517. [DOI] [PubMed] [Google Scholar]
- 40. Schnolzer M, Alewood P, Jones A, Alewood D, Kent SB (1992) In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int J Pept Protein Res 40: 180–193. [DOI] [PubMed] [Google Scholar]
- 41. Wu Z, Powell R, Lu W (2003) Productive folding of human neutrophil α-defensins in vitro without the pro-peptide. J Am Chem Soc 125: 2402–2403. [DOI] [PubMed] [Google Scholar]
- 42. Pace CN, Vajdos F, Fee L, Grimsley G, Gray T (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci 4: 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Otwinowski Z, Minor W (1997) Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- 44. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, et al. (2007) Phaser crystallographic software. Journal of applied crystallography 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta crystallographica Section D, Biological crystallography 53: 240–255. [DOI] [PubMed] [Google Scholar]
- 46. Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, et al. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta crystallographica Section D, Biological crystallography 58: 1948–1954. [DOI] [PubMed] [Google Scholar]
- 47. Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- 48. Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, et al. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic acids research 35: W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst 24: 946–950. [Google Scholar]
- 50. Chang TL, Vargas J Jr, DelPortillo A, Klotman ME (2005) Dual role of alpha-defensin-1 in anti-HIV-1 innate immunity. J Clin Invest 115: 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klotman ME, Chang TL (2006) Defensins in innate antiviral immunity. Nat Rev Immunol 6: 447–456. [DOI] [PubMed] [Google Scholar]
- 52. Demirkhanyan LH, Marin M, Padilla-Parra S, Zhan C, Miyauchi K, et al. (2012) Multifaceted mechanisms of HIV-1 entry inhibition by human alpha-defensin. The Journal of biological chemistry 287: 28821–28838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lay FT, Mills GD, Poon IK, Cowieson NP, Kirby N, et al. (2012) Dimerization of plant defensin NaD1 enhances its antifungal activity. The Journal of biological chemistry 287: 19961–19972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hoover DM, Rajashankar KR, Blumenthal R, Puri A, Oppenheim JJ, et al. (2000) The structure of human beta-defensin-2 shows evidence of higher order oligomerization. The Journal of biological chemistry 275: 32911–32918. [DOI] [PubMed] [Google Scholar]
- 55. Schibli DJ, Hunter HN, Aseyev V, Starner TD, Wiencek JM, et al. (2002) The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J Biol Chem 277: 8279–8289. [DOI] [PubMed] [Google Scholar]
- 56. Daly NL, Chen YK, Rosengren KJ, Marx UC, Phillips ML, et al. (2007) Retrocyclin-2: structural analysis of a potent anti-HIV theta-defensin. Biochemistry 46: 9920–9928. [DOI] [PubMed] [Google Scholar]
- 57. Zhang XL, Selsted ME, Pardi A (1992) NMR studies of defensin antimicrobial peptides. 1. Resonance assignment and secondary structure determination of rabbit NP-2 and human HNP-1. Biochemistry 31: 11348–11356. [DOI] [PubMed] [Google Scholar]
- 58. Zhang Y, Doherty T, Li J, Lu W, Barinka C, et al. (2010) Resonance assignment and three-dimensional structure determination of a human alpha-defensin, HNP-1, by solid-state NMR. J Mol Biol 397: 408–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pazgier M, Wei G, Ericksen B, Jung G, Wu Z, et al. (2012) Sometimes it takes two to tango: contributions of dimerization to functions of human alpha-defensin HNP1 peptide. The Journal of biological chemistry 287: 8944–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bai Y, Liu S, Jiang P, Zhou L, Li J, et al. (2009) Structure-dependent charge density as a determinant of antimicrobial activity of peptide analogues of defensin. Biochemistry 48: 7229–7239. [DOI] [PubMed] [Google Scholar]
- 61. Hong M, Su Y (2011) Structure and dynamics of cationic membrane peptides and proteins: insights from solid-state NMR. Protein science : a publication of the Protein Society 20: 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Szyk A, Wu Z, Tucker K, Yang D, Lu W, et al. (2006) Crystal structures of human alpha-defensins HNP4, HD5, and HD6. Protein Sci 15: 2749–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gounder AP, Wiens ME, Wilson SS, Lu W, Smith JG (2012) Critical determinants of human alpha-defensin 5 activity against non-enveloped viruses. The Journal of biological chemistry 287: 24554–24562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lehrer RI, Jung G, Ruchala P, Andre S, Gabius HJ, et al. (2009) Mutilvalent binding of carbohydrates by the human α-defensin HD5. J Immunol 183: 480–490. [DOI] [PubMed] [Google Scholar]
- 65. Chu H, Pazgier M, Jung G, Nuccio SP, Castillo PA, et al. (2012) Human alpha-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science 337: 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Selsted ME, Brown DM, DeLange RJ, Harwig SS, Lehrer RI (1985) Primary structures of six antimicrobial peptides of rabbit peritoneal neutrophils. The Journal of biological chemistry 260: 4579–4584. [PubMed] [Google Scholar]
- 67. Wimley WC, White SH (1996) Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nature structural biology 3: 842–848. [DOI] [PubMed] [Google Scholar]
- 68. Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, et al. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta crystallographica Section D, Biological crystallography 54: 905–921. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Stereo view of the backbone (A) and ribbon (B) traces of superimposed HNP1 mutant monomers with the monomers of wild type HNP1 (PDB:3GNY, [27] ). Wild type HNP1 is shown in green, I20A-HNP1 in turquoise, Y16A/F18A-HNP1 in gold, I20A/L25A-HNP1 in violet, and Y16A/I20A/L25A/F28A-HNP1 in coral. Disulfide bonds are shown in yellow, with sulfurs in yellow, nitrogens in blue, and oxygens in red. The crystal of the I20A-HNP1 mutant contained two defensin molecules in the asymmetric unit, and the I20A/L25A, Y16A/F28A and Y16A/I20A/L25A/F28A mutants crystallized with four defensin molecules in the asymmetric unit. Pairwise superposition of the crystallographically independent copies of I20A-HNP1, I20A/L25A-HNP1, Y16A/F28A-HNP1 and Y16A/I20A/L25A/F28A-HNP1 yielded average Cα RMDS values of 0.09, 0.66, 0.20, and 0.10 Å for 30 atoms, respectively.
(TIF)
Quaternary structure of Y16AF28A-HNP1. (A) Stereo view of the structural alignment of the Y16A/F28A-HNP1 and F28A-HNP1 dimers. Dimers were aligned based on monomer A and colored cyan and green (Y16A/F28A-HNP1) and pink and grey (F28A-HNP1, PDB:3LOE, [28]). Both dimers are stabilized by the same network of main chain H-bonds (shown as blue dashes). The H-bond formed between hydroxyl groups of Tyr3 in the F28A-HNP1 dimer (shown as magenta dashes) is replaced by the H-bond formed between the main chain nitrogen of Ala1 and the hydroxyl of Tyr3 in Y16A/F28A-HNP1 dimer (shown as light blue dashes). The molecular surface buried within the Y16A/F28A-HNP1 dimer is 494 Å2 per monomer, which compares to 500 Å2 for the F28A-HNP1 dimer. (B) Structural alignment of Y16A/F28A-HNP1 and wild type HNP1 tetramers. Tetramers were aligned based on the AB dimer and residues involved in tetramer formation are shown as balls and sticks. The Y16A/F28A-HNP1 dimers are colored as in (A) and the HNP1 dimers (PDB:3GNY, [27]) are colored red and blue. The molecular surface buried within the Y16A/F28A-HNP1 tetramer is 1420 Å2 per dimer, which compares to 1140 Å2 for the HNP1 tetramer.
(TIF)