

Abstract
Osteoblastoma is a bone forming tumor with histological features highly similar to osteoid osteoma; the discrimination between the tumor types is based on size and growth pattern. The vast majority of osteoblastomas are benign but there is a group of so-called aggressive osteoblastomas that can be diagnostically challenging at the histopathological level. The genetic aberrations required for osteoblastoma development are not known and no genetic difference between conventional and aggressive osteoblastoma has been reported. In order to identify recurrent genomic aberrations of importance for tumor development we applied cytogenetic and/or SNP array analyses on nine conventional and two aggressive osteoblastomas. The conventional osteoblastomas showed few or no acquired genetic aberrations while the aggressive tumors displayed heavily rearranged genomes. In one of the aggressive osteoblastomas, three neighboring regions in chromosome band 22q12 were homozygously deleted. Hemizygous deletions of these regions were found in two additional cases, one aggressive and one conventional. In total, 10 genes were recurrently and homozygously lost in osteoblastoma. Four of them are functionally involved in regulating osteogenesis and/or tumorigenesis. MN1 and NF2 have previously been implicated in the development of leukemia and solid tumors, and ZNRF3 and KREMEN1 are inhibitors of the Wnt/beta-catenin signaling pathway. In line with deletions of the latter two genes, high beta-catenin protein expression has previously been reported in osteoblastoma and aberrations affecting the Wnt/beta-catenin pathway have been found in other bone lesions, including osteoma and osteosarcoma.
Introduction
Osteoblastoma is a bone forming tumor that is usually located in the medullary cavity of the bone [1]. The disease can affect patients at any age but has a predilection for males in their teens and young adulthood. At the cellular level, the tumor is identical to osteoid osteoma; both tumor types show rich vascularization, irregular osteoid with osteoblasts and often osteoclast-type multinucleated giant cells. Differentiation between the two tumor types is based on size [2]. Osteoid osteoma has a limited growth potential and seldom exceeds 1 cm in largest diameter. In contrast, lesions larger than 2 cm are not considered to have a restricted growth potential and are referred to as osteoblastomas (also known as giant osteoid osteomas). Osteoblastoma typically shows a non-infiltrative growth pattern and when resected with free margins recurrences are uncommon. The treatment is therefore based on surgery alone and the prognosis is excellent [1]. However, there is a group of intra-osseous osteoblastic tumors that can be diagnostically challenging at the histopathological level [3]. These tumors have been referred to as aggressive, epithelioid or malignant osteoblastoma. Currently, they are considered within the morphological spectrum of osteoblastoma [1], and have the same clinical behavior. A very rare subtype of osteosarcoma exists, so-called osteoblastoma-like osteosarcoma, which shares some morphological features with osteoblastoma, but clinically behaves like conventional high-grade osteosarcoma [4]. Osteosarcomas, including these rare variants, generally present highly complex karyotypes with multiple aberrations [5]. Low-grade central osteosarcomas do not pose a histological differential diagnosis and are characterized at the genetic level by frequent gains of MDM2. In osteoblastoma the genetic findings are heterogeneous ranging from single balanced structural rearrangements to multiple and complex changes. No recurrent, tumor-associated aberration has been described.
In the present study, we have applied cytogenetic and single nucleotide polymorphism (SNP) array analyses on osteoblastomas in order to identify recurrent genomic aberrations of importance for tumor development. We have also re-analyzed previously published global gene expression data on osteoblastoma [6], in order to evaluate the possible impact of genomic alterations.
Materials and Methods
Ethics statement
All samples were obtained after informed written consent from patients or from patient’s parents. The study was approved by the Regional Ethics Committee of Lund University.
Patient information and tumor material
Tumor material was available from 11 osteoblastoma patients, treated at the Leiden University Medical Center, Leiden, The Netherlands, the Skåne University Hospital, Lund, Sweden and the Karolinska Hospital, Stockholm, Sweden. The age of the patients ranged from 10-44 years and the majority (9 of 11) of patients were male. Two of the tumors were classified as epithelioid or aggressive and the remaining cases were conventional osteoblastomas. Three of the tumors recurred and in the remaining patients there was no evidence of disease 25-253 months after diagnosis. Detailed patient information can be found in Table 1.
Table 1. Clinical and cytogenetic features of eleven osteoblastomas.
| Case a | Age/Sex | Location | Follow-up b | Karyotype c | SNP array analysis |
|---|---|---|---|---|---|
| 1d | 35/M | Skull | R 21, 53, NED 60 | 43,X,-Y,der(1)t(1;?22)(p3?;q?),add(2)(q3?),-6,del(8)(p12),der(9)t(9;17)(p12;q11),-12,der(14)t(?1;14)(?;q3?),-17,der(19)t(19;22)t(1;22)t(12;22)t(12;17),der(22)t(1;22)(?;q1?)[23]/46,XY[7] | Aberrant |
| 2e 13848:85 | 23/M | Proximal humerus | NED 168 | 44,X,-Y,add(1)(p34),del(1)(q21),del(2)(p21p23),+3,del(3)(p21)x2,del(6)(q15),der(6)t(6;13)(q27;q12),+10,der(10)t(8;10)(q11;q26)x2,del(12)(p11),-13,+der(15)t(1;15)(q21;p13)ins(1;?)(q32;?),+16,-17,add(17)(q11),-18,-22,-22[16]/43,idem,-15[3] | Aberrant |
| 3 | 14/M | Vertebra | NED 48 | 44-45,XY,-22[cp 43]/46,XY[25] | No |
| 4 | 17/F | Proximal femur | NED 25 | 48,XX,+2mar[9]/46,XX[17] | No |
| 5 | 20/M | Proximal humerus | R 18, NED 21 | 46,XY,add(18)(q2?3)[7]/46,XY[17] | No |
| 6 | 16/M | Proximal femur | NED 43 | 46,XY[25] | Normal |
| 7 | 10/F | Sacrum | R 9, NED 18 | 46,XX[25] | Normal |
| 8 | 37/M | Os ilium | NED 253 | 46,XY[25] | Normal |
| 9 | 44/M | Vertebra | NED 156 | No | Normal |
| 10 | 42/M | Vertebra | NED 36 | 46,XY[18] | Normal |
| 11 | 30/M | Aceta-bulum | NED156 | No | Normal |
Previously published karyotypes are indicated by their reference and case numbers in the Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (http://cgap.nci.nih.gov/Chromosomes/Mitelman).
Follow-up time given in months. R = recurrence; NED = no evidence of disease.
Karyotypes in cases 1 and 10 are based on data from COBRA FISH analysis.
Epithelioid osteoblastoma.
Aggressive osteoblastoma.
Cytogenetic analyses
Chromosome banding and COBRA fluorescence in situ hybridization analyses were performed at the Departments of Clinical Genetics, Lund and Molecular Cell Biology, Leiden, and karyotypes were described according to the recommendations in ISCN 2009 [7]. Chromosome preparations were prepared as described [8].
Genomic copy number and loss of heterozygosity analyses
SNP array analysis was used for combined DNA copy number and loss of heterozygosity investigation. In eight of the cases (cases 1, 2 and 6-11), fresh frozen tumor biopsies were available. DNA was extracted according to standard procedures [9], and hybridized onto Illumina Human Omni-Quad BeadChips, containing more than 1 million reporters, following protocols supplied by the manufacturer (Illumina, San Diego, CA). Data analysis was performed using the GenomeStudio software (Illumina), detecting imbalances by visual inspection. Constitutional copy number variations were excluded through comparison with the Database of Genomic Variants, http://projects.tcag.ca/variation/ [10].
Global gene expression analyses
Global gene expression analyses was previously performed on osteoblastomas and osteosarcoma as well as the putative progenitor cells of the tumors, i.e., mesenchymal stem cells (MSC) and the same MSC differentiated into osteoblasts [6]. In brief, RNA was extracted from frozen tissue sections, labeled and hybridized onto Hu133A GeneChip Arrays according to the manufacturer’s protocol (Affymetrix, Santa Clara, CA). Gene expression data were normalized, background-corrected, and summarized by using the Robust Multichip Analysis algorithm implemented in the Expression Console version 1.1 software (Affymetrix). Correlation-based principal component analysis and hierarchical clustering analysis were performed using the Qlucore Omics Explorer version 2.3 (Qlucore AB, Lund, Sweden). Differences between tumor groups in log2 transformed expression data were calculated using a t-test, and corrections for multiple testing were based on the Benjamini-Hochberg method (Qlucore AB). Genes with p < 0.001 and a false discovery rate (FDR) < 0.01 were considered significantly altered.
Genomic sequencing of candidate target genes
Sanger sequencing was used to screen the coding regions of MN1, ZNRF3, KREMEN1 and NF2 for mutations. PCR primers and protocols for all four genes are available in Table S1.
Results
Recurrent deletions affect chromosome 22 in osteoblastoma
Cases 1 and 2 were diagnosed as epithelioid and aggressive osteoblastomas, respectively. In both cases, chromosome banding and COBRA fluorescent in situ hybridization analyses revealed near-diploid karyotypes with multiple and complex aberrations including deletions, gains, translocations and insertions (Table 1). In both cases, SNP array analyses showed acquired copy number alterations that were in line with the complex karyotypes. SNP array analysis of case 1 showed alterations in chromosomes 1, 2, 6, 8, 9, 12, 17 and 22, including homozygous deletions in chromosome 22 (Figure 1, Table 2). These deletions affected three distinct regions between 0.06-1.5 Mb in size and in total 10 genes, including MN1, ZNRF3, KREMEN1 and NF2. In case 2, SNP array analysis showed copy number aberrations affecting chromosomes 1, 2, 3, 6, 8, 10, 16, 17, 18 and 22 (Table 2). However, loss of both copies of chromosome 22, which was found by chromosome banding, could not be corroborated by SNP array analysis. This is in line with the fact that nullisomy is a very rare finding, even at G-banding analysis [5], and that such losses most likely result in cell death. Instead, a hemizygous deletion affecting major parts of chromosome 22 was found by SNP arrays (Figure 1, Table 2). This deletion was overlapping with hemi- and homozygous deletions detected in case 1 (Figure 1). Two additional recurrent aberrations were found by SNP arrays. In chromosome 1, five small deletions (<1 Mb) affecting case 1 overlapped with a 42 Mb deletion in case 2. Chromosome 6 was deleted in case 1 and the long arm of this chromosome was lost in case 2.
Figure 1. Recurrent deletions in osteoblastoma affect chromosome 22.

By SNP array analyses, hemi-and homozygous deletions affecting chromosome 22 were found in cases 1 and 2. The upper parts of the figure display SNP array plots of the respective case and the lower part show a summary of the losses where blue and green bars represent hemi- and homozygous deletions, respectively. The locations of four homozygously lost genes implicated in bone formation and/or tumorigenesis are shown.
Table 2. Acquired DNA copy number aberrations detected by SNP arrays in two osteoblastomas.
| Case | Chromosome band | Base pair position (Mb)a | Aberration | Gene |
|---|---|---|---|---|
| 1 | 1p35 | 29.616-30.277 | Deletion | |
| 1 | 1p35 | 30.479-30.597 | Deletion | |
| 1 | 1p35 | 30.899-31.179 | Deletion | |
| 1 | 1p34 | 39.887-39.967 | Deletion | |
| 1 | 1p34 | 41.552-41.615 | Deletion | |
| 1 | 1p34 | 45.067-45.139 | Deletion | |
| 1 | 1p34 | 45.853-45.962 | Deletion | |
| 1 | 2p12 | 80.608-82.511 | Gain | |
| 1 | Chromosome 6 | 0.090-170.919 | Deletion | |
| 1 | 8p21-p23 | 0.165-28.624 | Deletion | |
| 1 | 9p11-p24 | 0.047-47.203 | Deletion | |
| 1 | 12p11-p13 | 0.148-34.856 | Deletion | |
| 1 | 12q12-q13 | 38.922-52.786 | Deletion | |
| 1 | 12q13 | 55.311-56.366 | Deletion | |
| 1 | 12q24 | 124.643-133.779 | Deletion | |
| 1 | 17p11 | 17.271-21.528 | Deletion | |
| 1 | 22q11-q12 | 23.704-27.427 | Deletion | |
| 1 | 22q12 | 27.458-28.964 | Homozygous deletion | MN1, PITPNB, MIR3199-1 MIR3199-2, TTC28 |
| 1 | 22q12 | 29.055-29.319 | Deletion | |
| 1 | 22q12 | 29.361-29.607 | Homozygous deletion | ZNRF3, C22orf31, KREMEN1, EMID1 |
| 1 | 22q12 | 29.835-30.031 | Deletion | |
| 1 | 22q12 | 30.048-30.111 | Homozygous deletion | NF2 |
| 1 | 22q12 | 30.112-30.163 | Deletion | |
| 1 | 22q12 | 30.391-30.438 | Deletion | |
| 1 | 22q12 | 36.162-36.444 | Deletion | |
| 1 | 22q13 | 45.711-46.280 | Deletion | |
| 2 | 1p34-p36 | 0.082-42.085 | Deletion | |
| 2 | 2p23-p25 | 0.015-25.967 | Deletion | |
| 2 | 3p11-p21 | 46.961-90.494 | Deletion | |
| 2 | 3q11-q29 | 93.537-197.896 | Gain | |
| 2 | 6q11-q27 | 61.885-170.919 | Deletion | |
| 2 | 8q11-q24 | 46.846-146.293 | Gain | |
| 2 | Chromosome 10 | 0.082-135.523 | Gain | |
| 2 | Chromosome 14 | 18.398-107.288 | Copy number neutral loss of heterozygosity | |
| 2 | Chromosome 16 | 0.087-90.170 | Gain | |
| 2 | 17p11-p13 | 0.007-17.179 | Deletion | |
| 2 | 17p11 | 17.231-22.261 | Gain | |
| 2 | 17q11 | 25.264-27.505 | Deletion | |
| 2 | Chromosome 18 | 0.028-78.015 | Deletion | |
| 2 | 22p11-q13 | 14.676-38.206 | Deletion | |
| 2 | 22q13 | 47.241-51.667 | Gain |
Human Feb. 2009 (GRCh37/hg19) Assembly
Three additional cases displayed chromosome alterations by cytogenetic analysis (cases 3-5; Table 1). All three cases were conventional osteoblastomas and they showed simple karyotypes with few alterations: loss of chromosome 22 (case 3), two supernumerary marker chromosomes (case 4) and addition of material on chromosome 18 (case 5).
In summary, five out of eleven cases showed acquired DNA copy number alterations using chromosome banding and/or SNP array analyses; three of these showed homo- and/or hemizygous deletions in chromosome 22 (Tables 1 and 2). Four cases showed normal karyotypes, which could be attributed to the growth normal fibroblast.
Gene expression analysis supports active Wnt/beta-catenin signaling in osteoblastoma
By unsupervised principal component analysis, osteoblastoma displayed a distinct global gene expression signature (Figure 2). In total, 140 genes showed a significantly different expression between osteoblastoma and osteosarcoma (p < 0.001, FDR < 0.01; Figure 2, Tables S2 and S3). Many of the genes (>40) with a high expression level in osteoblastoma were related to bone metabolism. At least four of these - BMP2, BMP4, PTGS2 and MMP16 - are known to be induced by the canonical Wnt signaling pathway that controls beta-catenin. The osteosarcoma samples were chosen as the primary control tissue to avoid gene expression artifacts introduced by cell culturing. However, as the Wnt signaling pathway is known to be affected also in the control group of osteosarcomas [11], we compared the gene expression levels also between osteoblastoma and two different types of cultured cells (MSC and MSC differentiated into osteoblasts). We could confirm that the four genes mentioned above showed high expression levels in osteoblastoma, regardless of the reference group (Figure 2).
Figure 2. Gene expression signature of osteoblastoma.
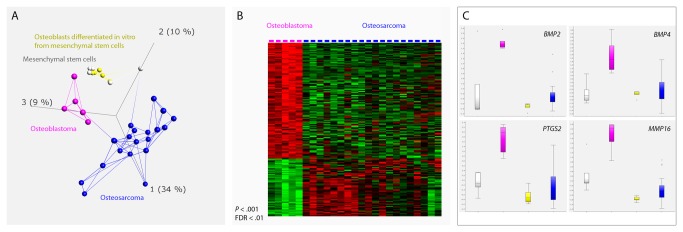
(A) Unsupervised principal component analysis based on the expression of the 1297 most variable genes (σ/σmax = 0.3) shows that the five osteoblastomas form a group that has an expression profile separate from the osteosarcomas, mesenchymal stem cells, and osteoblasts differentiated in vitro from mesenchymal stem cells. The first three principal components, representing 34%, 10%, and 9% of the variance, are displayed. Lines connect the three nearest neighbors. By subsequently comparing osteoblastoma and osteosarcoma, 140 genes showed a significantly different expression (p < 0.001, FDR < 0.01; Tables S2 and S3). (B) The differentially expressed genes are displayed in a heat map. Genes with high and low expression values are labeled in red and green, respectively. (C) Many of the highly expressed genes in osteoblastoma are known to be involved in bone metabolism and at least four of them are induced by the Wnt/beta-catenin signaling pathway; BMP2, BMP4, PTGS2 and MMP16. Boxes range from the 25th to the 75th percentile. The box whiskers are set at the lowest data point value still within 1.5 times the box range of the lower box limit, and at the highest data point value still within 1.5 times the box range of the upper box limit. The median is displayed as a dotted band. Outliers are defined as data point values falling outside of the box whisker limits.
Sanger sequencing did not identify any mutations in candidate targets
The coding regions of the genes MN1, ZNRF3, KREMEN1 and NF2 were sequenced in case 2 because this case had showed heterozygous deletions of these genes at SNP array analysis. No mutation was detected (data not shown). Exon 1 of ZNRF3 and KREMEN1, respectively, were not successfully amplified and were thus not evaluated. The lack of amplified material from parts of ZNRF3 and KREMEN1 could be due to technical failure and was not interpreted as acquired mutations.
Discussion
The genetic mechanisms underlying osteoblastoma development are largely unknown and no obvious genetic difference between conventional and so-called aggressive tumors has been identified [5]. Cytogenetic information is available from six published osteoblastomas and complex rearrangements are detected in some of them whereas others display simple karyotypes with few changes, irrespective of whether the tumors are termed conventional or aggressive (Table 3). In the present study, we found few or no genomic changes in nine conventional osteoblastomas while the two aggressive tumors displayed heavily rearranged genomes. The most common finding was deletion of whole or parts of the long arm of chromosome 22, which was detected by SNP array and/or cytogenetic analyses in three of the cases. Loss of chromosome 22 and rearrangement of 22q12, respectively, were reported in one case each of the previously published osteoblastomas. Furthermore, it could be noted that deletions affecting 22q13 has been described in two of three reported karyotypes from osteoid osteomas [12]. Available data thus indicate that a candidate target gene for osteoblastoma development may reside in the long arm of chromosome 22. In support of this, one of the aggressive tumors investigated here displayed homozygous deletions of three neighboring regions in 22q12. In total, ten genes were affected by the deletions and four of them may be particularly interesting due to their involvement in osteogenesis and/or tumorigenesis; ZNRF3, KREMEN1, MN1, and NF2.
Table 3. Clinical and cytogenetic features of previously published osteoblastomas.
| Casea | Age/Sex | Locationb | Diagnosisc | Karyotype |
|---|---|---|---|---|
| 5004:1 | 7/M | Vertebra | OB | 46,XY,+der(15)t(15;20)(p11;p11),der(17)t(17;20)(p11-12;q11),-20 |
| 7676:8 | 34/M | Os ilium | Aggr OB | 52,Y,t(X;11)(q22;p14),+2,del(5)(q22),der(6;8)(p10;q10),+del(9)(q31q33),add(12)(q24),-13,add(13)(p11),add(14)(p?),+16,add(18)(p11),+19,+add(19)(p13),-21,+3mar |
| 8024:16 | 62/F | N/A | OB | 39-40,XX,der(3;8)dic(3;8)(p25;p11)ins(3;?)(p25;?),der(4)t(4;?22)(p15;q12),r(7),-9,der(11)t(11;12)(p13;q12),-12,-14,add(19)(p13),dic(19;22)(q11;p13),-22/39-40,idem,add(19)(p13) |
| 8416:1 | 14/F | Mandible | LCET OB | 46,XX,del(1)(q42),t(1;5;17;22)(p32-33;p13;q21;q12) |
| 13047:1 | 23/F | Femur | OB | 46,XX,t(1;2;14)(q42;q13;q24) |
| 13302:1 | 12/F | Femur | Aggr OB | 46,XX,t(4;7;14)(q23-25;q31;q31) |
Previously published karyotypes are indicated by their reference and case numbers in The Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (http://cgap.nci.nih.gov/Chromosomes/Mitelman).
N/A = not available.
OB = osteoblastoma; Aggr OB = aggressive osteoblastoma; LCET OB = large cell, epithelioid, telangiectatic OB.
ZNRF3 and KREMEN1 are negative regulators of Wnt signaling transduction [13,14]. Wnt normally acts through different pathways to regulate cell proliferation, cell polarity and cell fate during embryogenesis and adult tissue homeostasis [15]. The different Wnt signaling pathways include the canonical and the non-canonical pathways, the former is also known as the beta-catenin-dependent pathway. In this pathway, cytoplasmic beta-catenin is constantly phosphorylated leading to ubiquitination and degradation when Wnt is absent [15]. In contrast, when Wnt protein is present it assembles its receptors - the Frizzled family of receptors and various co-receptors including the LDL receptor-related proteins 5 and 6 (LRP5/6) - which in turn prevents phosphorylation and degradation of beta-catenin. Stabilized beta-catenin will accumulate and translocate to the nucleus to form complexes with transcription factors and activate Wnt target gene expression. The Wnt/beta-catenin pathway regulates, among other things, bone mass and aberrations in this pathway has been found in e.g. osteodegenerative conditions and osteosarcoma [11,15]. To adjust and restrict Wnt signaling activity there are several negative regulators of this pathway. ZNRF3 encodes a cell-surface transmembrane ubiquitin ligase which reduces Wnt signals by promoting degradation of Frizzled and LRP6 receptors [13,16]. In absence of ZNRF3, membrane levels of Wnt receptors increase and this enhances Wnt signaling through both the canonical and non-canonical pathways [13]. KREMEN1 is a transmembrane receptor that inhibits the Wnt pathway by forming a ternary complex with Dickkopf1 (Dkk1) and LRP5/6 [14]. When assembled, this complex is removed from the plasma membrane by endocytosis, thereby blocking Wnt signaling through LRP5/6. Taken together, both loss of ZNRF3 and KREMEN1 would theoretically result in increased accumulation of beta-catenin that will translocate to the nucleus and activate Wnt target gene expression. In line with this, loss of ZNRF3 has been shown to result in accumulation of beta-catenin and loss of KREMEN1 has been implicated in increased bone formation [13,17,18]. Here, we hypothesized that if loss of genes such as ZNRF3 and KREMEN1 and subsequent activation of beta-catenin is important for osteoblastoma development we would find high expression levels of genes activated by Wnt/beta-catenin in these tumors. Indeed, Wnt/beta-catenin target genes involved in bone metabolism, such as BMP2, BMP4, PTGS2 and MMP16, showed high expression levels in osteoblastoma [19-25]. In line with this, both membranous and nuclear beta-catenin has been found to be strongly expressed in osteoblastoma [11]. Furthermore, constitutional APC mutations leading to accumulation of beta-catenin is associated with the development of osteoma, a benign bone lesion that shares some morphological features with osteoid osteoma and osteoblastoma [26,27].
ZNRF3 and KREMEN1 may thus be potential target genes of one of the homozygously deleted regions. Possible targets of the two adjacent, homozygously deleted regions may be MN1 and NF2. MN1 encodes a transcription regulator and was originally identified as a gene disrupted in meningioma and as part of a fusion gene in leukemia. MN1 has been shown to be involved in osteoblast proliferation and differentiation [28]. More specifically, osteoblasts derived from MN1 knock-out mice are defective in osteoblast proliferation, migration, differentiation and mineralization. The skeletal defects of the MN1 knock-out seem to primarily affect cranial skeletal elements while long bones of the appendicular skeleton appear to develop normally. Mutations in NF2 are associated with the autosomal dominant disorder neurofibromatosis type 2 [29]. Affected patients primarily develop tumours of the nervous system, including schwannomas, meningiomas and ependymomas. NF2 has not been specifically associated with bone formation. Instead, in patients with craniofacial dysmorphism and concomitant deletions affecting NF2, the deletions have been shown to encompass also the MN1 gene, supporting the role of MN1 in human craniofacial development [30]. The protein product of NF2 is also known to inhibit many signaling pathways at the membrane and in the nucleus, including the Wnt/beta-catenin pathway [29].
Conclusions
Most of the tumors investigated in the present study were conventional osteoblastomas and they showed very few or no acquired genetic aberrations. This was in contrast to the two epithelioid or aggressive osteoblastomas, which both displayed heavily rearranged genomes. Few aberrations were recurrent and the most intriguing findings were three neighboring homozygous deletions affecting 22q12 in one of the aggressive cases. Loss or rearrangement of the long arm of chromosome 22 was found in two additional cases investigated here, as well as two previously published osteoblastomas and two reported osteoid osteomas. These aberrations are thus not specific for the aggressive subtype of osteoblastoma but rather seem to affect osteoblastoma regardless of subtype as well as the closely related tumor type osteoid osteoma. The pathogenetic importance of these aberrations is not known but four of the homozygously deleted genes are involved in osteogenesis and/or tumorigenesis. Three of them - ZNRF3, KREMEN1 and NF2 - are inhibitors of the Wnt/beta-catenin pathway. Losses of these genes and subsequent accumulation of beta-catenin are in line with the high protein expression of beta-catenin previously detected in osteoblastoma.
Supporting Information
Primers and PCR protocols for mutation screening of MN1, NF2, KREMEN1 and ZNRF3.
(PDF)
Genes with a high expression value in osteoblastoma compared to osteosarcoma. Significance level was set to p<0.001 and FDR (q-value)<0.01.
(XLSX)
Genes with a low expression value in osteoblastoma compared to osteosarcoma. Significance level was set to p<0.001 and FDR (q-value)<0.01.
(XLSX)
Acknowledgments
We acknowledge the help with the microarray analyses from the Swegene Centre for Integrative Biology at Lund University.
Funding Statement
This work was supported by the Magnus Bergvall Foundation, the Royal Physiographic Society (Lund, Sweden) and the Swedish Cancer Society. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. de Andrea CE, Bridge JA, Schiller A (2013) Osteoblastoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. WHO classification of tumours of soft tissue and bone. Lyon: International Agency for Research on Cancer; pp. 279-280. [Google Scholar]
- 2. Horvai A, Klein M (2013) Osteoid osteoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. WHO classification of tumours of soft tissue and bone. Lyon: International Agency for Research on Cancer; pp. 277-278. [Google Scholar]
- 3. Radig K, Schneider-Stock R, Mittler U, Neumann HW, Roessner A (1998) Genetic instability in osteoblastic tumors of the skeletal system. Pathol Res Pract 194: 669-677. doi: 10.1016/S0344-0338(98)80125-8. PubMed: 9820862. [DOI] [PubMed] [Google Scholar]
- 4. Rosenberg AE, Cleton-Jansen AM, de Pinieux G, Deyrup AT, Hauben E et al. (2013) Conventional osteosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. WHO classification of tumours of soft tissue and bone. Lyon: International Agency for Research on Cancer; pp. 282-288. [Google Scholar]
- 5. Mitelman F, Johansson B, Mertens F, editors (2013) Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. Retrieved from http://cgap.nci.nih.gov/Chromosomes/Mitelman. Accessed 2013 April 2.
- 6. Cleton-Jansen AM, Anninga JK, Briaire-de Bruijn IH, Romeo S, Oosting J et al. (2009) Profiling of high-grade central osteosarcoma and its putative progenitor cells identifies tumourigenic pathways. Br J Cancer 101: 1909-1918. doi: 10.1038/sj.bjc.6605405. PubMed: 19888226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shaffer LG, Slovak ML, Campbell LJ (2009) An International System for Human Cytogenetic Nomenclature. Basel: S. Karger Publishing House. [Google Scholar]
- 8. Mertens F, Mandahl N, Örndahl C, Baldetorp B, Bauer HCF et al. (1993) Cytogenetic findings in 33 osteosarcomas. Int J Cancer 55: 44-50. doi: 10.1002/ijc.2910550109. PubMed: 8344751. [DOI] [PubMed] [Google Scholar]
- 9. Nord KH, Magnusson L, Isaksson M, Nilsson J, Lilljebjörn H et al. (2010) Concomitant deletions of tumor suppressor genes MEN1 and AIP are essential for the pathogenesis of the brown fat tumor hibernoma. Proc Natl Acad Sci U S A 107: 21122-21127. doi: 10.1073/pnas.1013512107. PubMed: 21078971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK et al. (2004) Detection of large-scale variation in the human genome. Nat Genet 36: 949-951. doi: 10.1038/ng1416. PubMed: 15286789. [DOI] [PubMed] [Google Scholar]
- 11. Cai Y, Mohseny AB, Karperien M, Hogendoorn PCW, Zhou G et al. (2010) Inactive Wnt/β-catenin pathway in conventional high-grade osteosarcoma. J Pathol 220: 24-33. doi: 10.1002/path.2628. PubMed: 19882675. [DOI] [PubMed] [Google Scholar]
- 12. Baruffi MR, Volpon JB, Barbieri Neto J, Casartelli C (2001) Osteoid osteomas with chromosome alterations involving 22q. Cancer Genet Cytogenet 124: 127-131. doi: 10.1016/S0165-4608(00)00327-7. PubMed: 11172903. [DOI] [PubMed] [Google Scholar]
- 13. Hao HX, Xie Y, Zhang Y, Charlat O, Oster E et al. (2012) ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 485: 195-200. doi: 10.1038/nature11019. PubMed: 22575959. [DOI] [PubMed] [Google Scholar]
- 14. Mao B, Wu W, Davidson G, Marhold J, Li M et al. (2002) Kremen proteins are Dickkopf receptors that regulate Wnt/β-catenin signalling. Nature 417: 664-667. doi: 10.1038/nature756. PubMed: 12050670. [DOI] [PubMed] [Google Scholar]
- 15. MacDonald BT, Tamai K, He X (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9-26. doi: 10.1016/j.devcel.2009.06.016. PubMed: 19619488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koo BK, Spit M, Jordens I, Low TY, Stange DE et al. (2012) Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488: 665-669. doi: 10.1038/nature11308. PubMed: 22895187. [DOI] [PubMed] [Google Scholar]
- 17. Ellwanger K, Saito H, Clément-Lacroix P, Maltry N, Niedermeyer J et al. (2008) Targeted disruption of the Wnt regulator Kremen induces limb defects and high bone density. Mol Cell Biol 28: 4875-4882. doi: 10.1128/MCB.00222-08. PubMed: 18505822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morvan F, Boulukos K, Clément-Lacroix P, Roman Roman S, Suc-Royer I et al. (2006) Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res 21: 934-945. doi: 10.1359/jbmr.060311. PubMed: 16753024. [DOI] [PubMed] [Google Scholar]
- 19. Zhang R, Oyajobi BO, Harris SE, Chen D, Tsao C et al. (2012) Wnt/β-catenin signaling activates bone morphogenetic protein 2 expression in osteoblasts. Bone 52: 145-156. PubMed: 23032104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujimori S, Novak H, Weissenböck M, Jussila M, Gonçalves A et al. (2010) Wnt/β-catenin signaling in the dental mesenchyme regulates incisor development by regulating Bmp4. Dev Biol 348: 97-106. doi: 10.1016/j.ydbio.2010.09.009. PubMed: 20883686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee YC, Cheng CJ, Bilen MA, Lu JF, Satcher RL et al. (2011) BMP4 promotes prostate tumor growth in bone through osteogenesis. Cancer Res 71: 5194-5203. doi: 10.1158/1538-7445.AM2011-5194. PubMed: 21670081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nuñez F, Bravo S, Cruzat F, Montecino M, De Ferrari GV (2011) Wnt/β-catenin signaling enhances cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer cells. PLOS ONE 6: e18562. doi: 10.1371/journal.pone.0018562. PubMed: 21494638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee HW, Kim SY, Kim AY, Lee EJ, Choi JY et al. (2009) Adiponectin stimulates osteoblast differentiation through induction of COX2 in mesenchymal progenitor cells. Stem Cells 27: 2254-2262. doi: 10.1002/stem.144. PubMed: 19522015. [DOI] [PubMed] [Google Scholar]
- 24. Lowy AM, Clements WM, Bishop J, Kong L, Bonney T et al. (2006) β-Catenin/Wnt signaling regulates expression of the membrane type 3 matrix metalloproteinase in gastric cancer. Cancer Res 66: 4734-4741. doi: 10.1158/0008-5472.CAN-05-4268. PubMed: 16651426. [DOI] [PubMed] [Google Scholar]
- 25. Tamamura Y, Otani T, Kanatani N, Koyama E, Kitagaki J et al. (2005) Developmental regulation of Wnt/β-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J Biol Chem 280: 19185-19195. doi: 10.1074/jbc.M414275200. PubMed: 15760903. [DOI] [PubMed] [Google Scholar]
- 26. Gómez García EB, Knoers NVAM (2009) Gardner's syndrome (familial adenomatous polyposis): a cilia-related disorder. Lancet Oncol 10: 727-735. doi: 10.1016/S1470-2045(09)70167-6. PubMed: 19573802. [DOI] [PubMed] [Google Scholar]
- 27. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (2013) WHO classification of tumours of soft tissue and bone. Lyon: International Agency for Research on Cancer. [Google Scholar]
- 28. Zhang X, Dowd DR, Moore MC, Kranenburg TA, Meester-Smoor MA et al. (2009) Meningioma 1 is required for appropriate osteoblast proliferation, motility, differentiation, and function. J Biol Chem 284: 18174-18183. doi: 10.1074/jbc.M109.001354. PubMed: 19386590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou L, Hanemann CO (2012) Merlin, a multi-suppressor from cell membrane to the nucleus. FEBS Lett 586: 1403-1408. doi: 10.1016/j.febslet.2012.03.016. PubMed: 22595235. [DOI] [PubMed] [Google Scholar]
- 30. Davidson TB, Sanchez-Lara PA, Randolph LM, Krieger MD, Wu SQ et al. (2012) Microdeletion del(22)(q12.2) encompassing the facial development-associated gene, MN1 (meningioma 1) in a child with Pierre-Robin sequence (including cleft palate) and neurofibromatosis 2 (NF2): a case report and review of the literature. BMC Med Genet 13: 19. doi: 10.1186/1471-2350-13-19. PubMed: 22436304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers and PCR protocols for mutation screening of MN1, NF2, KREMEN1 and ZNRF3.
(PDF)
Genes with a high expression value in osteoblastoma compared to osteosarcoma. Significance level was set to p<0.001 and FDR (q-value)<0.01.
(XLSX)
Genes with a low expression value in osteoblastoma compared to osteosarcoma. Significance level was set to p<0.001 and FDR (q-value)<0.01.
(XLSX)