

Summary
Tuberculosis (TB) has plagued mankind for millennia yet is classified as an emerging infectious disease, because its prevalence in the human population continues to increase. Immunity to TB depends critically on the generation of effective CD4+ T-cell responses. Sterile immunity has not been achieved through vaccination, although early T-cell responses are effective in controlling steady-state infection in the lungs. Although such early T-cell responses are clearly protective, the initiation of the Mycobacterium tuberculosis (Mtb) T-cell response occurs much later than is the case following other aerogenic infections. This fact suggests that there is a critical period, before the activation of the T-cell response, in which Mtb is able to establish infection. An understanding of the factors that regulate early T-cell activation should, therefore, lead to better control of the disease. This review discusses recent work that has investigated the early development of T-cell immunity following Mtb infection in the mouse.
Keywords: T cells, mycobacteria, immunity
Introduction
Although tuberculosis (TB) has afflicted the human population for millennia, it is nonetheless considered an emerging infectious disease, in part because disease incidence in human populations is increasing and because new forms of the pathogen pose additional threats to human health. Over two billion people are currently infected with Mycobacterium tuberculosis (Mtb), and many will experience active TB in their lifetimes (1). It is estimated that three million people will die each year from infection, making TB one of the major infectious diseases worldwide (1). TB infection rates are rising globally, particularly in Third World countries, where the combined factors of human immunodeficiency virus (HIV) infection, political instability, and breakdown of government infrastructures have contributed to declines in public health services. The problem has been further exacerbated by the increasing prevalence of multiple drug-resistant and extensively drug-resistant TB strains (2). Owing to the increasing mobility of the human population, these problems, in turn, increase the likelihood that the disease will become re-established in developed countries, where it has not been a serious problem since the onset of effective antibiotic therapies.
There is an urgent need for more effective controls of TB worldwide. Unfortunately, currently available vaccines have only limited efficacies (3–5). Chemotherapeutic regimens are effective against drug-susceptible TB but are typically long term, leading to patient non-compliance. Although a number of promising new vaccines are under development (3, 5), real progress will require more detailed knowledge of the host immune responses. Specific areas of that require elucidation include the exact roles of various immune cell subsets, the identities of the antigens recognized by immune effector cells, the effector mechanisms involved, the mechanisms of pathogen avoidance or subversion of immunity, and the regulation of the innate and adaptive immune responses.
Only a small percentage of humans known to have been exposed to Mtb will develop clinical manifestations and/or symptoms of the disease (6). This peculiarity suggests that the majority of individuals exhibit innate immunity sufficient to control naturally encountered infections. It is presumed that those who show immunological evidence of exposure, in the form of skin test reactivity, harbor the bacilli in a latent form yet are able to maintain active control of the bacteria. Similarly, only a small proportion of those who harbor latent infection will develop active disease (6). Although it is clear that immunosuppression caused by HIV or by immunosuppressive or immunotherapeutic drugs is a major factor in TB recrudescence, the underlying causes of susceptibility to active disease are not known; indeed, the contributions of host and/or bacterial genetics and host–bacterial cell interactions are not well understood.
Studies of the human cellular and humoral responses during infection and disease have been undertaken using peripheral blood and cells from the lung. A substantial humoral response to Mtb occurs, although the significance of this response, in terms of control of the disease, is unknown (7). The cellular response in both the blood and lungs is largely inflammatory in nature, and antigen-specific cells are capable of producing macrophage-activating cytokines (8). There is also some evidence of regulatory activity [i.e. interleukin-10 (IL-10) production (9)], although the relative contributions of inflammatory and regulatory activities to the development of disease are as yet undefined. While the nature of the cellular response in exposed and diseased individuals has been characterized, the very earliest events in T-cell activation have not been studied in depth either in humans or in animal models. Because it is likely that these early immunological events strongly affect long-term cellular responses and are therefore likely to modulate disease progression, the study of these early events form the basis of the present review.
Such a characterization of early immunity will be particularly valuable for understanding events subsequent to known exposure. Cellular responses are slow to develop in humans, i.e. skin tests are not useful before 5–6 weeks after exposure (10). If we can develop tools to examine early events in humans and/or to identify early cellular events in animal models, we will be able to better understand how the cellular response is induced in humans upon exposure. One area of intense interest in this regard is the response to vaccination with live-attenuated M. bovis Bacille Calmette-Guérin (BCG). While this vaccine is at best only modestly effective against adult pulmonary disease, it is administrated to neonates around the world and protects against early childhood disease (11). Recent studies in South Africa demonstrate that a multi-faceted cellular response to this vaccine occurs within 10 weeks post-vaccination (12). It remains to be seen, however, whether the nature of the initial response to vaccination bears any relation to the ability of the vaccine to protect against disease.
In addition to extensive and careful studies of vaccinated infants, a range of novel approaches are being applied to the investigation of TB immunity in adults. One recent advance is the development of multi-parameter flow cytometry, which has highlighted a heretofore unappreciated complexity of the cellular response to mycobacterial exposure (12). Another advance is the availability of major histocompatibility complex (MHC) tetramer reagents, which allow direct measurements of antigen-specific T-cell numbers in infected individuals (13, 14). Experimental approached involving animal models serve to identify mechanisms and pathways that will help to focus the work performed in human studies. A range of animal models, including mice, guinea pigs, zebrafish, and primates, have generated recent advances (15–18), and the exciting development of in vivo-imaging technologies in both the zebrafish and the mouse models has provided novel three-dimensional insights into cellular dynamics during mycobacterial infections (16, 19).
In this review, we focus on recent work using the highly tractable mouse model to study an issue that has long remained an enigma. As discussed above, the very earliest events in the stimulation of the immune response to aerogenically delivered Mtb have not been delineated. Currently, only the mouse model allows this question to be addressed experimentally. It is hoped that the recent progress made with this model will facilitate examination of the cellular response in humans, thereby leading to better insights into both disease development and immunity.
The mouse as an experimental model
Features of Mtb infection in the mouse have been studied in depth, as summarized in several recent reviews (20, 21). Inbred mouse strains exhibit a wide range of susceptibility to Mtb infections (22, 23) and have been used to investigate genetic contributions to disease susceptibilities (24–26). BALB/c and C57BL/6 mice have been commonly used for studies of immunity, even though these strains are relatively resistant to intravenous and aerosol Mtb infection (23). The C57BL/6 strain offers a major advantage for TB research, because many immunologically relevant gene-targeted mice are available on this genetic background. A general concern with regard to the mouse model, however, has been the lack of standardization among different laboratories, where variability in such key parameters as bacterial strain, inoculation route, and inoculum dose may be responsible for disparate findings. Many investigators have used intravenous and intratracheal inoculations, although it is now widely accepted that low-dose aerosol inoculation provides the most realistic model for normal human infection (27–30). Via this route, low-dose inoculations are typically designed to deliver 75–100 bacterial CFU to the lower respiratory tract (27, 29, 31, 32).
Innate immunity
Once delivered to the lungs, the bacilli attach to and enter host cells via any of several host receptors, including the mannose receptor, the dendritic-cell-specific intercellular adhesion molecule-3 (ICAM-3)-grabbing non-integrin (DC-SIGN), surfactant protein A, mannose-binding lectin, and the class A scavenger receptor (33). The significance of this exploitation of multiple receptors and the relevance to pathogenicity are not yet resolved, although it has been suggested that differential receptor utilization by the bacterium may affect disease susceptibility (34). However, the ability to interact with a wide number of receptors on various cell types may also be reflective of an evolutionary strategy, which has enabled Mtb to infect many differing cell types. Alveolar macrophages are generally considered to be the first cells infected after aerosol infection (35), and depletion of activated macrophages can lead to a dramatically altered disease progression (36). These cells presumably provide a first line of host defense, for example, they express antimicrobial peptides, i.e. cathelicidin, that may function in the early immune response (37).
As in most other types of infections, many components of the innate response are activated in Mtb-infected host cells. A number of studies have investigated roles for Toll-like receptors (TLRs), in particular TLR2 (a heterodimer of TLRs 1 and 6), TLR9, and TLR4 (38–43, 44). Recent work has suggested that interaction of the bacillus with TLR2 is due in part to effects of secreted ESAT-6, via its direct interaction with TLR2 on macrophages (45). A number of chemokines are also produced in the lungs and other tissues during infection and contribute to innate immune responses (46, 47). Although there is ample evidence that Mtb can trigger innate immunity, this triggering does not occur until several days after infection, at about the time when T-cell responses are elicited (48). Thus, although Mtb is clearly capable of activating the innate response, it does not do so for a prolonged period following aerosol infection; this delay likely provides a critical window of opportunity in which Mtb can establish infection in the lungs. The delay in the activation of the innate response in the lung may be a result of a number of factors, including low pathogen exposure (i.e. 50–200 bacteria in experimental models), naturally low inflammatory responses in the lung, or active inhibition of innate immunity by Mtb. There is no clear evidence to promote any one of these ideas over the other at present, in part because the lack of tools that are sufficiently sensitive to detect early innate responses following low-dose aerosol infection.
In addition to alveolar macrophages, dendritic cells (DCs) are likely to become infected upon aerosol exposure (34, 49, 50), and these cells probably play a major role in triggering T-cell responses (51). Some investigators have proposed that the Mtb infection compromises DC function (52), perhaps providing a means of immune evasion. After aerosol exposure, bacteria multiply in the lungs but disseminate to lymphoid tissues and other organs. How these early growth and dissemination phases impact the generation of the T-cell response was not well studied, before some significant recent advances described below.
Cellular immunity
In the mouse, Mtb grows in an apparently uncontrolled fashion for about the first 21 days (53), at which time further increase in steady-state bacterial numbers come under the control of the immune system (31, 54). A stationary phase of bacterial colonization ensues; immunity during this period is maintained by αβ T cells until the animal succumbs to infection, an event which in C57BL/6 mice occurs between 200 and 250 days after infection (31, 54). The precise cause of death is not known, although it could be a consequence of a loss of cellular immunity or a result of chronic T-cell-mediated pathology.
In the mouse model, the roles of particular immune cell subsets, cytokines, and chemokines during host adaptive immunity have been studied in depth. It is now well established that CD4+ T cells are instrumental in the control of TB infections in both humans and mice (21, 55–58). Chief among the functions of CD4+ T cells is the production of interferon γ (IFNγ), which is required for resistance to fatal infection, given that both class II MHC- and IFNγ-deficient mice rapidly succumb to infection (59, 60–63). An important function of IFNγ is the induction of nitric oxide synthesis in macrophages; IFNγ in turn controls bacterial growth (63–66). Although CD4+ T cells are widely assumed to provide an essential source of IFNγ, this relationship has not been formally demonstrated. Some studies have suggested that immune control is mediated independent of IFNγ (60, 67). Moreover, although CD4+ T cells are required for control of infection in the mouse, these immune cells are unable to eliminate the pathogen or to prevent fatal disease. T-helper 1 (Th1) responses are nevertheless a major component of protective immunity, because they clearly limit bacterial expansion. CD4+ T cells are well known to be required for immunity to Mtb, and recruitment of T cells to the lung is necessary for containment of Mtb within granulomas (68).
How can we explain bacterial persistence in the context of apparently robust innate and adaptive immune responses? One possibility is that the bacterium, perhaps due to its impervious cell wall, is resistant to otherwise damaging anti-microbial compounds produced by the host cell (i.e. reactive nitrogen and oxygen intermediates). Alternatively, the bacterium could actively subvert or modify effector functions of macrophages, DCs, T cells, or other host cells (69–71). It is also possible that the inflammatory consequences of infection produce an internal or external environment that is not conducive to bacterial-killing activity by the immune response. Currently, there are insufficient data to support any one of the above hypotheses over the others.
While CD4+ T cells are known to be essential, other T cells and lymphocytes also play important roles in host defense, even when their functions have been shown to be redundant (21, 72). A number of investigators have investigated the role of CD8+ T cells. In contrast to the dramatic impact of the absence of CD8 coreceptor-expressing cells on the development of disease following a high-dose intravenous infection (73), the absence of CD8+ T cells in the low-dose aerosol model had little impact disease progression, until after as long as 200 days (54, 74–76). Moreover, although transfer of Lyt-2+ (CD8+) cells was not particularly effective against low-dose aerosol infection, CD8+ T-cell transfer was more effective than were CD4+ T cells against high-dose infection (77). Thus, while CD4+ T cells are able to control low-dose infection, CD8+ T cells possess a unique activity that makes them essential when a high-dose infection occurs.
A difficulty in resolving the exact role of class I MHC-restricted CD8+ T cells is in part due to the fact that a variety of strains of gene-targeted mice have been used to assess infection in the absence of CD8+ T cells. Moreover, many gene-targeted strains exhibit incomplete elimination of the targeted cell populations, and/or they exhibit pleiotropic effects of the targeted mutations. For example, β2-microglobulin (β2m)-deficient mice lack both MHC class Ia- and MHC class Ib-restricted CD8+ T cells, and CD8-deficent mice contain class I MHC-restricted, CD8− T cells. Studies in the β2m-deficient mice are further complicated by an iron overload defect that renders this strain more susceptible to Mtb infection (78). Furthermore, the absence of β2m results in an early but limited reduction in the ability of the mice to control bacterial growth (75). However, no difference in disease development was noted in mice lacking another β2m-dependent class Ib molecule, CD1d (75, 79). Urdahl et al. (80) addressed roles for class Ia- and class Ib-restricted CD8+ T cell; using class I MHC KbDb-deficient mice, they demonstrated that class Ib-restricted CD8+ T cells, although present in infected lungs, were unable to fully compensate for the lack of class Ia-restricted T cells. These data revealed a non-redundant role for class Ia-restricted CD8+ T cells. Like CD4+ T cells, CD8+ T cells have been shown to function by producing IFNγ and by targeting infected cells for cytolysis (81–84). More recently, it has been demonstrated that CD8+ T cells recognized Mtb antigens generated by cross priming in vitro (85).
Mtb antigens recognized by CD4+ T cells
Early studies of T-cell immunity focused on polyclonal T-cell populations that were elicited by Mtb infection and re-stimulated in vitro with either live bacteria or crude proteins preparations. In these types of analyses, detection of antigen-specific cells was not possible (81, 86). Ex vivo stimulation of cells from humans, guinea pigs, and mice, using purified proteins demonstrated that immunodominant responses were associated with Mtb proteins, and the responses were not limited to a single Mtb protein (87, 88). Identification of the Mtb antigens and epitopes recognized by both mouse and humans (89–92) has been facilitated by the search for novel T-cell antigens relevant to vaccine development. This has led to the discovery of a wide array of Mtb antigens that activate CD4+ T cells, CD8+ T cells, non-classical class I-restricted CD8+ T cells and γδ T cells (92). These T-cell ligands include not only conventional peptide antigens that bind to classical MHC class I and class II molecules but also a number of abundant mycobacterial lipids that are presented by the non-classical class I molecule CD1 (93, 94).
Early studies suggested that immunity to Mtb is generated by viable but not killed bacteria (95). One explanation for these findings is that killed bacteria do not secrete Mtb antigens in vivo. Other infection models have shown antigen secretion to be an important requirement for the generation of T-cell ligands (96). Indeed, many Mtb proteins are released during bacterial culture (92), suggesting that such proteins are also secreted while the bacteria are residing within host cells in vivo. These culture filtrate proteins (CFPs) were found to be mixtures of secreted proteins and outer cell wall proteins that have been shed or released from dead bacteria (97), and considerable effort has been expended on their characterization. Such work identified many Mtb antigens among the CFPs that could elicit responses from T cells in both mice (87, 98) and humans (99). Importantly, CFPs have been shown to be the first antigens recognized during infection (100, 101), and lymphocyte responses are hypothesized to be directed largely against these proteins (90, 100, 102). Of considerable importance are studies in which immunization with CFPs isolated from short-term cultures elicited at least partial protection to Mtb infection in experimental mouse models (87, 101).
Many CFPs have now been shown to be recognized by T cells (87). One of the major CFP antigens is Ag85B (103, 104), a member of the p85 complex encoded by fbpB (105). The concentration of Ag85B within CFPs was 100-fold higher than concentrations in bacterial sonicates (106). Activated CD4+ T cells have been shown to respond to a particular Ag85B peptide epitope in C57BL/6 mice (residues 240–254) (107). Another major CFP antigen that has been characterized is the early secreted antigenic target-6 (ESAT-6). ESAT-6 is a low molecular mass (6 kDa) protein found in abundance in Mtb culture filtrates (91). Because ESAT-6 is a major target of both CD4+ and CD8+ T cells in mice and humans, it is a major vaccine candidate (108). Immunization of mice with an ESAT-6 peptide induced protection equivalent to that obtained by the use of BCG (109). Modified BCG strains are now available that express ESAT-6; re-introduction of the genetic region of difference RD1, which includes ESAT-6, into BCG restored pathogen virulence [the ESAT-6 gene is deleted in the attenuated BCG strain (111)] (110).
The identification of new Mtb T-cell epitopes is proceeding at a rapid pace, and the new antigens will be of use for studies of cellular immunity. A recent study has cataloged all currently known Mtb T- and B-cell epitopes identified in a wide range of mammalian species (112). Given that only a small portion of the Mtb proteome has been studied, many additional T-cell epitopes in different host species will undoubtedly be identified. Nevertheless, the epitope database, along with other studies of Mtb T-cell epitopes (72, 89), will be an important tool for both researchers and clinicians.
Expression of Mtb antigens in vivo
Given the identification of multiple Mtb T-cell antigens, it will be important to ascertain when and where these antigens are produced during infection. This information is critical for understanding the genesis and maintenance of protective immunity, especially because antigen expression likely varies temporally and spatially during infection. One study investigated the expression of ESAT-6 and Ag85B following virulent Mtb (H37Rv) infection (113). mRNAs for both antigens were expressed during acute and chronic phases of infection, although the magnitude and kinetics of antigen mRNA expression differed. Both mRNAs were expressed early following infection, but ESAT-6 mRNA expression persisted at 10-fold higher levels through day 20 post-infection. Expression of both antigens declined after day 20; based on the ratio of transcripts to Mtb CFUs, only a few of the bacteria expressed these antigens in vivo. These data also suggest that the bacilli enter into a quiescent, non-dividing state in vivo, once their expansion has been controlled by the cellular immune response. These data raise an important question: how are protective long-term effector T-cell responses, which likely require a stable source of antigen, maintained during chronic and/or latent infections. Although the study by Rogerson et al. (113) showed evidence of major changes in mRNA expression that could contribute to differing T-cell responses during acute and chronic infection, the work needs to be extended through study of additional Mtb antigens.
Analyses of antigen-specific T cells
The identification of the ESAT-6 peptide epitope recognized in C57BL/6 mice made available a powerful tool that allowed us to examine when and where IAb/ESAT-6-specific CD4+ T cells are located during acute and chronic phases of infection (114). In our studies, we used enzyme-linked immunospot (ELISpot) analyses to enumerate IAb/ESAT-61 – 20-specific CD4+ T cells in vivo. (Note that because the secretion of IFNγ is antigen dependent, this approach does not reveal whether IFNγ is actually expressed in vivo but rather measures the capacity of the cells to produce cytokines following antigen stimulation). We observed that IAb/ESAT-61 – 20-specific T cells were detectable no earlier than 14 days post-infection in the lungs and the mediastinal lymph node (MLN). High frequencies of responder cells were not detected until day 21, and the response was maximal on day 28 post-infection in the lungs. At that time point, the frequency of antigen-specific T cells was approximately 1.5% of total lung CD4+ T cells, corresponding to 1–5 × 105 cells in the lungs. Thereafter, the frequency of responding cells stabilized in the lungs at approximately 1%, a frequency maintained at least through day 160 post-infection. Similar findings were obtained in the MLN and spleen, except that the frequencies of T cells in these latter tissues were lower. These data revealed that the antigen-specific effector CD4+ T-cell response did not become detectable until relatively late following infection, that the CD4+ T cells underwent major expansion, and that chronic infection was associated with a persistent T-cell presence in the lungs. The antigen-specific cells identified in this study exhibited characteristics of fully activated effector cells: high expression of CD44, low expression of CD62L, and the ability to secrete IFNγ upon contact with antigen (114). While we concluded that cellular immune responses must be present within the lung, the extent to which the lung CD4+ T cells play a role in maintaining control of bacterial burden during the chronic phase is unclear.
Several other investigators drew similar conclusions regarding the generation of antigen-specific effector CD4+ T cells. Chackerian et al. (115) observed IFNγ production by a spleen cell population in response to a sonicate of Mtb following aerosol infection, as early as day 11 post-infection in C57BL/6 mice. The particularly early response in that study could have resulted from the use of the complex antigen preparation and/or the production of IFNγ by non-T lymphocytes. Lazarevic et al. (116) reported that IAb/ESAT-6-specific T cells undergo dynamic changes during acute and chronic infection. The above studies have made important contributions to the study of early T-cell immunity, by revealing that the onset of the T-cell response during Mtb infection is delayed, compared with infections mediated by other pulmonary pathogens such as the influenza virus (117).
Antigen-specific CD8+ T-cell responses
Several investigators have examined antigen-specific CD8+ T-cell responses. In one study, CD8+ T-cell responses were shown to be initiated in the lung-draining lymph node (LN) within 11 days after infection (118). The T-cell response was also interpreted to be initiated only after the dissemination of Mtb to the MLN, given that bacterial infection was first detected on day 9 post-infection. Thus, the authors concluded that bacterial dissemination precedes the development of the T-cell response. However, a caveat relates to the sensitivity of the IFNγ enzyme-linked immunosorbent assay (ELISA) used to detect T-cell responses; possibly the T cells had undergone activation before day 9 post-infection, but IFNγ was present at levels below the limits of detection.
Other studies have examined CD8+ T-cell responses to defined class I MHC peptides, including Kd/TB10.3/10.420 – 28 (119) and Kk/CFP32 – 39 (83). The Kk/CFP32 – 39- specific cytolytic CD8+ T cells were first detected 2–3 weeks post-infection and persisted in the lungs for as long as 260 days post-infection (83). These studies of antigen-specific effector CD8+ T cells were extended after the development of class I MHC tetramers that recognize Kk/CFP32 – 39- and Kd/TB10.3.4/10.420 – 28-specific CD8+ T cells (83). Remarkably, Kd/TB10.3.4/10.420 – 28-specific T cells were found at frequencies as high as 30% in the lungs of chronically infected BALB/c mice, demonstrating that TB10.3 and TB10.4 are immunodominant antigens in this strain. An additional MHC class I tetramer has been described that recognizes the Mtb72F polyprotein in the context of the MHC class II protein IAb (120). In that study, the tetramer was used to identify IAb/Mtb72F92 – 102-specific CD8+ T cells in aerosol-infected mice within 30 days and as late as 100 days post-infection. The IAb/Mtb72F92 – 102-specific CD8+ T cells were present at frequencies as high as 7.5% in low-dose-infected mice, indicating that Mtb72 is also an immunodominant antigen and that the antigen is expressed during chronic infection. The availability of mouse MHC class I tetramer reagents is thus a significant advance and will be particularly useful for future analysis of the antigen-specific lymphocyte responses during the adaptive immune response to Mtb.
T-cell responses are delayed in Mtb infections
The relatively late initiation of the CD4+ and CD8+ T-cell responses to aerosol Mtb infection contrast with the timing of the responses to several acute viral infections (121, 122) as well as with the timing of responses to other intracellular bacterial infections (123–125). In these other infection models, antigen-specific CD4+ and CD8+ T-cell responses were typically detected much earlier than are detected during Mtb infection, often as early as 5 days post-infection. Although they occur late following aerosol inoculation, Mtb-specific T-cell responses are not inherently delayed, because experimental intravenous inoculation can generate robust T-cell activation within 1–3 days post-infection (51, 126, 127). Our recent studies, in which 2 × 105 bacteria were administered intravenously, revealed that priming occurred at as early as 18 h (W. Reiley, G. Winslow and D. Woodland, unpublished data). Such early T-cell priming was observed by other investigators, who examined T-cell responses to an ovalbumin epitope expressed by a recombinant BCG (128). In those studies, transgenic OVA-specific CD4+ T cells initiated cell division within 5 days of infection, supporting the idea that early CD4+ T-cell responses can occur early following BCG infection, if sufficient antigen is available. A limitation of the above studies of early antigen-specific T cells responses is that none had been able to identify the exact time, after infection, when T-cell responses are initiated. The inability has been in part due to the lack of tools that would allow the direct detection of T-cell responses in vivo. For example, studies have been limited by reliance on indirect assays of T-cell activation (i.e. cytokine secretion); these effector responses require 3–4 days to develop after the T cells first encounter antigen.
Why, then, are T-cell responses to Mtb detected relatively late after infection? This question is an important one, because a delay in the generation of cellular immunity is likely a critical factor in allowing the bacteria to establish persistent infection. A simple explanation is that the apparent delay in T-cell activation is due to the lack of MHC/peptide complexes able to prime T cells. Although the population doubling times of Mtb strains vary, the doubling time within the lungs in the first 10 days following intravenous infection is approximately 36 h for strain H37Rv (53). Aerosol inoculation with 200 CFUs of H37Rv would in this case be expected to yield approximately 2000 live bacteria within the lungs within the first 5 days and 20 000 bacteria in the first 10 days post-infection. Although T cells require as few as 10–100 MHC/peptide complexes to be presented on an antigen-presenting cell (APC) before they can undergo priming (129), it is not yet possible to correlate the number of bacteria to the antigen loads on APCs; tools to quantitate MHC/peptide complexes on the surface of APCs are not available for Mtb antigens. Nevertheless, one study reported that effector T-cell responses in the spleen did not occur when fewer than 250 CFUs were detected (118), suggesting that the bacteria were unable to generate sufficient MHC/peptide complexes to trigger T cells. A caveat is that it is not known whether the T cells that initiate the response need to be activated by DCs that are infected by bacteria or whether DCs can acquire antigens passively, e.g. by acquiring soluble antigens or antigens acquired from apoptotic cells (85). Nevertheless, antigen availability within secondary lymphoid tissue is a likely factor that limits T-cell priming. If that is the case, it should be possible to accelerate T-cell activation by increasing the size of the experimental inoculation. Experiments designed to test this idea are described below.
It is alternatively possible that the apparent delay in the priming of the T-cell response occurs because Mtb avoids immune detection in infected alveolar macrophages and is, therefore, able to replicate to high numbers. Infected alveolar macrophages are unlikely to migrate to secondary lymphoid organs to initiate T-cell priming events in the lung-draining LN. It is also possible that bacteria administered during low-dose aerosol infection are deposited in a location that does not allow host cell detection and transport to the LN by DCs that are sampling the airways. Thus, an understanding of the kinetics and location(s) of the first peptide/MHC complexes available for recognition is key to deciphering the early events in T-cell activation that follow aerosol exposure to Mtb.
A third explanation for the apparent delay in T-cell priming is that Mtb suppresses early antigen presentation in secondary LNs or influences the capacity of T cells to undergo activation. For example, cells loaded with mycobacterial antigens may not be capable of initiating T-cell activation after they have migrated from the lung to the draining LNs. DCs, which are infected by Mtb, are well known to be the most important APCs in initiation of T-cell responses in vivo; in this hypothesis, DC function is impaired following Mtb infection. Such impairment could arise, for example, through compromised DC trafficking from the lungs to the draining LNs or through a compromised ability of DCs to provide proper signals for T-cell activation. Evidence is available to support both mechanisms. Several studies have demonstrated that antigen presentation can be suppressed in macrophages and by extension DCs, following Mtb infection of APCs (70, 130–132). It has also been suggested that that infected in vitro-generated DCs can be impaired in their ability to prime T cells (52), although other studies suggest that DCs are highly effective APCs during Mtb infection in vivo (133).
Early events in T-cell activation
To understand how protective T-cell responses are initiated during Mtb infection, it is helpful to first review what is known from other infection models regarding early T-cell activation. Precedents are available from several studies of early T-cell responses following acute viral infections (117, 121). Naive antigen-specific T cells migrate throughout the circulation in mice, and encounter with antigen typically occurs in LNs that drain infected tissues. The antigen-specific T cells form stable contacts, engage DCs, and undergo an orchestrated series of interactions with DCs (134–136). The initial contact, duration, and quality of the interaction made with antigen-laden DC cells are likely important factors that influence T-cell responses (137, 138). Within 4–8 h, antigen-activated T cells express a number of important cell surface receptors, including CD69 and CD25 (the IL-2 receptor α chain). CD69 is the earliest T-cell activation marker expressed and is first detected within 3 h of contact with antigen (139). After the expression of CD69, activated T cells become less mobile, losing their ability to migrate out of the LNs, due to loss of the expression of sphingosine-1-phosphate receptor-1 (S1P)(140). During the following 2 days, a programmed series of events occur within the activated T cell, leading to the upregulation of CD44, the downregulation of CD62L, and the onset of cell division. As activated T cells begin to proliferate, they become motile and again encounter DCs (134). CD69 expression is then lost from the cell surface, and S1P1 is re-expressed, enabling migration of the T cell. In the periphery, the activated T cells again encounter antigen, most likely via interactions with macrophages and DCs, and elaborate effector activities that contribute to pathogen clearance. Thereafter, most T cells die; at the population level, this results in a contraction of the response. Some T cells, in a process governed by signals that are not well defined, develop into memory T cells that can provide long-lived protection (141).
Early T-cell activation during Mtb infection
The processes of T-cell activation, differentiation, and migration during Mtb infection have only begun to be studied. An important advance is the availability of tools that allow the detection of endogenous antigen-specific T cells. Mtb-specific class I MHC tetramers, for example, described above, offer significant promise, especially if incorporated into methods that enhance the detection of very low numbers of endogenous antigen-specific cells (142).
An alternative approach to the study of T-cell activation involves the use of T cells from T-cell receptor transgenic mice. T cells from these mice form a homogenous pool of identical antigen-specific cells and have long been used to study naive T-cell responses in vivo. One well-described experimental technique involves the seeding of mice with naive transgenic T cells, at cell numbers sufficient for detection by flow cytometry (143, 144). Although a number of concerns have been raised recently regarding the effects of introduction of non-physiological numbers of T cells in such experimental models, the approach can be powerful, as long as possible artefacts associated with relatively high precursor cell frequencies are taken into account. Although high precursor frequencies are well known to influence effector T-cell differentiation and memory development (142, 145), transgenic T cells are valuable tools in the study of early T-cell activation in vivo.
An Mtb Ag85B-specific T-cell receptor transgenic mouse has been used by Wolf et al. (146) to monitor early T-cell expansion after aerosol infection. This CD4+ T-cell receptor transgenic mouse (P25 TCR-Tg), which produces T cells that recognize IAb/Ag85240 – 254, was developed by Tamura et al. (147). In the studies of these Ag85-specific transgenic T cells, Wolf et al. (146) adoptively transferred 3 × 106 IAb/Ag85240 – 254-specific CD4+ T cells into naive C57BL/6 mice before aerosol Mtb infection. The T cells then underwent significant cell division between days 11 and 14 post-aerosol infection in the MLN, demonstrating that T-cell response to Mtb is initiated in this lymphoid tissue. T cells were not detected in the lungs until day 17 post-infection, and, because most of these cells had undergone many cycles of cell division, the authors concluded that those T cells were first activated in the draining LN and only thereafter migrated to the lungs. These data, in contrast to previous findings, suggested that the lungs are not a site of T-cell priming; instead, cells are activated and undergo cell division only in the MLN (148). These latter studies support the notion that recognition of antigen in the draining LNs is the critical event in the emergence of immunity to Mtb.
In these same studies by Wolf et al. (146), a strict correlation was observed between the number of colonizing bacteria in the mediastinal and inguinal LNs and the percentage of proliferating T cells, revealing that T-cell activation is contemporaneous with bacterial dissemination. This conclusion supports data by Chackerian et al. (118), discussed above. On the basis of these studies, it was suggested by Wolf et al. (146) that bacterial dissemination is essential for T-cell activation and that Mtb peptide/MHC antigens must be produced in the local draining LN. This proposal argues against the hypothesis that uninfected DCs acquire antigen passively in the lungs and migrate to the draining LN, where they initiate T-cell responses. An alternative explanation is that infected host cells (i.e. DCs or other APCs) traffic the bacteria to the LNs, where the migrating cells either present antigen to T cells directly or else indirectly via another APC. Although DCs were shown to migrate to the draining LNs during Mtb infection, Wolf et al. (146) argued that this process is impaired at an early stage after infection. In their interpretation, T-cell responses are delayed, not due to lack of antigen but rather to a lack of antigen-bearing DCs within the draining LNs. Such a mechanism, if correct, would be highly significant for our understanding of early T-cell immunity and protection from Mtb infection.
One prediction of the ‘impaired DC’ hypothesis is that impairment would occur in a fashion largely independent of changes in bacterial numbers and/or antigen load. With this in mind, Wolf et al. (146) addressed whether the early onset of the T-cell response was dependent on the size of the aerosol Mtb inoculum. They found that a reduction of the Mtb inoculum had no effect on the timing of T-cell activation (above a minimal inoculum threshold). In contrast, increasing the inoculum sevenfold (from 100 to 700 CFU) led to an increase in the frequency of cells that had undergone cell division on day 14. The authors argued on this basis that Mtb actively subverts early effector T-cell responses and in this way gains a foothold, before the development of T-cell immunity.
Although the studies described above made important contributions to our understanding of early T-cell activation following Mtb infection, a limitation was that T-cell activation was not directly examined. Instead, indirect readouts of T-cell activation (i.e. T-cell proliferation and cytokine secretion) were monitored. These effector T-cell responses typically are initiated only several days after T cells encounter antigen and are often accompanied by T-cell migration. Thus, in the experiments by Wolf et al. (146), it was necessary to infer the time of T-cell activation on the basis of the number of cell divisions undergone by the T cells.
To directly identify the earliest T-cell activation events following low-dose aerosol infection, our laboratories utilized another CD4+ T-cell receptor transgenic mouse strain that produces T cells that recognize the major MHC class II IAb T-cell epitope in ESAT-6 (149). The epitope (IAb/ESAT-61 – 20) was first identified by Brandt et al. (149). We generated T-cell hybridomas that recognized this epitope, characterized, and cloned the T-cell receptor α and β chain genes, and generated transgenic mice. The IAb/ESAT-61–20-specific T cells recognized the antigen in vitro and in vivo during Mtb infection (149).
We used adoptive transfer of transgenic T cells to address exactly when CD4+ T cells first become activated in vivo. This approach required that sufficient numbers of T cells be present in the LNs, lungs, and other organs, to allow detection and characterization of the T cells by flow cytometry. For this reason, we transferred 0.25 × 106 and 2 × 106 highly purified transgenic CD4+ T cells to Thy-1 congenic mice and examined activation marker expression on the donor cells daily, beginning on day 8 following aerosol infection with 75 CFU of Mtb strain H37Rv. On the basis of CD69 upregulation, we determined that the donor T cells first encountered antigen on day 10 post-infection in the MLN. CD69 expression was never detected in transgenic T cells in the lung, suggesting that these T cells migrated to the lungs after their initial activation in the MLN (149). Given that the timing of the earliest T-cell activation was coincident with the arrival of bacteria in the MLN, our data support the conclusions of Wolf et al. (146), that early T-cell priming to Mtb occurs exclusively in the MLN. In contrast, our direct measurement of IAb/ESAT-61 – 20-specific T cells revealed that activation occurred by day 10 post-infection, 1 day earlier than had been reported for the IAb/Ag85-specific T cells. The disparity in the findings may be due to the differences in the experimental approaches between the two studies or to differences in the kinetics of antigen presentation between Ag85B and ESAT-6.
The transgenic IAb/ESAT-61 – 20-specific T cells used in our studies subsequently upregulated CD44 and downregulated CD62L, findings that are indicative of T-cell activation (149). These two markers were also expressed in the lungs beginning on about day 14 in both the MLN and lungs, indicating that about 4 days were required for cell division to be initiated after antigen encounter. We were unable to detect any increase above background in CD69 expression by endogenous CD4+ T cells in the MLN, lungs, or spleen, on day 10–15 post-infection; the inability to detect endogenous T-cell priming validates the use of the transgenic T cells for detecting early T-cell activation (149). A schematic that summarizes the kinetics of early CD4 T-cell activation in different tissues following aerosol Mtb infection is shown in Fig. 1.
Fig. 1. Schematic of early T-cell responses after aerosol Mtb infection.
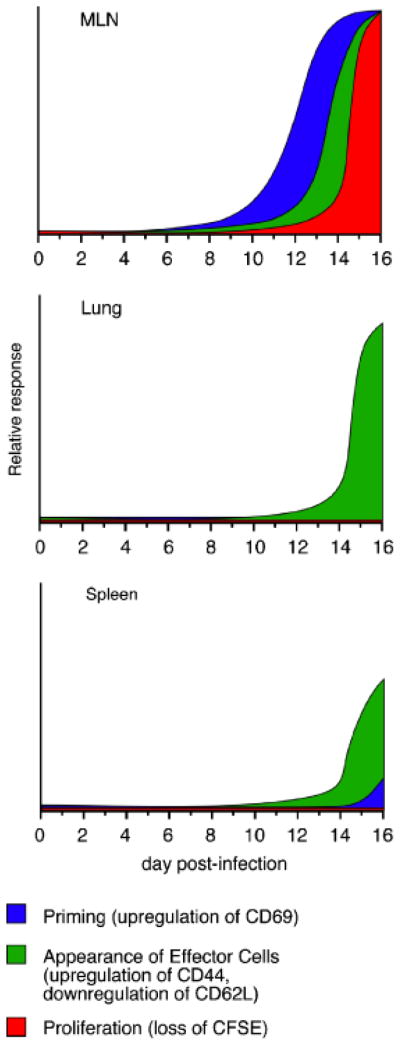
Idealized representations of T-cell priming, effector cell generation, and proliferation, in three different tissues, are shown.
In contrast to the findings of Wolf et al. (146), we (149) observed a direct correlation between the size of the Mtb CFU inoculum and the timing of early activation of the transgenic T cells. Increasing the size of the aerosol inoculation approximately 15-fold (from 75 to 1200 CFU) resulted in a 2-day acceleration of T-cell activation within the lung-draining LN (149). Because the larger inoculum likely increased the available antigen in the lung, this finding suggests that antigen is a critical factor that limits the early activation of CD4+T cells. The implication of these findings is that early T-cell priming is dependent on available antigen and is not necessarily affected by inhibition of DC function and T-cell priming by the bacterium. An alternative explanation is that the higher bacterial burden induced more rapid DC migration.
On the basis of the studies discussed above (118, 146), it has been proposed that Mtb dissemination from the lung to the draining LN precedes and is essential for the initiation of the T-cell response. Our studies corroborate the findings of these groups, as we have observed that T-cell priming in the MLN occurs at the same time that bacteria are found in this tissue. In vivo depletion of DCs using CD11c-promoter diptheria toxin receptor transgenic mice delayed the spleen CD4+ T-cell response and exacerbated mycobacterial infection following intravenous inoculation (51), suggesting that DC migration from the lungs to the draining LN is a critical step in T-cell priming. By comparing CFUs and the time of early T-cell activation, Wolf et al. proposed that T-cell activation requires that at least 1500 bacteria be present in the MLN (52). It is not known, however, if DCs or other cells are responsible for delivering the bacteria to the draining LNs or whether uninfected DCs can also acquire antigen from infected cells in the lungs before trafficking to the LN. Thus, the relationship between DC antigen delivery and bacterial dissemination is not yet resolved.
Temporal and spatial arrangements of lymphocytes
While flow cytometric studies provide critical information on the phenotypes, numbers, and functions of cells in different tissues, they cannot generate information on the three-dimensional interactions of cells within inflamed tissues. To begin to acquire knowledge of immune responses to Mtb in situ, several investigators have begun to study where T cells and APCs localize within infected lungs (150–152). For example, CD4+ and CD8+ T cells exhibit different spatial and temporal distributions in the lungs during infection that may contribute to or dictate cellular functions (150). We have begun to characterize the spatial localization of the transgenic IAb/ESAT-61 – 20-specific T cells in our experimental model. Within 17 days post-infection, the transgenic cells were visualized in the lung airways in infected mice (Fig. 2). Other investigators have reported that chronic infection in both mice and humans is associated with the development of chemokine-dependent lymphoid-like structures within the lungs (153, 154). Such studies are likely to be important for understanding the interaction of T cells with APCs and other cells in vivo.
Fig. 2. Detection of transgenic CD4+ T cells in the lungs.
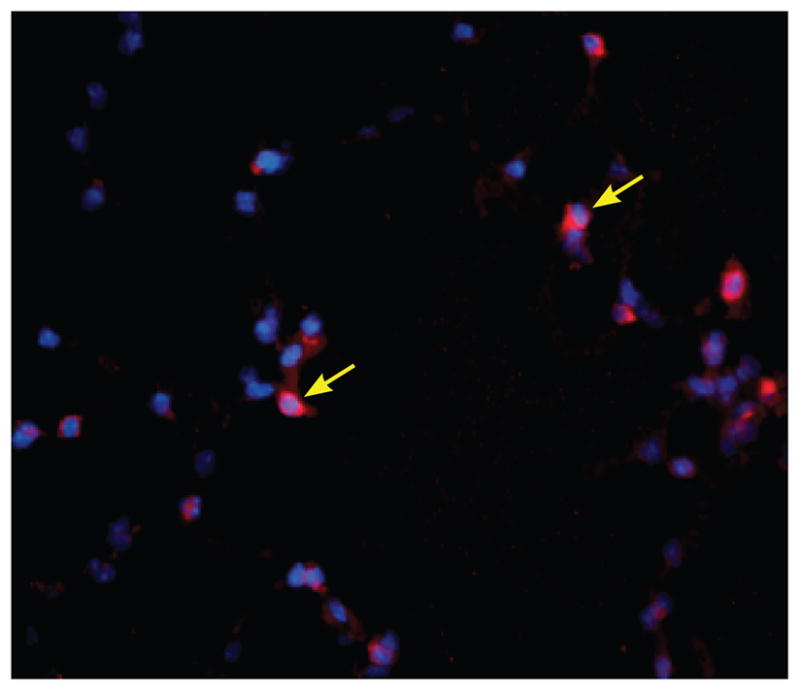
Transgenic IAb/ESAT-6-specific CD4+ T-donor cells were visualized in a mouse lung on day 17 post-aerosol inoculation. The transgenic CD4+ T cells (yellow arrows) were detected using an anti-CD90.2 antibody (visible in red). The cell nuclei were stained using DAPI. The image was captured using a Zeiss fluorescence microscope at × 40 magnification.
Events following T-cell activation
Once activated, the IAb/ESAT-6-specific T cells regulated the expression of several markers associated with T-cell differentiation, including CD62L, CD27, CD43, and CD127, consistent with the differentiation of the newly activated T cells into effector T cells (W. Reiley, G. Winslow and D. Woodland, unpublished data). The CD4+ T cells exhibited a normal pace of T-cell differentiation, suggesting that CD4+ T-cell responses are unabated during Mtb infection. Other investigators have reported similar findings from studies of polyclonal populations of CD4+ T cells during Mtb infection (155). These data suggest that Mtb does not impair T-cell responses, once the T cells have been activated. However, two recent studies have reported, in contrast, that early CD4+ T-cell responses in fact may be impaired, due to the activity of forkhead box protein 3 (Foxp3) and/or CD25+ T-regulatory cells (156, 157). In the study by Urdahl et al. (156), Foxp3+ T-regulatory cells were detected in the lungs and LNs of Mtb infected mice within 21 days post-infection, although T-cell numbers increased significantly later, during chronic infection. In the study by Kursar et al. (157), naive CD4+ T cells were transferred to recombination-activating gene (RAG)-deficient mice, with or without putative CD25+ T-regulatory cells, before Mtb infection. Expansion of the naive CD4+ T cells was inhibited in the presence of the co-transferred CD25+ T cells. In both studies, bacterial infection was higher in mice that contained putative T-regulatory cells, a fact that argues for a role of these cells in attenuating T-cell responses during both acute and chronic infection. Remarkably, transfer of CD25− CD4+ T cells to RAG-deficient mice in the absence of the CD25+T-cell population resulted in a 1–2 log10 reduction of bacterial infection in lung and spleen. These studies together suggest that T-regulatory cells play a critical role in attenuating T-cell responses during Mtb infection, although clearly additional studies are warranted.
Chronic Mtb infection
Although our focus in the present review is on early T-cell activation, mouse models are also valuable for the study of chronic Mtb infection. As discussed above, bacterial infection is stabilized at about 1 × 106 bacteria per lung after about 28 days post-infection, at least in part due to the action of CD4+ T cells. During this period of chronic infection, which can last as long as 250 days, antigen-specific effector CD4+ T cells are actively recruited and maintained in the lungs (114, 158). Our study showed that the steady-state CD4+ T-cell population under went continual turnover, during which the entire population was replaced within 7 days; the rate of turnover declined once control of infection was established (114). These data suggest that CD4+T cells are either maintained by proliferation, within the lungs or the lymphoid organs, or else are continually recruited from naive thymus-derived T cells. The former possibility was suggested by our finding that the lung IAb/ESAT-6-specific T-cell population was maintained in thymectomized mice, although a caveat is that conventional thymectomy may have not eliminated all thymic tissue in that study (159). The data nevertheless reveal that sufficient IAb/ESAT-61–20 was present throughout chronic infection to maintain long-term effector T-cell responses. This observation does not rule out a contribution from newly generated T cells to immunity during chronic infection, however. In normal mice and humans, naive T cells are produced lifelong by the thymus, albeit at age-dependent rates. These newly formed T cells may be recruited to chronically Mtb-infected tissues and/or granulomas and may contribute to the control of infection, although this idea has not been formally investigated. We have begun to model a possible contribution of naive CD4+ T cells during chronic infection, by transferring IAb/ESAT-61 – 20-specific transgenic T cells to mice during various stages of infection.
Memory T cells and vaccination
Investigators have used a range of different immunization strategies to address vaccine efficacy in mice; in a number of cases, a degree of protection has been achieved (typically 1 log10 reduction in bacterial load) (20, 127, 160). Such studies have provided an experimental rationale for human vaccine trials (3), although researchers have only begun to identify human memory phenotype T or B cells generated by vaccination (161). In the mouse model, administration of defined antigens and adjuvants has been shown to result in the early recruitment of CD4+ T cells to the lungs upon challenge, resulting in better control of infection (152, 158, 162). Those latter studies have suggested that effector T cells can be elicited earlier than in unimmunized mice following Mtb inoculation and that these earlier responses are critical in establishing immune control of infection. Early T-cell responses act to limit Mtb growth and positively affect the outcome of the infection, by limiting steady-state bacterial infection in the lungs (the bacterial ‘set-point’). In no case, however, has immunization prevented infection or achieved sterilizing immunity. These data suggest that pre-exposure vaccination strategies will be successful in limiting early Mtb colonization, but they may not succeed in preventing infection.
It is also apparent that prior Mtb infection does not generate highly effective immunity to re-infection. A retrospective examination of a number of human studies has shown that Mtb re-infections are not uncommon, suggesting that at least some humans do not generate protective immunity following infection (163, 164). In mice, this question has been addressed experimentally following antibiotic treatment of infected animals (28, 165). Mtb infection of drug-treated mice resulted in earlier IFNγ production by effector T cells, and the treated mice exhibited a lower the bacterial set-point (126, 152). Another recent study has shown that antigen-specific memory phenotype CD8+ T cells were present in lungs, LNs, and spleen 12 weeks after antibiotic treatment, suggesting that memory T cells are generated during Mtb infection (166). Nevertheless, the apparently relatively poor protective value of prior Mtb infection has led some investigators to question whether effective vaccination is attainable against TB (152, 164).
Why is it, then, that neither immunization nor prior infection has been able to achieve a better outcome? One possibility, discussed above, is that Mtb, once established in host cells, is impervious to defenses triggered by T cells. This would explain why early T-cell responses can sometimes limit but cannot prevent infection. Another possibility is that natural infection and/or vaccination does not generate effective T-cell memory. Mtb infection may induce abortive memory T-cell response, perhaps due to inflammatory conditions that do not favor memory generation. Alternatively, vaccination strategies attempted thus far may not have succeeding in generating sufficient numbers of memory T cells sufficient to elaborate effector functions that will control TB. In this regard, correlates of protective T-cell-mediated immunity are unavailable in either mice or humans that would allow us to evaluate what constitutes a protective memory response. Thus, much more work will be required to first understand and then to harness early memory T-cell responses during both primary and secondary infections.
Conclusions
Available evidence indicates that early innate and adaptive responses to Mtb are of critical importance for successful containment of infection. These events are poorly understood, however, in part because low-dose aerogenic inoculation does not trigger a robust early inflammatory response and because very early T-cell responses have been difficult to study. Important questions remain regarding whether Mtb delays the onset of early immunity, either by accident or design, and whether true memory T lymphocytes can be elicited by vaccination. New tools are now available to address these questions and have the potential to improve our understanding of TB immunity.
Acknowledgments
The authors wish to thank Susan Wittmer, John Pearl, Dr Shabanna Khader, and Jeff Fountain, for excellent scientific and technical expertise relevant to the studies conducted in our laboratories, and Dr Joanne Turner (Ohio State University) for critical reading of the manuscript. We also apologize for any failure to acknowledge work in the field contributed by many excellent investigators. This work was supported by U.S. Public Health Service grants R015R01AI073564 to G. M. W. and D. L. W. and AI067723 and AI46530 to A. M. C., and by a Pott’s Medical Foundation grant to G. M. W.
References
- 1.World Health Organization. Global Tuberculosis Control: Surveillance, Planning, Financing. Geneva, Switzerland: World Health Organization; 2007. [Google Scholar]
- 2.Raviglione MC, Smith IM. XDR tuberculosis – implications for global public health. N Engl J Med. 2007;356:656–659. doi: 10.1056/NEJMp068273. [DOI] [PubMed] [Google Scholar]
- 3.Andersen P. Tuberculosis vaccines – an update. Nat Rev Microbiol. 2007;5:484–487. doi: 10.1038/nrmicro1703. [DOI] [PubMed] [Google Scholar]
- 4.Young D, Dye C. The development and impact of tuberculosis vaccines. Cell. 2006;124:683–687. doi: 10.1016/j.cell.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Kaufmann SH. Envisioning future strategies for vaccination against tuberculosis. Nat Rev Immunol. 2006;6:699–704. doi: 10.1038/nri1920. [DOI] [PubMed] [Google Scholar]
- 6.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 7.Verma RK, Jain A. Antibodies to mycobacterial antigens for diagnosis of tuberculosis. FEMS Immunol Med Microbiol. 2007;51:453–461. doi: 10.1111/j.1574-695X.2007.00302.x. [DOI] [PubMed] [Google Scholar]
- 8.Scriba TJ, et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J Immunol. 2008;180:1962–1970. doi: 10.4049/jimmunol.180.3.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giacomini E, et al. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol. 2001;166:7033–7041. doi: 10.4049/jimmunol.166.12.7033. [DOI] [PubMed] [Google Scholar]
- 10.Huebner RE, Schein MF, Bass JB., Jr The tuberculin skin test. Clin Infect Dis. 1993;17:968–975. doi: 10.1093/clinids/17.6.968. [DOI] [PubMed] [Google Scholar]
- 11.Andersen P, Doherty TM. The success and failure of BCG – implications for a novel tuberculosis vaccine. Nat Rev Microbiol. 2005;3:656–662. doi: 10.1038/nrmicro1211. [DOI] [PubMed] [Google Scholar]
- 12.Soares AP, et al. Bacillus Calmette-Guerin vaccination of human newborns induces T cells with complex cytokine and phenotypic profiles. J Immunol. 2008;180:3569–3577. doi: 10.4049/jimmunol.180.5.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith SM, et al. Human CD8+ CTL specific for the mycobacterial major secreted antigen 85A. J Immunol. 2000;165:7088–7095. doi: 10.4049/jimmunol.165.12.7088. [DOI] [PubMed] [Google Scholar]
- 14.Tully G, et al. Highly focused T cell responses in latent human pulmonary Mycobacterium tuberculosis infection. J Immunol. 2005;174:2174–2184. doi: 10.4049/jimmunol.174.4.2174. [DOI] [PubMed] [Google Scholar]
- 15.Ordway D, et al. The cellular immune response to Mycobacterium tuberculosis infection in the guinea pig. J Immunol. 2007;179:2532–2541. doi: 10.4049/jimmunol.179.4.2532. [DOI] [PubMed] [Google Scholar]
- 16.Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. Real-time visualization of mycobacterium–macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity. 2002;17:693–702. doi: 10.1016/s1074-7613(02)00475-2. [DOI] [PubMed] [Google Scholar]
- 17.Lewinsohn DM, et al. High resolution radiographic and fine immunologic definition of TB disease progression in the rhesus macaque. Microbes Infect. 2006;8:2587–2598. doi: 10.1016/j.micinf.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Chege GK, Williamson AL, Passmore JS, Bourn W, Ryffel B, Shephard EG. The immune response of the Chacma baboon to Bacille Calmette Guerin: development of a primate model for BCG-based vaccine research. Vaccine. 2005;23:5783–5791. doi: 10.1016/j.vaccine.2005.07.106. [DOI] [PubMed] [Google Scholar]
- 19.Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity. 2008;28:271–284. doi: 10.1016/j.immuni.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.North RJ, Jung YJ. Immunity to tuberculosis. Annu Rev Immunol. 2004;22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635. [DOI] [PubMed] [Google Scholar]
- 21.Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 22.Turner J, et al. Immunological basis for reactivation of tuberculosis in mice. Infect Immun. 2001;69:3264–3270. doi: 10.1128/IAI.69.5.3264-3270.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medina E, North RJ. Resistance ranking of some common inbred mouse strains to Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunology. 1998;93:270–274. doi: 10.1046/j.1365-2567.1998.00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fortin A, Abel L, Casanova JL, Gros P. Host genetics of mycobacterial diseases in mice and men: forward genetic studies of BCG-osis and tuberculosis. Annu Rev Genom Hum Genet. 2007;8:163–192. doi: 10.1146/annurev.genom.8.080706.092315. [DOI] [PubMed] [Google Scholar]
- 25.Hill AV. Aspects of genetic susceptibility to human infectious diseases. Annu Rev Genet. 2006;40:469–486. doi: 10.1146/annurev.genet.40.110405.090546. [DOI] [PubMed] [Google Scholar]
- 26.Lyadova IV, et al. Comparative analysis of T lymphocytes recovered from the lungs of mice genetically susceptible, resistant, and hyperresistant to Mycobacterium tuberculosis-triggered disease. J Immunol. 2000;165:5921–5231. doi: 10.4049/jimmunol.165.10.5921. [DOI] [PubMed] [Google Scholar]
- 27.Orme IM, Collins FM. Aerogenic vaccination of mice with Mycobacterium bovis BCG. Tubercle. 1986;67:133–140. doi: 10.1016/0041-3879(86)90007-3. [DOI] [PubMed] [Google Scholar]
- 28.Orme IM, Collins FM. Crossprotection against nontuberculous mycobacterial infections by Mycobacterium tuberculosis memory immune T lymphocytes. J Exp Med. 1986;163:203–208. doi: 10.1084/jem.163.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardona PJ, et al. The intravenous model of murine tuberculosis is less pathogenic than the aerogenic model owing to a more rapid induction of systemic immunity. Scand J Immunol. 1999;49:362–366. doi: 10.1046/j.1365-3083.1999.00522.x. [DOI] [PubMed] [Google Scholar]
- 30.North RJ, LaCourse R, Ryan L. Vaccinated mice remain more susceptible to Mycobacterium tuberculosis infection initiated via the respiratory route than via the intravenous route. Infect Immun. 1999;67:2010–2012. doi: 10.1128/iai.67.4.2010-2012.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rhoades ER, Frank AA, Orme IM. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuber Lung Dis. 1997;78:57–66. doi: 10.1016/s0962-8479(97)90016-2. [DOI] [PubMed] [Google Scholar]
- 32.North RJ. Mycobacterium tuberculosis is strikingly more virulent for mice when given via the respiratory than via the intravenous route. J Infect Dis. 1995;172:1550–1553. doi: 10.1093/infdis/172.6.1550. [DOI] [PubMed] [Google Scholar]
- 33.Ernst JD. Macrophage receptors for Mycobacterium tuberculosis. Infect Immun. 1998;66:1277–1281. doi: 10.1128/iai.66.4.1277-1281.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neyrolles O, Gicquel B, Quintana-Murci L. Towards a crucial role for DC-SIGN in tuberculosis and beyond. Trends Microbiol. 2006;14:383–387. doi: 10.1016/j.tim.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Gaynor CD, McCormack FX, Voelker DR, McGowan SE, Schlesinger LS. Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J Immunol. 1995;155:5343–5351. [PubMed] [Google Scholar]
- 36.Leemans JC, et al. Macrophages play a dual role during pulmonary tuberculosis in mice. J Infect Dis. 2005;191:65–74. doi: 10.1086/426395. [DOI] [PubMed] [Google Scholar]
- 37.Rivas-Santiago B, et al. Expression of cathelicidin LL-37 during Mycobacterium tuberculosis infection in human alveolar macrophages, monocytes, neutrophils, and epithelial cells. Infect Immun. 2008;76:935–941. doi: 10.1128/IAI.01218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heldwein KA, Fenton MJ. The role of Toll-like receptors in immunity against mycobacterial infection. Microbes Infect. 2002;4:937–944. doi: 10.1016/s1286-4579(02)01611-8. [DOI] [PubMed] [Google Scholar]
- 39.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edwards AD, et al. Microbial recognition via Toll-like receptor-dependent and -independent pathways determines the cytokine response of murine dendritic. J Immunol. 2002;169:3652–3660. doi: 10.4049/jimmunol.169.7.3652. [DOI] [PubMed] [Google Scholar]
- 41.Pompei L, et al. Disparity in IL-12 release in dendritic cells and macrophages in response to Mycobacterium tuberculosis is due to use of distinct TLRs. J Immunol. 2007;178:5192–5199. doi: 10.4049/jimmunol.178.8.5192. [DOI] [PubMed] [Google Scholar]
- 42.Reiling N, et al. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. 2002;169:3480–3484. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 43.Jang S, Uematsu S, Akira S, Salgame P. IL-6 and IL-10 induction from dendritic cells in response to Mycobacterium tuberculosis is predominantly dependent on TLR2-mediated recognition. J Immunol. 2004;173:3392–3397. doi: 10.4049/jimmunol.173.5.3392. [DOI] [PubMed] [Google Scholar]
- 44.Bhatt K, Salgame P. Host innate immune response to Mycobacterium tuberculosis. J Clin Immunol. 2007;27:347–362. doi: 10.1007/s10875-007-9084-0. [DOI] [PubMed] [Google Scholar]
- 45.Pathak SK, et al. Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol. 2007;8:610–618. doi: 10.1038/ni1468. [DOI] [PubMed] [Google Scholar]
- 46.Algood HM, Lin PL, Yankura D, Jones A, Chan J, Flynn JL. TNF influences chemokine expression of macrophages in vitro and that of CD11b+cells in vivo during Mycobacterium tuberculosis infection. J Immunol. 2004;172:6846–6857. doi: 10.4049/jimmunol.172.11.6846. [DOI] [PubMed] [Google Scholar]
- 47.Orme IMCA. Cytokine/chemokine cascades in immunity to tuberculosis. Immunol Today. 1999;20:307–312. doi: 10.1016/s0167-5699(98)01438-8. [DOI] [PubMed] [Google Scholar]
- 48.Rhoades E, Cooper A, Orme I. Chemokine response in mice infected with Mycobacterium tuberculosis. Infect Immun. 1995;63:3871–3877. doi: 10.1128/iai.63.10.3871-3877.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geijtenbeek TB, et al. Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med. 2003;197:7–17. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tailleux L, et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J Exp Med. 2003;197:121–127. doi: 10.1084/jem.20021468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tian T, Woodworth J, Skold M, Behar SM. In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. J Immunol. 2005;175:3268–3272. doi: 10.4049/jimmunol.175.5.3268. [DOI] [PubMed] [Google Scholar]
- 52.Wolf AJ, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol. 2007;179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 53.Dunn PL, North RJ. Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology, and cause mortality in mice. Infect Immun. 1995;63:3428–3237. doi: 10.1128/iai.63.9.3428-3437.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med. 2001;193:271–280. doi: 10.1084/jem.193.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Orme IM, Collins FM. Adoptive protection of the Mycobacterium tuberculosis-infected lung. Dissociation between cells that passively transfer protective immunity and those that transfer delayed-type hypersensitivity to tuberculin. Cell Immunol. 1984;84:113–120. doi: 10.1016/0008-8749(84)90082-0. [DOI] [PubMed] [Google Scholar]
- 56.North RJ. Importance of thymus-derived lymphocytes in cell-mediated immunity to infection. Cell Immunol. 1973;7:166–176. doi: 10.1016/0008-8749(73)90193-7. [DOI] [PubMed] [Google Scholar]
- 57.Lefford MJ. Transfer of adoptive immunity to tuberculosis in mice. Infect Immun. 1975;11:1174–1181. doi: 10.1128/iai.11.6.1174-1181.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raupach B, Kaufmann SH. Immune responses to intracellular bacteria. Curr Opin Immunol. 2001;13:417–428. doi: 10.1016/s0952-7915(00)00236-3. [DOI] [PubMed] [Google Scholar]
- 59.Ladel CH, Daugelat S, Kaufmann SH. Immune response to Mycobacterium bovis bacille Calmette Guerin infection in major histocompatibility complex class I- and II-deficient knock-out mice: contribution of CD4 and CD8 T cells to acquired resistance. Eur J Immunol. 1995;25:377–384. doi: 10.1002/eji.1830250211. [DOI] [PubMed] [Google Scholar]
- 60.Caruso AM, Serbina N, Klein E, Triebold K, Bloom BR, Flynn JL. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J Immunol. 1999;162:5407–5416. [PubMed] [Google Scholar]
- 61.Muller I, Cobbold SP, Waldmann H, Kaufmann SHE. Impaired resistance to Mycobacterium tuberculosis infection after selective in vivo depletion of of L3T4+ and Lyt-2+ T cells. Infect Immun. 1987;55:2037–2041. doi: 10.1128/iai.55.9.2037-2041.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon γ gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon γ in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pearl JE, Saunders B, Ehlers S, Orme IM, Cooper AM. Inflammation and lymphocyte activation during mycobacterial infection in the interferon-gamma-deficient mouse. Cell Immunol. 2001;211:43–50. doi: 10.1006/cimm.2001.1819. [DOI] [PubMed] [Google Scholar]
- 65.Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect Immun. 1995;63:736–740. doi: 10.1128/iai.63.2.736-740.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dalton DK, Pitts-Meek S, Keshav TA, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-γ genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 67.Cowley SC, Elkins KL. CD4+ T cells mediate IFN-gamma-independent control of Mycobacterium tuberculosis infection both in vitro and in vivo. J Immunol. 2003;171:4689–4699. doi: 10.4049/jimmunol.171.9.4689. [DOI] [PubMed] [Google Scholar]
- 68.Lande R, et al. IFN-alpha beta released by Mycobacterium tuberculosis-infected human dendritic cells induces the expression of CXCL10: selective recruitment of NK and activated T cells. J Immunol. 2003;170:1174–1182. doi: 10.4049/jimmunol.170.3.1174. [DOI] [PubMed] [Google Scholar]
- 69.Fortune SM, et al. Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. J Immunol. 2004;172:6272–6280. doi: 10.4049/jimmunol.172.10.6272. [DOI] [PubMed] [Google Scholar]
- 70.Gehring AJ, Rojas RE, Canaday DH, Lakey DL, Harding CV, Boom WH. The Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits gamma interferon-regulated HLA-DR and Fc gamma R1 on human macrophages through Toll-like receptor 2. Infect Immun. 2003;71:4487–4497. doi: 10.1128/IAI.71.8.4487-4497.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ting LM, Kim AC, Cattamanchi A, Ernst JD. Mycobacterium tuberculosis inhibits IFN-gamma transcriptional responses without inhibiting activation of STAT1. J Immunol. 1999;163:3898–3906. [PubMed] [Google Scholar]
- 72.Woodworth JS, Behar SM. Mycobacterium tuberculosis-specific CD8+ T cells and their role in immunity. Crit Rev Immunol. 2006;26:317–352. doi: 10.1615/critrevimmunol.v26.i4.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Flynn JL, Goldstein MM, Triebold KJ, Koller B, Bloom BR. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc Natl Acad Sci USA. 1992;89:12013–12017. doi: 10.1073/pnas.89.24.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Turner J, et al. CD8- and CD95/95L-dependent mechanisms of resistance in mice with chronic pulmonary tuberculosis. Am J Respir Cell Mol Biol. 2001;24:203–209. doi: 10.1165/ajrcmb.24.2.4370. [DOI] [PubMed] [Google Scholar]
- 75.D’Souza CD, et al. A novel nonclassic beta2-microglobulin-restricted mechanism influencing early lymphocyte accumulation and subsequent resistance to tuberculosis in the lung. Am J Respir Cell Mol Biol. 2000;23:188–193. doi: 10.1165/ajrcmb.23.2.4063. [DOI] [PubMed] [Google Scholar]
- 76.Sousa AO, et al. Relative contributions of distinct MHC class I-dependent cell populations in protection to tuberculosis infection in mice. Proc Natl Acad Sci USA. 2000;97:4204–4208. doi: 10.1073/pnas.97.8.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Orme IM. The kinetics of emergence and loss of mediator T lymphocytes acquired in response to infection with Mycobacterium tuberculosis. J Immunol. 1987;138:293–298. [PubMed] [Google Scholar]
- 78.Schaible UE, Collins HL, Priem F, Kaufmann SHE. Correction of the iron overload defect in {beta}-2-Microglobulin knockout mice by lactoferrin abolishes their increased susceptibility to tuberculosis. J Exp Med. 2002;196:1507–1513. doi: 10.1084/jem.20020897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Behar SM, Dascher CC, Grusby MJ, Wang CR, Brenner MB. Susceptibility of mice deficient in CD1d or TAP1 to infection with Mycobacterium tuberculosis. J Exp Med. 1999;189:1973–1980. doi: 10.1084/jem.189.12.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Urdahl KB, Liggitt D, Bevan MJ. CD8+T Cells accumulate in the lungs of Mycobacterium tuberculosis-infected Kb −/− Db−/− Mice, but provide minimal protection. J Immunol. 2003;170:1987–1994. doi: 10.4049/jimmunol.170.4.1987. [DOI] [PubMed] [Google Scholar]
- 81.Serbina NV, Flynn JL. Early emergence of CD8(+) T cells primed for production of type 1 cytokines in the lungs of Mycobacterium tuberculosis-infected mice. Infect Immun. 1999;67:3980–3988. doi: 10.1128/iai.67.8.3980-3988.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Serbina NV, Liu CC, Scanga CA, Flynn JL. CD8+ CTL from lungs of Mycobacterium tuberculosis-infected mice express perforin in vivo and lyse infected macrophages. J Immunol. 2000;165:353–363. doi: 10.4049/jimmunol.165.1.353. [DOI] [PubMed] [Google Scholar]
- 83.Kamath AB, Woodworth J, Xiong X, Taylor C, Weng Y, Behar SM. Cytolytic CD8+ T cells recognizing CFP10 are recruited to the lung after Mycobacterium tuberculosis infection. J Exp Med. 2004;200:1479–1489. doi: 10.1084/jem.20041690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Orme IM, et al. T lymphocytes mediating protection and cellular cytolysis during the course of Mycobacterium tuberculosis infection. Evidence for different kinetics and recognition of a wide spectrum of protein antigens. J Immunol. 1992;148:189–196. [PubMed] [Google Scholar]
- 85.Winau F, et al. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity. 2006;24:105–117. doi: 10.1016/j.immuni.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 86.Griffin JP, Orme IM. Evolution of CD4 T-cell subsets following infection of naive and memory immune mice with Mycobacterium tuberculosis. Infect Immun. 1994;62:1683–1690. doi: 10.1128/iai.62.5.1683-1690.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Andersen P, Askgaard D, Ljungqvist L, Bentzon MW, Heron I. T-cell proliferative response to antigens secreted by Mycobacterium tuberculosis. Infect Immun. 1991;59:1558–1563. doi: 10.1128/iai.59.4.1558-1563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haslov K, Andersen A, Nagai S, Gottschau A, Sorensen T, Andersen P. Guinea pig cellular immune responses to proteins secreted by Mycobacterium tuberculosis. Infect Immun. 1995;63:804–810. doi: 10.1128/iai.63.3.804-810.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lewinsohn DA, et al. Immunodominant tuberculosis CD8 antigens preferentially restricted by HLA-B. PLoS pathogens. 2007;3:1240–1249. doi: 10.1371/journal.ppat.0030127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andersen P, Askgaard D, Gottschau A, Bennedsen J, Nagai S, Heron I. Identification of immunodominant antigens during infection with Mycobacterium tuberculosis. Scand J Immunol. 1992;36:823–831. doi: 10.1111/j.1365-3083.1992.tb03144.x. [DOI] [PubMed] [Google Scholar]
- 91.Sorensen AL, Nagai S, Houen G, Andersen P, Andersen AB. Purification and characterization of a low-molecular-mass T-cell antigen secreted by Mycobacterium tuberculosis. Infect Immun. 1995;63:1710–1717. doi: 10.1128/iai.63.5.1710-1717.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Andersen P. Host responses and antigens involved in protective immunity to Mycobacterium tuberculosis. Scand J Immunol. 1997;45:115–131. doi: 10.1046/j.1365-3083.1997.d01-380.x. [DOI] [PubMed] [Google Scholar]
- 93.Moody DB, Sugita M, Peters PJ, Brenner MB, Porcelli SA. The CD1-restricted T-cell response to mycobacteria. Res Immunol. 1996;147:550–559. doi: 10.1016/s0923-2494(97)85221-2. [DOI] [PubMed] [Google Scholar]
- 94.Jayawardena-Wolf J, Bendelac A. CD1 and lipid antigens: intracellular pathways for antigen presentation. Curr Opin Immunol. 2001;13:109–113. doi: 10.1016/s0952-7915(00)00190-4. [DOI] [PubMed] [Google Scholar]
- 95.Orme IM. Induction of nonspecific acquired resistance and delayed-type hypersensitivity, but not specific acquired resistance in mice inoculated with killed mycobacterial vaccines. Infect Immun. 1988;56:3310–3312. doi: 10.1128/iai.56.12.3310-3312.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shen H, Miller JF, Fan X, Kolwyck D, Ahmed R, Harty JT. Compartmentalization of bacterial antigens: differential effects on priming of CD8 T cells and protective immunity. Cell. 1998;92:535–545. doi: 10.1016/s0092-8674(00)80946-0. [DOI] [PubMed] [Google Scholar]
- 97.Andersen P, Askgaard D, Ljungqvist L, Bennedsen J, Heron I. Proteins released from Mycobacterium tuberculosis during growth. Infect Immun. 1991;59:1905–1910. doi: 10.1128/iai.59.6.1905-1910.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rook GA, Steele J, Barnass S, Mace J, Stanford JL. Responsiveness to live M. tuberculosis, and common antigens, of sonicate-stimulated T cell lines from normal donors. Clin Exp Immunol. 1986;63:105–110. [PMC free article] [PubMed] [Google Scholar]
- 99.Havlir DV, Wallis RS, Boom WH, Daniel TM, Chervenak K, Ellner JJ. Human immune response to Mycobacterium tuberculosis antigens. Infect Immun. 1991;59:665–670. doi: 10.1128/iai.59.2.665-670.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Orme IM, et al. T lymphocytes mediating protection and cellular cytolysis during the course of Mycobacterium tuberculosis infection. Evidence for different kinetics and recognition of a wide spectrum of protein antigens. J Immunol. 1992;148:189–196. [PubMed] [Google Scholar]
- 101.Roberts AD, et al. Characteristics of protective immunity engendered by vaccination of mice with purified culture filtrate protein antigens of Mycobacterium tuberculosis. Immunology. 1995;85:502–508. [PMC free article] [PubMed] [Google Scholar]
- 102.Orme IM, Roberts AD, Griffin JP, Abrams JS. Cytokine secretion by CD4 T lymphocytes acquired in response to Mycobacterium tuberculosis infection. J Immunol. 1993;151:518–525. [PubMed] [Google Scholar]
- 103.Yanagisawa S, Koike M, Kariyone A, Nagai S, Takatsu K. Mapping of V beta 11+helper T cell epitopes on mycobacterial antigen in mouse primed with Mycobacterium tuberculosis. Int Immunol. 1997;9:227–237. doi: 10.1093/intimm/9.2.227. [DOI] [PubMed] [Google Scholar]
- 104.Pais TF, Silva RA, Smedegaard B, Appelberg R, Andersen P. Analysis of T cells recruited during delayed-type hypersensitivity to purified protein derivative (PPD) versus challenge with tuberculosis infection. Immunology. 1998;95:69–75. doi: 10.1046/j.1365-2567.1998.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wiker HG, Harboe M. The antigen 85 complex: a major secretion product of Mycobacterium tuberculosis. Microbiol Rev. 1992;56:648–661. doi: 10.1128/mr.56.4.648-661.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. 1997;276:1420–1422. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 107.Kariyone A, et al. Identification of amino acid residues of the T-cell epitope of Mycobacterium tuberculosis alpha antigen critical for Vbeta11(+) Th1 cells. Infect Immun. 1999;67:4312–4319. doi: 10.1128/iai.67.9.4312-4319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ulrichs T, Anding P, Porcelli S, Kaufmann SH, Munk ME. Increased numbers of ESAT-6- and purified protein derivative-specific gamma interferon-producing cells in sub-clinical and active tuberculosis infection. Infect Immun. 2000;68:6073–6076. doi: 10.1128/iai.68.10.6073-6076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Olsen AW, Hansen PR, Holm A, Andersen P. Efficient protection against Mycobacterium tuberculosis by vaccination with a single sub-dominant epitope from the ESAT-6 antigen. Eur J Immunol. 2000;30:1724–1732. doi: 10.1002/1521-4141(200006)30:6<1724::AID-IMMU1724>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 110.Pym AS, et al. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat Med. 2003;9:533–539. doi: 10.1038/nm859. [DOI] [PubMed] [Google Scholar]
- 111.Harboe M, Oettinger T, Wiker HG, Rosenkrands I, Andersen P. Evidence for occurrence of the ESAT-6 protein in Mycobacterium tuberculosis and virulent Mycobacterium bovis and for its absence in Mycobacterium bovis BCG. Infect Immun. 1996;64:16–22. doi: 10.1128/iai.64.1.16-22.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Blythe MJ, et al. An analysis of the epitope knowledge related to Mycobacteria. Immunome Res. 2007;3:10. doi: 10.1186/1745-7580-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rogerson BJ, Jung YJ, LaCourse R, Ryan L, Enright N, North RJ. Expression levels of Mycobacterium tuberculosis antigen-encoding genes versus production levels of antigen-specific T cells during stationary level lung infection in mice. Immunology. 2006;118:195–201. doi: 10.1111/j.1365-2567.2006.02355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Winslow GM, Roberts AD, Blackman MA, Woodland DL. Persistence and turnover of antigen-specific CD4 T cells during chronic tuberculosis infection in the mouse. J Immunol. 2003;170:2046–2052. doi: 10.4049/jimmunol.170.4.2046. [DOI] [PubMed] [Google Scholar]
- 115.Chackerian AA, Perera TV, Behar SM. Gamma interferon-producing CD4+ T lymphocytes in the lung correlate with resistance to infection with Mycobacterium tuberculosis. Infect Immun. 2001;69:2666–2674. doi: 10.1128/IAI.69.4.2666-2674.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lazarevic V, Nolt D, Flynn JL. Long-term control of Mycobacterium tuberculosis infection is mediated by dynamic immune responses. J Immunol. 2005;175:1107–1117. doi: 10.4049/jimmunol.175.2.1107. [DOI] [PubMed] [Google Scholar]
- 117.Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 1998;8:683–691. doi: 10.1016/s1074-7613(00)80573-7. [DOI] [PubMed] [Google Scholar]
- 118.Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun. 2002;70:4501–4509. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Majlessi L, Rojas MJ, Brodin P, Leclerc C. CD8+ T-cell responses of Mycobacterium-infected mice to a newly identified major histocompatibility complex class I-restricted epitope shared by proteins of the ESAT-6 family. Infect Immun. 2003;71:7173–7177. doi: 10.1128/IAI.71.12.7173-7177.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Irwin SM, et al. Tracking antigen-specific CD8 T lymphocytes in the lungs of mice vaccinated with the Mtb72F polyprotein. Infect Immun. 2005;73:5809–5816. doi: 10.1128/IAI.73.9.5809-5816.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lawrence CW, Braciale TJ. Activation, differentiation, and migration of naive virus-specific CD8+T cells during pulmonary influenza virus infection. J Immunol. 2004;173:1209–1218. doi: 10.4049/jimmunol.173.2.1209. [DOI] [PubMed] [Google Scholar]
- 122.Zimmermann C, Prevost-Blondel A, Blaser C, Pircher H. Kinetics of the response of naive and memory CD8 T cells to antigen: similarities and differences. Eur J Immunol. 1999;29:284–290. doi: 10.1002/(SICI)1521-4141(199901)29:01<284::AID-IMMU284>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 123.Busch DH, Pilip IM, Vijh S, Pamer EG. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 1998;8:353–362. doi: 10.1016/s1074-7613(00)80540-3. [DOI] [PubMed] [Google Scholar]
- 124.Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early programming of T cell populations responding to bacterial infection. J Immunol. 2000;165:6833–6839. doi: 10.4049/jimmunol.165.12.6833. [DOI] [PubMed] [Google Scholar]
- 125.Corbin GA, Harty JT. Duration of infection and antigen display have minimal influence on the kinetics of the CD4+ T cell response to Listeria monocytogenes infection. J Immunol. 2004;173:5679–5687. doi: 10.4049/jimmunol.173.9.5679. [DOI] [PubMed] [Google Scholar]
- 126.Cooper AM, Callahan JE, Keen M, Belisle JT, Orme IM. Expression of memory immunity in the lung following re-exposure to Mycobacterium tuberculosis. Tuber Lung Dis. 1997;78:67–73. doi: 10.1016/s0962-8479(97)90017-4. [DOI] [PubMed] [Google Scholar]
- 127.Jung YJ, Lacourse R, Ryan L, North RJ. Immunization against airborne tuberculosis by an earlier primary response to a concurrent intravenous infection. Immunology. 2008;124:514–521. doi: 10.1111/j.1365-2567.2007.02803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.van Faassen H, Dudani R, Krishnan L, Sad S. Prolonged antigen presentation, APC-, and CD8+ T cell turnover during mycobacterial infection: comparison with Listeria monocytogenes. J Immunol. 2004;172:3491–3500. doi: 10.4049/jimmunol.172.6.3491. [DOI] [PubMed] [Google Scholar]
- 129.Demotz S, Grey HM, Sette A. The minimal number of class II MHC-antigen complexes needed for T cell activation. Science. 1990;249:1028–1030. doi: 10.1126/science.2118680. [DOI] [PubMed] [Google Scholar]
- 130.Tobian AA, et al. Alternate class I MHC antigen processing is inhibited by Toll-like receptor signaling pathogen-associated molecular patterns: Mycobacterium tuberculosis 19-kDa lipoprotein, CpG DNA, and lipopolysaccharide. J Immunol. 2003;171:1413–1422. doi: 10.4049/jimmunol.171.3.1413. [DOI] [PubMed] [Google Scholar]
- 131.Pai RK, Convery M, Hamilton TA, Boom WH, Harding CV. Inhibition of IFN-gamma-induced class II transactivator expression by a 19-kDa lipoprotein from Mycobacterium tuberculosis: a potential mechanism for immune evasion. J Immunol. 2003;171:175–184. doi: 10.4049/jimmunol.171.1.175. [DOI] [PubMed] [Google Scholar]
- 132.Noss EH, Harding CV, Boom WH. Mycobacterium tuberculosis inhibits MHC class II antigen processing in murine bone marrow macrophages. Cell Immunol. 2000;201:63–74. doi: 10.1006/cimm.2000.1633. [DOI] [PubMed] [Google Scholar]
- 133.Khader SA, et al. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med. 2006;203:1805–1815. doi: 10.1084/jem.20052545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 135.Bousso P, Robey E. Dynamics of CD8+T cell priming by dendritic cells in intact lymph nodes. Nat Immunol. 2003;4:579–585. doi: 10.1038/ni928. [DOI] [PubMed] [Google Scholar]
- 136.Miller MJ, Hejazi AS, Wei SH, Cahalan MD, Parker I. T cell repertoire scanning is promoted by dynamic dendritic cell behavior and random T cell motility in the lymph node. Proc Natl Acad Sci USA. 2004;101:998–1003. doi: 10.1073/pnas.0306407101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Busch DH, Pamer EG. T cell affinity maturation by selective expansion during infection. J Exp Med. 1999;189:701–710. doi: 10.1084/jem.189.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rees W, et al. An inverse relationship between T cell receptor affinity and antigen dose during CD4(+) T cell responses in vivo and in vitro. Proc Natl Acad Sci USA. 1999;96:9781–9786. doi: 10.1073/pnas.96.17.9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Testi R, Phillips JH, Lanier LL. T cell activation via Leu-23 (CD69) J Immunol. 1989;143:1123–1128. [PubMed] [Google Scholar]
- 140.Shiow LR, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 141.Stockinger B, Bourgeois C, Kassiotis G. CD4+ memory T cells: functional differentiation and homeostasis. Immunol Rev. 2006;211:39–48. doi: 10.1111/j.0105-2896.2006.00381.x. [DOI] [PubMed] [Google Scholar]
- 142.Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science. 2006;312:114–116. doi: 10.1126/science.1124228. [DOI] [PubMed] [Google Scholar]
- 143.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T-cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 144.Garside P, et al. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 145.Badovinac VP, Haring JS, Harty JT. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity. 2007;26:827–841. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wolf AJ, et al. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med. 2008;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tamura T, et al. The role of antigenic peptide in CD4+ T helper phenotype development in a T cell receptor transgenic model. Int Immunol. 2004;16:1691–1699. doi: 10.1093/intimm/dxh170. [DOI] [PubMed] [Google Scholar]
- 148.Anis MM, Fulton SA, Reba SM, Harding CV, Boom WH. Modulation of naive CD4+ T-cell responses to an airway antigen during pulmonary mycobacterial infection. Infect Immun. 2007;75:2260–2268. doi: 10.1128/IAI.01709-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Reiley WW, Calayag MD, Wittmer ST, Huntington JL, Pearl JE, Fountain JJ, et al. ESAT-6-specific CD4 T cell responses to aerosol M. tuberculosis infection are initiated in the mediastinal lymph nodes. Proc Natl Acad Sci USA. 2008;105:10961–10966. doi: 10.1073/pnas.0801496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gonzalez-Juarrero M, Turner OC, Turner J, Marietta P, Brooks JV, Orme IM. Temporal and spatial arrangement of lymphocytes within lung granulomas induced by aerosol infection with Mycobacterium tuberculosis. Infect Immun. 2001;69:1722–1728. doi: 10.1128/IAI.69.3.1722-1728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pedroza-Gonzalez A, et al. In situ analysis of lung antigen-presenting cells during murine pulmonary infection with virulent Mycobacterium tuberculosis. Int J Exp Pathol. 2004;85:135–145. doi: 10.1111/j.0959-9673.2004.00381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jung YJ, Ryan L, Lacourse R, North RJ. Properties and protective value of the secondary versus primary T helper type 1 response to airborne Mycobacterium tuberculosis infection in mice. J Exp Med. 2005;201:1915–1924. doi: 10.1084/jem.20050265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kahnert A, Hopken UE, Stein M, Bandermann S, Lipp M, Kaufmann SH. Mycobacterium tuberculosis triggers formation of lymphoid structure in murine lungs. J Infect Dis. 2007;195:46–54. doi: 10.1086/508894. [DOI] [PubMed] [Google Scholar]
- 154.Ulrichs T, et al. Human tuberculosis granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J Pathol. 2004;204:217–228. doi: 10.1002/path.1628. [DOI] [PubMed] [Google Scholar]
- 155.Kapina MA, et al. CD27 low CD4 T lymphocytes that accumulate in the mouse lungs during mycobacterial infection differentiate from CD27 high precursors in situ, produce IFN-γ, and protect the host against tuberculosis infection. J Immunol. 2007;178:976–985. doi: 10.4049/jimmunol.178.2.976. [DOI] [PubMed] [Google Scholar]
- 156.Scott-Browne JP, et al. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J Exp Med. 2007;204:2159–2169. doi: 10.1084/jem.20062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kursar M, et al. Cutting edge: regulatory T cells prevent efficient clearance of Mycobacterium tuberculosis. J Immunol. 2007;178:2661–2665. doi: 10.4049/jimmunol.178.5.2661. [DOI] [PubMed] [Google Scholar]
- 158.Serbina NV, Flynn JL. CD8(+) T cells participate in the memory immune response to Mycobacterium tuberculosis. Infect Immun. 2001;69:4320–4328. doi: 10.1128/IAI.69.7.4320-4328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Terszowski G, et al. Evidence for a functional second thymus in mice. Science. 2006;312:284–287. doi: 10.1126/science.1123497. [DOI] [PubMed] [Google Scholar]
- 160.Goonetilleke NP, McShane H, Hannan CM, Anderson RJ, Brookes RH, Hill AV. Enhanced immunogenicity and protective efficacy against Mycobacterium tuberculosis of bacille Calmette-Guerin vaccine using mucosal administration and boosting with a recombinant modified vaccinia virus Ankara. J Immunol. 2003;171:1602–1609. doi: 10.4049/jimmunol.171.3.1602. [DOI] [PubMed] [Google Scholar]
- 161.Beveridge NE, et al. Immunisation with BCG and recombinant MVA85A induces long-lasting, polyfunctional Mycobacterium tuberculosis-specific CD4+ memory T lymphocyte populations. Eur J Immunol. 2007;37:3089–3100. doi: 10.1002/eji.200737504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Khader SA, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 163.Chiang CY, Riley LW. Exogenous reinfection in tuberculosis. Lancet Infect Dis. 2005;5:629–636. doi: 10.1016/S1473-3099(05)70240-1. [DOI] [PubMed] [Google Scholar]
- 164.Kaufmann SH. Is the development of a new tuberculosis vaccine possible? Nat Med. 2000;6:955–960. doi: 10.1038/79631. [DOI] [PubMed] [Google Scholar]
- 165.Orme IM. Characteristics and specificity of acquired immunologic memory to Mycobacterium tuberculosis infection. J Immunol. 1988;140:3589–3593. [PubMed] [Google Scholar]
- 166.Kamath A, Woodworth JS, Behar SM. Antigen-specific CD8+ T cells and the development of central memory during Mycobacterium tuberculosis infection. J Immunol. 2006;177:6361–6369. doi: 10.4049/jimmunol.177.9.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]