

Abstract
Tau proteins are the building blocks of neurofibrillary tangles (NFTs) found in a range of neurodegenerative tauopathies, including Alzheimer's disease. Recently, we demonstrated that tau is extensively post-translationally modified by lysine acetylation, which impairs normal tau function and promotes pathological aggregation. Identifying the enzymes that mediate tau acetylation could provide targets for future therapies aimed at reducing the burden of acetylated tau. Here, we report that mammalian tau proteins possess intrinsic enzymatic activity capable of catalyzing self-acetylation. Functional mapping of tau acetyltransferase activity followed by biochemical analysis revealed that tau uses catalytic cysteine residues in the microtubule-binding domain to facilitate tau lysine acetylation, thus suggesting a mechanism similar to that employed by MYST-family acetyltransferases. The identification of tau as an acetyltransferase provides a framework to further understand tau pathogenesis and highlights tau enzymatic activity as a potential therapeutic target.
Tau proteins are expressed primarily in the central nervous system (CNS) and comprise six isoforms containing either three (3R-tau) or four (4R-tau) repeat domains that mediate microtubule binding, thereby regulating microtubule stability1,2. Regulation of tau function occurs by post-translational mechanisms including phosphorylation, which occurs primarily in regions flanking the repeats3. In Alzheimer's disease and related tauopathies, tau becomes abnormally hyperphosphorylated, which results in abrogated binding to microtubules. This leads to marked accumulation of cytoplasmic tau aggregates, termed NFTs, which represent a major pathological hallmark that characterizes neurodegenerative tauopathies4. We and others previously demonstrated that tau is extensively acetylated on lysine residues directly within the microtubule-binding repeats, thus providing a new regulatory modification controlling normal and abnormal tau properties5–7. Indeed, tau acetylation was shown to functionally impair normal tau-microtubule interactions, prevent physiological tau-mediated stabilization of microtubules and promote pathological tau fibril formation that is predominantly associated with insoluble, thioflavin-positive tau aggregates5,7. Neuropathological analysis of a panel of human tauopathy cases indicated that acetylation on a single lysine residue (Lys280) within the second repeat represents a distinctly pathological signature marking mature tau lesions in several major tauopathies including Alzheimer's disease, corticobasal degeneration and progressive supranuclear palsy but is rarely observed in control brain tissue or cultured wild-type neurons6. Thus, acetylation has emerged as a critical post-translational tau modification that occurs directly within the microtubule-binding repeats, probably acting in conjunction with phosphorylation, to impair normal tau functions and promote aggregation in Alzheimer's disease and related tauopathies.
Prior studies have implicated CREB-binding protein (CBP or p300) as a putative tau acetyltransferase5,7, and we confirmed that CBP has strong activity toward tau substrate both in vitro and in a cell-culture model (Supplementary Fig. 1a–c). However, CBP is localized in the nucleus, whereas tau is predominantly cytoplasmic, which suggests that other acetyltransferase activities probably exist that mediate tau acetylation. Unexpectedly, our previous findings hinted that tau autoacetylation can occur upon incubation with acetyl-CoA substrate alone5, raising the possibility that tau itself possesses a previously unrecognized acetyltransferase activity. Here, we sought to uncover the dominant mechanisms that mediate tau acetylation, which could potentially open up new therapeutic avenues to reducing tau aggregation and ameliorating disease pathogenesis. We identify tau itself as a bona fide acetyltransferase with sequence and functional similarities to the MYST family of acetyltransferases. Biochemical and kinetic studies indicate that tau catalyzes self-acetylation (autoacetylation) mediated by a pair of catalytic cysteine residues residing within the microtubule-binding domain. Our study suggests that prolonged activation of tau acetyltransferase activity could represent a new pathway that mediates tau pathogenesis and highlights tau enzymatic function as a potential therapeutic target for Alzheimer's disease and related tauopathies characterized by the pathological accumulation of acetylated tau.
Results
Tau proteins possess autoacetyltransferase activity
To characterize a putative tau acetyltransferase activity, we purified full-length tau proteins containing either three or four microtubule-binding repeats (that is, 3R-tau and 4R-tau), tau fragments containing only three or four repeats (that is, tau-K19 and tau-K18) or tau protein lacking the repeats (tau-K18(−)) and measured acetyltransferase activity in the presence of [14C]acetyl-CoA. (Fig. 1a shows a schematic of all tau proteins used in this study.) All 3R-tau and 4R-tau proteins possessed autoacetylation activity, which was prominent with repeat-containing tau-K19 and tau-K18 fragments (Fig. 1b). Notably, the tau-K18(−) protein lacking repeat regions had no detectable activity (Fig. 1b), which suggests that enzymatic activity resides within the microtubule-binding regions. Acetyltransferase activity was similarly detected upon co-incubation of 3R-tau or 4R-tau isoforms separately or co-incubation of all six tau isoforms together (Fig. 1c).
Figure 1.

Tau proteins containing microtubule-binding repeats possess acetyltransferase activity. (a) Schematic representation of all the purified tau proteins used in this study: tau proteins with three or four microtubule-binding repeats (3R- or 4R-tau) and a variable number of N-terminal inserts (0−2N) and tau lacking all microtubule-binding repeats but containing both N-terminal inserts (no-repeat tau). Locations of C291A and C322A mutations in 3R- and 4R-tau are highlighted. (b) SDS-PAGE with Coomassie blue staining (top) of tau proteins incubated with [14C]-labeled acetyl-CoA followed by autoradiography (bottom) to detect acetylated tau proteins. MW, molecular weight. (c) As in b, with 3R-tau or 4R-tau proteins. (d) Immunoblot of 3R- or 4R-tau isoforms incubated in the absence or presence of cold CoA or acetyl-CoA, using antibodies to acetyl-lysine, Ac-K280 and total tau (T46). Ac, acetyl. (e) In-gel acetyltransferase assay (described in Online Methods) with Tau-K18, 0N4R-tau, 2N4R-tau and tau-K18(−), followed by Commassie staining (top) and autoradiography (bottom).
To confirm autoacetylation of lysine residues, tau proteins were incubated with CoA or acetyl-CoA followed by immunoblotting using antibodies to pan-acetyl-lysine and acetyl-tau-K280 (Ac-K280)5,6. Consistent with the autoradiography results, all six tau isoforms as well as the tau-K18 fragment showed marked autoacetylation, as revealed by anti–acetyl-lysine immunoblotting (Fig. 1d). As expected, the anti–Ac-K280 antibody detected only autoacetylated 4R-tau isoforms, as residue Lys280 within the second repeat is present in 4R-tau but not 3R-tau. To identify the full extent of autoacetylated lysine residues within the repeat region, we performed MS on autoacetylated tau-K18 and identified a subset of acetylated lysines that overlap with previously identified CBP-mediated acetylated lysines5,7 (data not shown). Thus, both 3R- and 4R-tau proteins possess acetyltransferase activity.
To rule out the possibility that a contaminating acetyltransferase accounts for the observed acetyltransferase activity in our highly purified recombinant tau proteins, we performed an in-gel assay previously used to assess histone acetyltransferase activity8,9. A gel containing two different concentrations of the active tau-K18 fragment, 0N4R- or 2N4R-tau proteins or inactive tau-K18(−) was incubated with radiolabeled [3H]acetyl-CoA and followed by autoradiography (Fig. 1e). In-gel tau acetyltransferase activity comigrated prominently with tau-K18 and to a lesser extent with both full-length tau proteins, whereas tau-K18(−) showed minimal detectable acetyltransferase activity with this assay (Fig. 1e). Therefore, the in-gel assay supports our interpretation that tau autoacetyltransferase activity is mediated by the microtubule-binding repeat regions.
To evaluate whether brain-derived tau can undergo de novo autoacetylation, we boiled extracts from cortical brain homogenates of wild-type and tau-knockout mice, which inactivated any potentially contaminating acetyltransferases and simultaneously enriched for heat-stable tau proteins. Wild-type or tau-knockout brain extracts were incubated with CoA or acetyl-CoA, and tau acetylation was evaluated by immunoblotting using acetylation-specific antibodies (Fig. 2a). Notably, incubation of wild-type extracts with acetyl-CoA but not CoA led to a pronounced accumulation of acetylated mouse tau, as detected with anti–Ac-K280, and a modest increase in total acetylated proteins (Fig. 2a). As expected, acetyl-CoA–dependent tau acetylation was not detected in tau-knockout brain extracts, although a modest increase in total acetylated proteins was also observed (Fig. 2a). Finally, we evaluated soluble tau proteins derived from frontal-cortex homogenates of control and Alzheimer's disease–affected brain, which were similarly capable of de novo autoacetylation, as detected by anti–Ac-K280, without detectable disease-specific differences (Fig. 2b).
Figure 2.

Mouse and human tau proteins possess acetyltransferase activity. (a) Immunoblot analysis of heat-stable brain extracts from wild-type (WT) and tau-knockout (KO) mice incubated with CoA or acetyl-CoA, using antibodies to acetyl-lysine, Ac-K280, total tau (T49) and GAPDH. Unboiled brain extracts and GAPDH immunoblotting were used to normalize protein loading (bottom). (b) Immunoblot analysis of highsalt–extracted tau proteins from control and Alzheimer's disease–affected brain, similar to a.
Tau acetyltransferase activity is mediated by cysteines
We next searched for sequence similarities between tau and known acetyltransferases. Several variants of the well-characterized motif A were identified within the repeat region (GXG motifs), which could participate in direct acetyl-CoA binding10–12. Additionally, we identified two regions in the second and third repeats with high sequence homology (∼73%) to the MYST-family acetyltransferases ESA1 and Tip60 (refs. 13,14; boxed regions in Fig. 3a). The minimal MYST region shown represents part of a larger conserved core acetyltransferase domain, which led us to speculate that a similar region in tau could facilitate autoacetylation. To investigate this possibility, we mapped tau acetyltransferase activity, using a cell-based in vitro acetylation assay in which HEK293 cells were transfected with a series of tau expression plasmids lacking individual repeats (ΔR1−R4) or lacking both repeats 2 and 3 in combination (ΔR2+3) (Fig. 3b). Overexpressed tau proteins were immunoprecipitated from cell lysates and incubated with [14C]acetyl-CoA. Deletion of R2 or R3 but not R1 or R4 led to a modest reduction in tau acetyltransferase activity (Fig. 3b). Consistent with the presence of two catalytic regions, deletion of both R2 and R3 in combination completely abrogated tau acetyltransferase activity (Fig. 3b), thereby mapping tau enzymatic activity to R2 and R3, which contain sequence homology to acetyl-CoA–binding regions of MYST acetyltransferases.
Figure 3.

Cysteine residues are required for tau-mediated acetyltransferase activity. (a) Amino acid sequence alignment of a minimal region of tau R2 and R3 with MYST-family acetyltransferases ESA1 and Tip60. Red, identical or conserved amino acid residues; blue triangle, putative tau catalytic cysteine residues; blue circles, disease-associated tau mutations (L315R, K317M and S320F). (b) Cell-based acetyltransferase assays using [14C]-labeled acetyl-CoA. Acetylated tau proteins are shown visualized by Coomassie staining (top) and autoradiography (bottom). Deletions of individual repeat regions correspond to the following amino acid residues in full-length 2N4R tau: ΔR1, 244–274; ΔR2, 275–305; ΔR3, 306–336; ΔR4, 337–372; ΔR2+3, 275336. IP, immunoprecipitation. Asterisk marks a nonspecific band that coelutes with immunoprecipitated tau proteins. (c) Wild-type 2N4R-tau, single C291A or C322A mutants, the double C291A C322A (2CA) mutant and the indicated pathogenic mutations, evaluated in acetyltransferase assays as in b. Asterisk marks a nonspecific band as in b. (d) Cysteine-mutant proteins in full-length 2N4R-tau, tau-K18 or tau-K19, analyzed in acetyltransferase activity assays. (e) Acetylation immunoblotting of wild-type and cysteine-mutant tau-K18 and tau-K19 fragments, using antibodies to Ac-K280, Ac-K369, acetyl-lysine and total tau (K9JA). (f) Acetyltransferase assays of wild-type and cysteine mutants, performed in the absence (lanes 1–4) or presence (lanes 5–8) of active recombinant CBP. (g) Immunoblot analysis using antibodies to Ac-K280 (ac-tau) and total tau (K9JA). Samples are lysates from QBI-293 cells transfected with tau-K18 containing a P301L mutation (K18-PL) or a comparable mutant lacking Cys291 and Cys322 (K18-PL-2CA) followed by treatment with 3MA or sodium arsenite. Quantification of acetylated tau-K18 is shown as a ratio of acetylated tau (ac-tau) to total tau (t-tau).
Previous structural and functional studies demonstrated that MYST-family acetyltransferases can use an active site acetylcysteine intermediate to facilitate acetyl-group transfer to terminal lysines14–16. Sequence analysis within R2 and R3 revealed an R2 cysteine (Cys291) and a comparable R3 cysteine (Cys322) that are notably absent in R1 and R4 (Fig. 3a, blue triangle), which suggests that tau cysteines may represent catalytic residues that mediate acetyltransferase activity. To evaluate this possibility, we mutated Cys291 and Cys322 to alanines individually or in combination and analyzed tau proteins by immunoprecipitation followed by acetyltransferase assays. Individual cysteine mutants retained detectable tau enzymatic activity, but mutation of both cysteine residues abolished autoacetylation activity (Fig. 3c). Analysis of several pathogenic FTDP-17 tau mutations within this region demonstrated that the S320F mutation, within proximity to Cys322, produced a modest reduction in autoacetylation activity (Fig. 3c). A panel of full-length and repeat-containing tau proteins lacking cysteines (2N4R-tau-2CA, tau-K18-2CA or tau-K19-1CA) showed abrogated acetyltransferase activity by both autoradiography (Fig. 3d) and immunoblotting using acetylated tau-specific antibodies (Fig. 3e). Although cysteine-deficient tau had impaired autoactivity, the tau-K18-2CA mutant was readily acetylated by active recombinant CBP (Fig. 3f), which suggests that direct lysine acetylation mediated by other acetyltransferases is not generally impaired in the absence of tau cysteines.
To evaluate tau acetyltransferase activity in cells, we developed a cell-based model of tau acetylation in the absence of ectopically expressed CBP. Given that Lys280-acetylated tau is only detectable under pathological conditions in the brain5,6 we devised a genetic and pharmacological approach to increase the cellular pool of acetylated tau. Cells expressing a tau-K18 fragment containing the pathogenic P301L mutation (K18-PL) were treated with the autophagy inhibitor 3-methyladenine (3MA), which impairs K18-PL degradation17, as well as with the oxidative stressor sodium arsenite, previously shown to increase tau phosphorylation18. In combination, 3MA-arsenite treatment resulted in prominent pathological tau hallmarks, including the accumulation of acetylated and biochemically insoluble K18-PL protein (Fig. 3g). Notably, the increased K18-PL acetylation and shift in solubility was dramatically abrogated by an enzyme-deficient K18-PL lacking Cys291 and Cys322 (Fig. 3g). Thus, tau cysteines are required for autoacetylation and insoluble tau accumulation in a cell-based model of tau pathogenesis. Supporting a minimal role for CBP in K18-PL acetylation, pharmacological inhibition of endogenous CBP with the compound C646 had no effect on K18-PL acetylation levels (Supplementary Fig. 1d).
We next used a pharmacological approach to confirm the importance of Cys291 and Cys322 in mediating tau acetyltransferase activity. Pretreatment of 2N4R-tau and tau-K18 with either N-ethylmaleimide (NEM) or iodoacetamide, compounds that bind free cysteine thiol groups, resulted in the abrogation of 2N4R-tau or tau-K18 autoacetylation activity (Fig. 4a). Because cysteine-containing R2 and R3 bind tubulin, we next asked whether the presence of tubulin affected tau enzymatic activity. Indeed, increasing concentrations of either monomeric tubulin or preformed microtubules markedly reduced tau acetyltransferase activity (Fig. 4b,c), which suggests that only free unbound tau proteins, but not those associated with tubulin, are capable of autoacetylation. These data suggest that tau acetyltransferase activity occurs under conditions that favor reduced tau binding to tubulin, which exposes free cysteine residues for catalysis.
Figure 4.
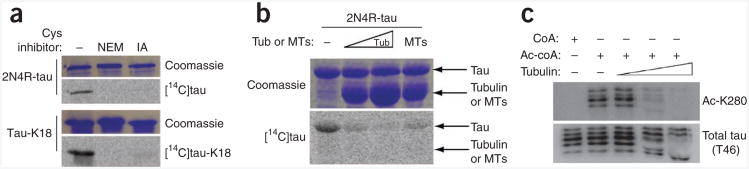
Cysteine blockers inhibit tau-mediated acetyltransferase activity. (a) Tau proteins preincubated with NEM or iodoacetamide (IA), showing minimal acetyltransferase activity. (b) 2N4R-tau preincubated with either increasing concentrations of tubulin (Tub) or preformed microtubules (MTs), showing inhibition of tau acetylation in acetyltransferase activity assays. (c) Acetyltransferase assays of bovine brain-derived microtubule-associated protein (MAP-rich) fractions containing all CNS tau isoforms in the presence of increased tubulin concentrations, followed by immunoblotting with antibodies to Ac-K280 or total tau (T46).
Kinetic analysis of tau acetyltransferase activity
To quantify tau acetyltransferase activity, we performed filter-binding assays using recombinant wild-type tau-K18 or cysteine mutants (tau-K18-2CA and tau-K18-2CS) containing either alanine or serine substitutions. (Fig. 5a shows tau-K18 sequence.) A saturation plot of reaction velocity against acetyl-CoA concentration revealed that tau-K18 acetyltransferase activity followed classic Michaelis-Menten behavior, whereas 2CA- and 2CS-mutant activity was impaired, as expected (Fig. 5b). Although the calculated turnover rate for tau-K18 (Kcat = 0.005 min−1) is quite low in comparison to those of known nuclear acetyltransferases, it is comparable to that recently reported for cytoplasmic α-tubulin acetyltransferase (αTAT-1, Kcat = 0.037 min−1)19, which indicates distinct kinetic differences between nuclear and cytoplasmic acetyltransferases. (Supplementary Table 1 shows kinetic comparisons among nuclear and cytoplasmic acetyltransferases.) Consistent with acetyl-CoA binding to tau, inhibition of tau acetyltransferase activity was readily observed by cofactor competition assays, in which titration of increasing amounts of CoA or the structurally related acetyl-CoA analog acetonyl-CoA inhibited acetyl-CoA–dependent tau-K18 acetyltransferase activity (Fig. 5c), with half-maximum inhibitory concentration (IC50) values of 59 μM and 668 μM for CoA and acetonyl-CoA, respectively. As a negative control, an acetyl-CoA precursor, pantothenic acid, did not show significant inhibition of tau activity.
Figure 5.

Kinetic and functional analysis of tau acetyltransferase activity. (a) Schematic of the tau-K18 microtubule-binding region. K18 mutants containing sequential deletions are numbered 1–13 above the solid black lines. Green, catalytic cysteine residues; red, phosphorylated KXGS serines; magenta, autoacetylated tau lysine-280 (K280). (b) Tau-K18 or cysteine-deficient 2CA and 2CS mutants, analyzed by filter-binding acetyltransferase assays in the presence of increasing concentrations of [3H]-labeled acetyl-CoA. Saturation-plot analysis, yielding Kcat and Km values for tau-K18 acetyltransferase activity is shown. (c) Cofactor inhibition analysis performed with increasing concentrations of CoA, acetonyl-CoA or pantothenic acid. Percentage remaining tau acetyltransferase activity and IC50 values for CoA and acetonyl-CoA-mediated inhibition are shown. (d) Filter-binding acetyltransferase assays of wild-type tau-K18 (black), individual S262E (light gray) or S356E (dark gray) phosphomimic mutants or the double S262E S356E mutant (2SE, white). The velocities at 800 μM acetyl-CoA were normalized to wild type to determine fold induction of tau acetyltransferase activity. (e) Immunoblot analysis of soluble and insoluble lysates, using antibodies to Ac-K280 (ac-tau) and total tau (K9JA). Samples are lysates from QBI-293 cells transfected with K18-P301L (K18-PL), the phosphomimic mutant (K18-PL-2SE) or a control K18-PL containing serine-to-alanine substitutions (K18-PL-2SA), treated with 3MA or sodium arsenite where indicated. Throughout figure, error bars are s.e.m. from n = 3 independent biological replicates. *P < 0.0125; ***P < 0.0002 by unpaired t test.
In addition to catalytic cysteines, we dissected the tau domains and regulatory elements that are critical for substrate binding or catalysis. We generated a panel of tau-K18 mutant proteins containing sequential 10–amino acid deletions and performed kinetic analysis to determine Km values relative to wild-type tau-K18 (Table 1). Although many of the tau-deletion proteins analyzed retained enzymatic function comparable to that of wild type (Km = 147 μM), mutants 3 and 10, located within repeats 1 and 4, respectively, had Km values elevated by ∼20- to 40-fold (Table 1, rows 3 and 10), which indicates that distinct structural regions spanning tau-K18 are probably required for substrate binding or catalysis. Given that pathological tau aggregates in diseased brains are both hyperphosphorylated and acetylated5,6, we also determined whether tau phosphorylation regulates tau acetyltransferase activity. Tau is phosphorylated on several KXGS motifs within the repeats, most notably at Ser262 and Ser356 comprised by the 12E8 epitope20, thus providing regulatory cross-talk that could activate tau acetyltransferase activity. To examine this possibility in vitro, we generated phosphomimic tau-K18 proteins containing single S262E or S356E mutations or a double S262E S356E mutation (2SE) and performed acetyltransferase assays. Notably, we observed increased acetyltransferase activity with all phosphomimic proteins, including the 2SE mutant that showed an approximately threefold activation (Fig. 5d and Supplementary Fig. 2). These results were confirmed in a cell-based model, in which expression of a phosphomimic K18-PL mutant (K18-PL-2SE), but not a control mutant containing serine-to-alanine substitutions (K18-PL-2SA), resulted in elevated acetylated tau in response to either 3MA alone or 3MA and arsenite in combination (Fig. 5e). Thus, pseudophosphorylation within the microtubule-binding domain is sufficient to increase tau-K18 acetyltransferase activity.
Table 1. Kinetic analysis of tau-K18 proteins containing sequential deletions.
| Km (μM) | |
|---|---|
| 1 | 332 |
| 2 | 370 |
| 3 | 2,758 |
| 4 | 1,215 |
| 5 | ND |
| 6 | 1,150 |
| 7 | 716 |
| 8 | 141 |
| 9 | 319 |
| 10 | 5,404 |
| 11 | 421 |
| 12 | 250 |
| 13 | 273 |
| WT | 147 |
WT, wild type; ND, not determined.
Mechanism of tau-mediated acetyltransferase activity
To examine putative tau substrates, we next determined whether tau directly acetylated microtubules, as described for the tubulin acetyltransferase αTAT-1 (refs. 19,21). However, in a PTK2 cell-culture model, tau was unable to mediate microtubule acetylation, in contrast with α-TAT-1 (Supplementary Fig. 3). Additionally, tau was unable to restore microtubule acetylation in HeLa cells lacking αTAT-1 (data not shown), which further supports a minimal role, if any, for direct tau-mediated acetylation of microtubules. We therefore focused on tau itself as a major autoacetylated substrate and determined whether tau autoacetylation occurred by an intra- or intermolecular reaction mechanism. To distinguish between these possibilities, we performed filter-binding assays to determine the initial rate of tau acetylation as a function of tau concentration. A linear correlation of the reaction rate (μM s−1) versus tau concentration (μM) was observed at all time points examined (Fig. 6a and Supplementary Fig. 4), which supports a predominantly first-order intramolecular reaction mechanism. However, these results did not exclude potential intermolecular interactions as a contributing factor to tau autoacetylation. Therefore, we used an enzyme-substrate acetylation assay in which increasing concentrations of enzymatically active tau-K18 or the enzyme-deficient tau-K18-2CA mutant were incubated with fulllength 2N4R-tau substrate (Fig. 6b). Autoacetylation of 2N4R-tau (relative molecular mass ∼65,000 (Mr 65K)) was not detected with anti–acetyl-lysine and anti–Ac-K280 antibodies at the low substrate concentrations used (Fig. 6c). However, increasing amounts of tau-K18 (Mr ∼15K) increased the acetylation of 2N4R-tau and 2N4R-tau-2CA substrates, an effect that was partially impaired in the presence of the enzyme-deficient tau-K18-2CA (Fig. 6c). Taken together, these results are consistent with tau autoacetylation occurring preferentially in cis (intramolecularly) but also partly by a trans mechanism (intermolecularly).
Figure 6.

Tau autoacetylation occurs by intra- and intermolecular mechanisms. (a) Plot of initial rate of autoacetylation versus tau-K18 concentration, for a 30-min reaction. Error bars, s.e.m. from n = 3 independent experiments. (Additional data are in Supplementary Fig. 4.) (b) Schematic of tau-K18-mediated acetylation of full-length 2N4R-tau, showing impaired acetyltransferase activity of the K18-2CA mutant toward full-length (FL) tau substrates. (c) Immunoblot analysis of 2N4R-tau and 2N4R-tau-2CA incubated with increasing concentrations of tau-K18 or tau-K18-2CA. Intermolecular tau acetylation products for full-length 2N4R-tau and tau-K18, analyzed with the indicated antibodies to tau, are shown. (d) Double-labeling immunofluorescence using antibodies to Ac-K280 or total mouse tau (mTau), assessing the ability of exogenously added 3R-tau to acetylate endogenous 4R-tau present in mature mouse neurons. Transacetylation immunofluorescence was performed by incubating purified tau-K19 (top and middle) or tau-K19-C322A mutant (bottom) with fixed and permeabilized hippocampal neurons. Immunofluorescence images are shown in low magnification (top and bottom; scale bars, 100 μm) and high magnification (middle; scale bar, 25 μm).
To evaluate whether tau autoacetylation can occur in neurons, we developed an in-trans immunofluorescence assay using cultured primary hippocampal neurons. Because mouse tau is not normally acetylated on Lys280 in primary neurons, we asked whether addition of purified tau-K19 or enzyme-deficient tau-K19-C322A proteins to fixed and permeabilized neurons would be sufficient to promote endogenous mouse tau acetylation. Immunofluorescence analysis using anti–Ac-K280, which detects acetylated mouse tau but not the exogenously added 3R-tau enzyme lacking residue Lys280, showed that active tau-K19 but not the enzyme-deficient tau-K19-C322A mutant markedly increased Ac-K280 immunoreactivity that colocalized with endogenous mouse tau (Fig. 6d). A similar in-trans immunofluorescence analysis using transfected HEK293 cells further demonstrated that exogenously added 3R-tau was sufficient to acetylate ectopically expressed 4R-tau present in fixed and permeabilized cells (Supplementary Fig. 5). Thus, an active tau acetyltransferase is capable of specifically acetylating cellular tau substrates in trans, which further supports tau autoacetylation activity.
Discussion
Here we have identified tau as a bona fide acetyltransferase with sequence and functional similarities to members of the MYST family of lysine acetyltransferases. Using biochemical methods, cell-based assays and kinetic analysis, we provide evidence that tau uses essential cysteine residues to catalyze tau autoacetylation by both intra- and intermolecular acetylation mechanisms. What might be the physiological role for tau acetyltransferase activity? Given that tau is predominantly cytoplasmic, we determined whether tau regulates tubulin acetylation and thereby modulates microtubule dynamics in ways similar to those recently reported for αTAT-1 (refs. 19,21). However, several experiments in vitro and in cell-based assays (Supplementary Fig. 3 and data not shown) precluded a dominant role for tau as a tubulin acetyltransferase, although we cannot currently exclude the possibility that particular cell types or additional cellular factors are required for direct tau-mediated acetylation of microtubules. Additionally, it remains plausible that tau acetylates unknown cytoplasmic or microtubule-associated substrates, the scope of which is slowly emerging with the development of new proteomic approaches to identify and characterize globally acetylated substrates22.
We propose that tau itself represents a major target of its own acetyltransferase activity as part of an autoinhibitory signaling mechanism to prevent tau-microtubule interactions. Supporting this possibility, acetylated tau showed reduced ability to bind and stabilize microtubules in vitro and in cell-based models5. We have been unable to detect acetylated tau on Lys280 under normal physiological conditions in the brain or in primary cultured neurons5,6, which suggests that the majority of tau is normally deacetylated at this residue and bound to microtubules. In fact, it is estimated that tau proteins are ∼99% bound to microtubules in mature neurons23, and therefore physiological binding to microtubules would inhibit tau acetyltransferase activity, probably owing to blocked catalytic cysteine residues that actively engage microtubules. Tau binding to the cytoskeleton may therefore serve as an off switch to inhibit tau acetyltransferase activity, which is supported by impaired tau autoacetylation in the presence of tubulin or microtubules (Fig. 4). In contrast, accumulation of acetylated tau aggregates in 4R and 3R-4R tauopathies is readily observed under distinctly pathological conditions in which tau is detached from microtubules and aggregated to form mature tau lesions5–7. We hypothesize that microtubule detachment and activation of tau autoacetyltransferase activity could represent a pathological event, in which continual self-acetylation gradually shifts the tau-microtubule binding equilibrium toward cytosolic tau accumulation, providing an increased pool of aggregation-prone tau species. Indeed, tau acetylation has the dual effect of both inhibiting tau-microtubule interactions and facilitating the formation of β-structure within the repeat regions5. The presence of free catalytic cysteine residues as well as a readily available pool of acetyl-CoA would trigger autoacetylation on lysines within the repeat regions, thereby promoting tau aggregation and NFT formation. Consistent with this model, the levels of acetyl-CoA are reported to be elevated in Alzheimer's disease brain24, which would support increased tau acetyltransferase activity during disease pathogenesis.
Our results indicate that regulatory cross-talk exists between phosphorylation and acetylation in regulating tau activity, a phenomenon that has been similarly demonstrated with several other acetyltransferases including Tip60 and ATF-2 (refs. 25,26). Tau phosphorylation within the microtubule-binding repeats, particularly at Ser262 comprised by the 12E8 epitope, has a pronounced inhibitory effect on the affinity of tau for microtubules27,28. Therefore, we asked whether phosphorylation could enhance tau autoacetylation activity. Indeed, in in vitro or cell-based approaches, individual phosphomimic mutants (S262E or S2356E) or, more prominently, the double mutant (S262E S356E) was sufficient to enhance tau acetyltransferase activity in a cysteine-dependent manner (Fig. 5). These findings suggest that tau phosphorylation within the repeats initiates a conformational change that enhances tau acetyltransferase activity, potentially by increased cofactor binding and catalysis. Supporting this possibility, a phosphorylation-induced conformational change was previously demonstrated by NMR spectroscopy, in which pseudophosphorylation on KXGS motifs within the repeats was capable of structurally modifying R1 and R2, resulting in conformational alterations that reduced tau binding to microtubules29. Given the uncertain role for phosphorylation itself in driving tau pathogenesis30,31, future biochemical studies could shed light on whether phosphorylation-induced autoacetylation of tau represents a new pathogenic mechanism that occurs during the onset or progression of tauopathies. Overall, the identification of tau acetyltransferase activity provides a new framework for understanding tau pathogenesis, which could provide therapeutic avenues to target tau enzymatic activity in Alzheimer's disease and related tauopathies.
Methods
Methods and any associated references are available in the online version of the paper.
Online Methods
Recombinant tau in vitro methods
Acetyltransferase assays using purified tau proteins were performed as previously described5 and subjected to autoradiography or immunoblotting with the following anti-tau antibodies: anti-tau polyclonal (Dako, A0024, 1:10,000), anti-acetyl lysine (Cell Signaling, 9441, 1:1,000), anti-tau monoclonal (T46, 1:1,000), anti-mouse tau (T49, 1:1,000), anti-acetylated tau (Ac-K280, 1:1,000) or anti-acetylated tau (Ac-K369, 1:1,000). For tau acetyltransferase-inhibition studies, tau proteins were preincubated with 0.5 mM N-ethyl-maleimide (NEM), 4 mM iodoacetamide (IA) or increasing concentrations of either tubulin (5–10 μM) or preformed microtubules (5 μM). MS (nanoLC nanospray MS-MS) analysis was performed at the University of Pennsylvania proteomics core facility, which identified tau autoacetylated lysine residues (data not shown). In-gel acetyltransferase assays were performed by using a variation of the in-gel histone acetyltransferase protocol described previously8. Briefly, tau proteins (5–50 μg total protein) were electrophoresed on a 15% SDS gel followed by extensive washing in Buffer 1 (50 mM Tris, pH 8.0, 20% isopropanol, 0.1 mM EDTA, 1 mM DTT). Gels were equilibrated in Buffer 4 (50 mM Tris, pH 8.0, 10% glycerol, 0.1 mM EDTA, 1 mM DTT) followed by incubation for 2 h with Buffer 4 containing 5 μCi [3H]acetyl-CoA (1–10 Ci mmol−1, PerkinElmer). Gels were then washed in 5% trichloroacetic acid several times to reduce background, dried down and exposed to film for 2 weeks. For tau transacetylation western blotting, 2N4R-tau and 2N4R-tau-2CA mutant (0.3 μM) were incubated with increasing concentrations of enzymatically active tau-K18 or deficient tau-K18-2CA (2–7 μM) for subsequent analysis of intermolecular tau acetylation. Reaction products were readily distinguished between 2N4R-tau (Mr ∼65K) and tau-K18 (Mr ∼15K) by immunoblotting using antibodies to acetylated tau (Ac-K280) and total tau (E10). We note that total tau immunoreactivity when using polyclonal anti-tau E10 is reduced upon tau acetylation.
Cell- and tissue-based acetylation assays
Cell-based tau-T40 (2N4R) acetyltransferase assays were performed by immunoprecipitation of tau proteins (tau mAb T46+T14) bound to protein-A or protein-G beads followed by addition of [14C]acetyl-CoA for 2 h. Acetylated tau proteins bound to beads were analyzed by SDS-PAGE and Coomassie staining followed by phosphorimaging with Storm software. For acetyltransferase assays of brain lysates, boiled mouse lysates or salt-extracted human lysates were dialyzed into PBS and supplemented with Tris, pH 8.0, to a final concentration of 50 mM. CoA or acetyl-CoA was added to 1 mM and incubated for 2 h at 37 °C, followed by immunoblotting using the indicated anti-tau antibodies. Cell-based K18 acetylation was performed, using K18-P301L (K18-PL), mutants lacking cysteines (K18-PL-C291-322A) or mutants containing phosphomimic mutations (K18-PL-S262-356E). Transfected cells were treated overnight with 10 μM 3-methyladenine (3MA) and 50 μM sodium arsenite, where indicated, followed by sequential biochemical extraction of soluble (RIPA-solubilized) and insoluble (UREA-solubilized) fractions, as previously described32. Immunoblotting of cell lysates was performed, using the following primary antibodies to the following proteins in addition to those indicated above for recombinant methods: total tubulin (Sigma, DM1A, 1:2,000), acetylated tubulin (Sigma, T6793, 1:1,000), acetylated histone H3K18 (Active Motif, 39755, 1:1,000), acetylated histone H3K9 (Millipore, 07-352, 1:1,000), phospho-tau 12E8, 1:1,000), GAPDH (Advanced Immunochemical, 2-RGM2, 1:3,000) or M2-FLAG (Sigma, F1804, 1:2,000). In-trans immunofluorescence (trans-IF) was performed by using primary hippocampal neurons isolated from CD1 embryos (Charles River), following the NIH Guide for the Care and Use of Experimental Animals, and was approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Neurons were grown on cover slips for 21 d in culture, followed by fixation in 4% paraformaldehyde and permeabilization in 0.2% Triton for 15 min. Cover slips were incubated with 0.5 mg ml−1 tau-K19 or tau-K19-C322A mutant proteins in the presence of 1 mM CoA or acetyl-CoA for 2 h, followed by extensive washing in PBS and immunostaining analysis using antibodies to Ac-K280 and total mouse anti-tau (T49). A similar analysis was performed by incubating purified 0N3R-tau with 2N4R-tau–transfected QBI-293 cells (Supplementary Fig. 5).
Antibody generation
Polyclonal anti-acetyl tau Lys369 antibodies were generated similarly to those for Lys280 as previously described5, using the tau peptide C-GGNKKIE (United Peptide) containing acetylated K369 to immunize rabbits (Pocono Rabbit Farm and Laboratory). Double affinity purification was performed with native and acetylated peptides sequentially, using Sulfolink columns (Pierce Biotechnology). Site specificity of Ac-K369 was confirmed in vitro and in cells using tau proteins lacking residue Lys369.
Tau autoacetylation filter-binding assays
Kinetic analysis
Reactions were assembled in 50 μl of reaction buffer containing 50 mM Tris, pH 8.0, 100 μM EDTA and 10% glycerol. Unless specifically noted, reactions contained 25 μM of either WT tau-K18 or tau-K18-2CA mutant. The reactions were initiated by the addition of [3H]acetyl-CoA (2.35 Ci mmol−1, PerkinElmer) at concentrations ranging from 2.5 μM to 1 mM, incubated at 37 °C and allowed to proceed for 1 h (unless otherwise noted). Reactions were stopped by the addition of 10 μl of 3 mM CoA, and 25 μl of the reaction was then spotted to a P81 filter paper (Whatman), washed in 10 mM HEPES, pH 7.5, three times and dried with acetone. Incorporated [3H]acetyl-CoA was then measured by using the PerkinElmer Tri-Carb 2800TR Liquid Scintillation Analyzer, and the molar amount of acetyl groups incorporated into tau were calculated from a standard curve of the radiolabeled acetyl-CoA.
Inhibition analysis
Reactions were performed in the buffer described above and at 25 μM tau. Reactions included between 1 and 4,000 μM of the inhibitors CoA, acetonyl-CoA or pantothenic acid. All reactions were initiated by the addition of 450 μM [3H]acetyl-CoA (2.35 Ci mmol−1, PerkinElmer), incubated for 1 h at 37 °C and stopped by spotting 25 μl of the reaction to the P81 filter paper. Radiolabel incorporation was measured as stated above. All data were analyzed by using GraphPad Prism to determine kinetic parameters and IC50 values.
Supplementary Material
Acknowledgments
We would like to thank T.P. Yao, J.Q. Trojanowski and L.K. Kwong for critical reading of this manuscript. We thank T.P. Yao (Duke University, Durham, North Carolina, USA) for kindly providing plasmids expressing a panel of histone acetyltransferases. We thank P. Seubert (Elan Pharmaceuticals, San Francisco, California, USA) for kindly providing anti-tau 12e8 antibody. We thank K. Brunden, A. Crowe, C. Li and other members of the Center for Neurodegenerative Disease Research for their technical support, helpful comments and critical suggestions. This study was supported by the US National Institutes of Health grants AG17586 (V.M.Y.L.) and GM060293 (R.M.) and the Association for Frontotemporal Degeneration (T.J.C.).
Footnotes
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html
Note: Supplementary information is available in the online version of the paper.
Author Contributions: T.J.C. and A.W.H. performed in vitro and cell-based acetylation experiments, mouse and human biochemical procedures and MS analysis, and T.J.C. was involved in the design and writing of this study. D.F. performed kinetic analysis of tau acetyltransferase activity and was involved in the writing of this study. V.M.Y.L. and R.M. supervised and designed the experiments and were involved in the writing of this study.
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Andreadis A, Brown WM, Kosik KS. Structure and novel exons of the human tau gene. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 2.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 3.Lee VM. Regulation of tau phosphorylation in Alzheimer's disease. Ann NY Acad Sci. 1996;777:107–113. doi: 10.1111/j.1749-6632.1996.tb34408.x. [DOI] [PubMed] [Google Scholar]
- 4.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 5.Cohen TJ, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irwin DJ, et al. Acetylated tau, a novel pathological signature in Alzheimer's disease and other tauopathies. Brain. 2012;135:807–818. doi: 10.1093/brain/aws013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Min SW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brownell JE, Allis CD. An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc Natl Acad Sci USA. 1995;92:6364–6368. doi: 10.1073/pnas.92.14.6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brownell JE, Mizzen CA, Allis CD. An SDS-PAGE-based enzyme activity assay for the detection and identification of histone acetyltransferases. Methods Mol Biol. 1999;119:343–353. doi: 10.1385/1-59259-681-9:343. [DOI] [PubMed] [Google Scholar]
- 10.Kawahara T, et al. Two essential MYST-family proteins display distinct roles in histone H4K10 acetylation and telomeric silencing in trypanosomes. Mol Microbiol. 2008;69:1054–1068. doi: 10.1111/j.1365-2958.2008.06346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sternglanz R, Schindelin H. Structure and mechanism of action of the histone acetyltransferase Gcn5 and similarity to other N-acetyltransferases. Proc Natl Acad Sci USA. 1999;96:8807–8808. doi: 10.1073/pnas.96.16.8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan Y, Barlev NA, Haley RH, Berger SL, Marmorstein R. Crystal structure of yeast Esa1 suggests a unified mechanism for catalysis and substrate binding by histone acetyltransferases. Mol Cell. 2000;6:1195–1205. doi: 10.1016/s1097-2765(00)00116-7. [DOI] [PubMed] [Google Scholar]
- 14.Yan Y, Harper S, Speicher DW, Marmorstein R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat Struct Biol. 2002;9:862–869. doi: 10.1038/nsb849. [DOI] [PubMed] [Google Scholar]
- 15.Decker PV, Yu DY, Iizuka M, Qiu Q, Smith MM. Catalytic-site mutations in the MYST family histone acetyltransferase Esa1. Genetics. 2008;178:1209–1220. doi: 10.1534/genetics.107.080135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang C, Wu J, Zheng YG. Function of the active site lysine autoacetylation in tip60 catalysis. PLoS ONE. 2012;7:e32886. doi: 10.1371/journal.pone.0032886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giasson BI, et al. The environmental toxin arsenite induces tau hyperphosphorylation. Biochemistry. 2002;41:15376–15387. doi: 10.1021/bi026813c. [DOI] [PubMed] [Google Scholar]
- 19.Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV. The major α-tubulin K40 acetyltransferase αTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci USA. 2010;107:21517–21522. doi: 10.1073/pnas.1013728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 21.Akella JS, et al. MEC-17 is an α-tubulin acetyltransferase. Nature. 2010;467:218–222. doi: 10.1038/nature09324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 23.Congdon EE, et al. Nucleation-dependent tau filament formation: the importance of dimerization and an estimation of elementary rate constants. J Biol Chem. 2008;283:13806–13816. doi: 10.1074/jbc.M800247200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corrigan FM, Horrobin DF, Skinner ER, Besson JA, Cooper MB. Abnormal content of n-6 and n-3 long-chain unsaturated fatty acids in the phosphoglycerides and cholesterol esters of parahippocampal cortex from Alzheimer's disease patients and its relationship to acetyl CoA content. Int J Biochem Cell Biol. 1998;30:197–207. doi: 10.1016/s1357-2725(97)00125-8. [DOI] [PubMed] [Google Scholar]
- 25.Kawasaki H, et al. ATF-2 has intrinsic histone acetyltransferase activity which is modulated by phosphorylation. Nature. 2000;405:195–200. doi: 10.1038/35012097. [DOI] [PubMed] [Google Scholar]
- 26.Lemercier C, et al. Tip60 acetyltransferase activity is controlled by phosphorylation. J Biol Chem. 2003;278:4713–4718. doi: 10.1074/jbc.M211811200. [DOI] [PubMed] [Google Scholar]
- 27.Drewes G, et al. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem. 1995;270:7679–7688. doi: 10.1074/jbc.270.13.7679. [DOI] [PubMed] [Google Scholar]
- 28.Mandelkow EM et al. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging. 1995;16:355–362. 362–363. doi: 10.1016/0197-4580(95)00025-a. discussion. [DOI] [PubMed] [Google Scholar]
- 29.Fischer D, et al. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48:10047–10055. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- 30.Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- 32.Cohen TJ, Hwang AW, Unger T, Trojanowski JQ, Lee VM. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012;31:1241–1252. doi: 10.1038/emboj.2011.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.