

Summary
The heart is a highly oxidative organ in which cardiomyocyte turnover is virtually absent, making it particularly vulnerable to accumulation of lipid peroxidation products (LPPs) formed as a result of oxidative damage.
Reactive oxygen and nitrogen species are the most common electrophiles formed during lipid peroxidation and lead to the formation of both stable and unstable lipid peroxidation products (LPPs). Of the LPPs formed, highly reactive aldehydes are a well-recognized causative factor in aging and age-associated diseases including cardiovascular disease and diabetes.
Recent studies have identified that the mitochondria are both a primary source and target of LPPs, with specific emphasis on aldehydes in cardiomyocytes, and how these affect the electron transport system and Ca2+ balance.
A number of studies have found that there are functional consequences in the heart as a consequence of exposure to specific aldehydes (acrolein, trans-2-hexanal, 4-hydroxynonenal, and acetaldehyde). Since these LPPs are known to form in heart failure, cardiac ischemia/reperfusion injury, and diabetes, they may have an underappreciated role in the pathophysiology of these disease processes.
LPPs are involved in transcriptionally regulating endogenous anti-oxidant systems. Recent evidence has demonstrated that transient increases in LPPs might be beneficial in cardioprotection by contributing to mito-hormesis (i.e. this induction of anti-oxidant systems) in cardiomyocytes. Thus, exploitation of cardioprotective actions of LPPs may represent a novel therapeutic strategy for future treatment of heart disease.
Keywords: Mitochondria, lipid peroxidation products, aldehydes, heart failure, ischemia, cardiac hypertrophy, diabetes
Introduction
The heart is a post-mitotic, highly oxidative organ in which cell turnover rates are virtually absent, so products formed from oxidative damage of tissue and cellular material accumulate with time. The accumulation of these oxidative products is now a well-recognized causative factor in aging and age-related diseases such as diabetes and cardiovascular disease1. In fact, for all diseases where acute and chronic oxidative stress is either a causative factor or deleterious consequence, lipid peroxides are starting to take centre stage as the most potent, persistent and physiologically relevant agents of this stress2–4.
Oxidation of membrane phospholipids, and other polyunsaturated fatty acids (PUFAs) that are stored or otherwise located in cardiomyocytes, is one of the most prominent manifestations of oxidative stress in the heart. With their long, chain-like structure and large number of unsaturated carbon-carbon bonds, PUFAs are some of the most readily oxidized chemicals in nature, occurring through enzymatic or non-enzymatic pathways. Non-enzymatic peroxidation of PUFAs, particularly those contained within membrane phospholipids, is typically initiated by electrophilic attack on one of the methylene carbons contained within the fatty acid side-chains. Reactive oxygen (ROS) and nitrogen species (RNS) are the electrophiles most commonly involved in lipid peroxidation, and if allowed to proceed unchecked this reaction results in formation of both stable and unstable lipid peroxidation products (LPPs). Of all the LPPs formed in this manner, reactive aldehydes have been shown to have regulatory roles in physiological systems, and have been implicated to play important roles in cardiovascular disease5.
Because of its insatiable appetite for nutrients and oxygen required to meet its energetic demands, the heart contains a very high mitochondrial content relative to other organs. Mitochondria are the largest source of intracellular ROS in cardiomyocytes6, as superoxide (O2•−) is continuously formed at sites within the electron transport system during oxidative phosphorylation. Furthermore, mitochondria are double membrane-bound organelles that have an enormous amount of unsaturated phospholipids contained within their inner and outer membranes, notably cardiolipin, a phospholipid exclusive to mitochondria that is highly unsaturated and prone to peroxidation7, 8. Thus, due to these characteristics mitochondria are ostensibly the largest endogenous source of LPPs in the heart. To offset the formation of LPPs, mitochondria have a vast network of antioxidant and detoxification systems, ensuring that lipid peroxidation and levels of reactive aldehydes are kept at sub-toxic levels. Over time, however, and under various pathological states, these antioxidant and aldehyde detoxification systems become compromised and/or lost, and the subsequent accumulation of LPPs and aldehydes has profound consequences for the function of mitochondria, the cardiomyocyte and the heart. The focus of this review will be on the role of mitochondria as both a primary source, and also a target, of LPPs and reactive aldehydes, and on the current state of knowledge regarding the role of LPPs and aldehydes in causing mitochondrial and cardiac dysfunction in pathological conditions. A brief overview of the potential role of LPPs in cardioprotection will also be presented.
I. Mitochondria as a primary source of LPPs in heart
Formation of LPPs and aldehydes in mitochondria
It has been known for decades that mitochondria generate reactive oxygen species (ROS) as a by-product of oxidative phosphorylation9, 10. Mitochondrial super oxide (O2•−) is formed when electrons ‘leak’ from the electron transfer system in the mitochondria and are taken on by molecular oxygen11. The exact sites of O2•− formation within the electron transport system are controversial but there is broad consensus that redox reactions within Complex I and Complex III are both capable of producing O2•− (shown in detail in Figure 1A). However, it is not clear which of these complexes is the predominant site of superoxide formation in vivo12, 13. Most superoxide (O2•−) is immediately converted to hydrogen peroxide (H2O2) by superoxide dismutase enzymes in either the mitochondrial matrix (by manganese superoxide dismutase (MnSOD)) or in the cytosol (by copper/zinc superoxide dismutase (Cu/ZnSOD)). In addition to the electron transport system, mitochondria also generate H2O2 from monoamine oxidase (MAO) bound to the outer membrane11, 14. MAO is the enzyme responsible for metabolism of catecholamines, and has recently been shown to be a substantial source of H2O2 and oxidative stress in heart failure15.
Figure 1.
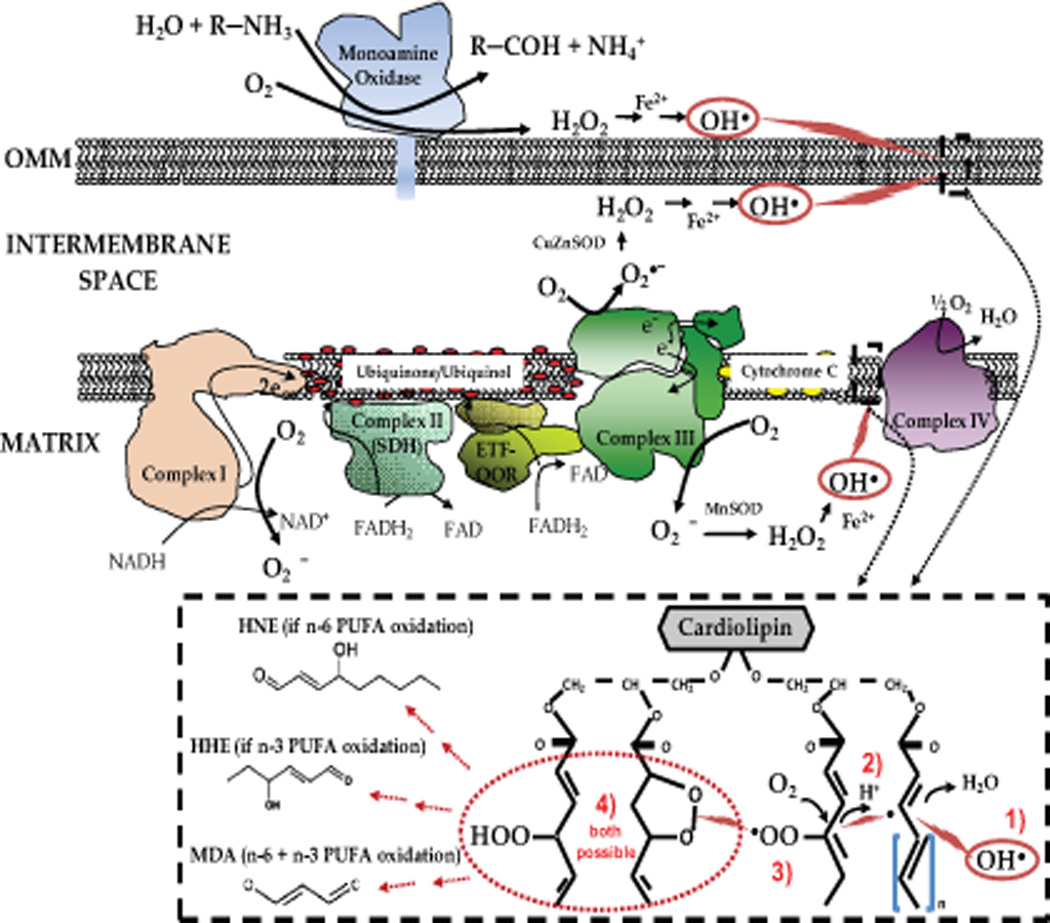
Top: Primary sites of ROS formation in mitochondria, and reaction scheme for peroxidation of cardiolipin in outer- and inner-mitochondrial membrane. Sites of ROS formation at Complex I and III are depicted, with the formation of hydroxyl radical (OH•−) circled in red. Electrophilic attack on cardiolipin from OH•− initiates lipid peroxidation by abstracting hydrogen from a methylene carbon in cardiolipin fatty acyl side-chain. Stepwise reaction scheme and products formed are outlined in dashed box. Dashed box: The general reaction scheme of non-enzymatic lipid peroxidation in mitochondria with initial step coming from OH• attack of unsaturated fatty acids contained within cardiolipin (step 1). Following initiation by OH•, an unstable lipid radical is formed, which can continue to abstract allylic hydrogens from nearby unsaturated fatty acids (step 2), or react with molecular O2 (step 3) to form a lipo-peroxyl radical that either continues on to react with another fatty acid forming a new radical, or reacts with itself to form a lipid peroxide (step 4).
Nitric oxide, as it exists with its unpaired electron (NO•), is also a free radical and can frequently react with superoxide (O2•−) to form peroxynitrite (ONOO•−). Peroxynitrite is considered to be the chief reactive nitrogen species formed in physiological systems16, being a highly reactive electrophile responsible for nitration and nitrosylation reactions with hydroxyl and thiol groups in proteins during periods of oxidative stress. Though still a matter of some debate, several studies have reported the presence of a nitric oxide synthase (NOS) isoform within mitochondria, distinct from other established isoforms of NOS such as eNOS, nNOS and iNOS17, 18. The presence of NOS within mitochondria, coupled to continuous O2•− formation by the electron transport system would suggest that tight regulation of ONOO•− formation and the presence of ONOO•− scavenging systems are critical to maintaining homeostasis.
Ultimately it is peroxynitrite (ONOO•−), in addition to hydroxyl radical OH•, formed via Fenton reaction of Fe2+ or Cu2+ with H2O2, that initiates lipid peroxidation by electrophilic attack on mitochondrial phospholipids, particularly the high unsaturated cardiolipin with is prone to peroxidation (see Figure 1 for details). Formation of lipid peroxides from cardiolipin through these reactions has been suggested to be partly responsible for the altered cardiac function seen in the aged heart19. These changes occur through disruption in the inner membrane of the mitochondria, where it constitutes about 20% of the total lipid composition, and by altering mitochondrial fission and fusion. This leads to destabilization of cytochrome c and the complexes within the electron transport system, all of which have profound effects on cell energetics and vitality20–22.
If they are not neutralized by endogenous antioxidants, lipid peroxides will fragment and decompose to form reactive aldehydes such as di-aldehydes (malondialdehyde, MDA) and α,β-unsaturated aldehydes (acrolein; 4-hydroxynonenal (HNE); and 4-hydroxyhexenal (HHE))23. The 4-hydroxyalkenals formed from PUFA oxidation (HNE from n-6 PUFAs, HHE from n-3 PUFAs) are highly reactive electrophiles capable of covalently modifying proteins, DNA and other macromolecules, similar to the ROS/RNS that spawned them, though they have unique properties endowing them with distinct roles in biological systems. These 4-hydroxyalkenals are uncharged, lipophilic and chemically stable molecules capable of readily diffusing through membranes. In addition, some of the less hydrophobic aldehydes such as MDA, acrolein and 4-hydroxy-hexanals (HHE) are able to diffuse fairly long distances from their sites of origin, enabling them to act as signalling mediators within cells and tissues under various physiological and pathological contexts5.
Mechanisms for scavenging LPPs and aldehydes
Mitochondria are endowed with such a highly concentrated and layered antioxidant network that they can be considered to not only be primary ‘sources’ of ROS/RNS within cells, but also primary ‘sinks’11, 24. This network includes the glutathione and thioredoxin systems, along with peroxidases, catalase, superoxide dismutase, glutaredoxin, sulfiredoxin, peroxiredoxin, and others (see recent comprehensive review24–26).
The major endogenous enzyme responsible for neutralizing lipid peroxides in the heart, as in other cell types, is glutathione peroxidase 4 (GPx4)27, 28. Glutathione peroxidase 4 resides in the cytosol, nucleus, and the inner membrane of mitochondria, where it utilizes glutathione (GSH) to reduce lipid peroxides to their corresponding alcohol. Glutathione not only provides the reducing power for a large number of redox enzymes capable of reducing reactive oxygen species29, but it is also capable of neutralizing electrophilic lipids, such as HNE, after they are formed30. Figure 2 illustrates the major enzymatic and non-enzymatic endogenous systems present in mitochondria and the cytosol specific for neutralizing LPPs and aldehydes (i.e. scavenging LPPs and aldehydes once they have already been formed). In the heart, the conjugation of glutathione to HNE is catalysed by the enzyme glutathione-transferase, resulting in export of this conjugate from the cell and into the systemic circulation31. The aldehyde dehydrogenase (ALDH) family of enzymes is also capable of neutralizing LPPs. In particular, the cytosolic enzyme ALDH3a1, also called fatty aldehyde dehydrogenase (FALDH)32, and ALDH233, a mitochondrial matrix enzyme, are the 2 isoforms of ALDH that are most likely to be the enzymes responsible for detoxification of LPPs in vivo (circled in blue in Figure 2), though other isoforms may also play a role. Aldehyde dehydrogenase (ALDH) converts aldehydes to acetate, rendering them far less reactive and virtually benign.
Figure 2.
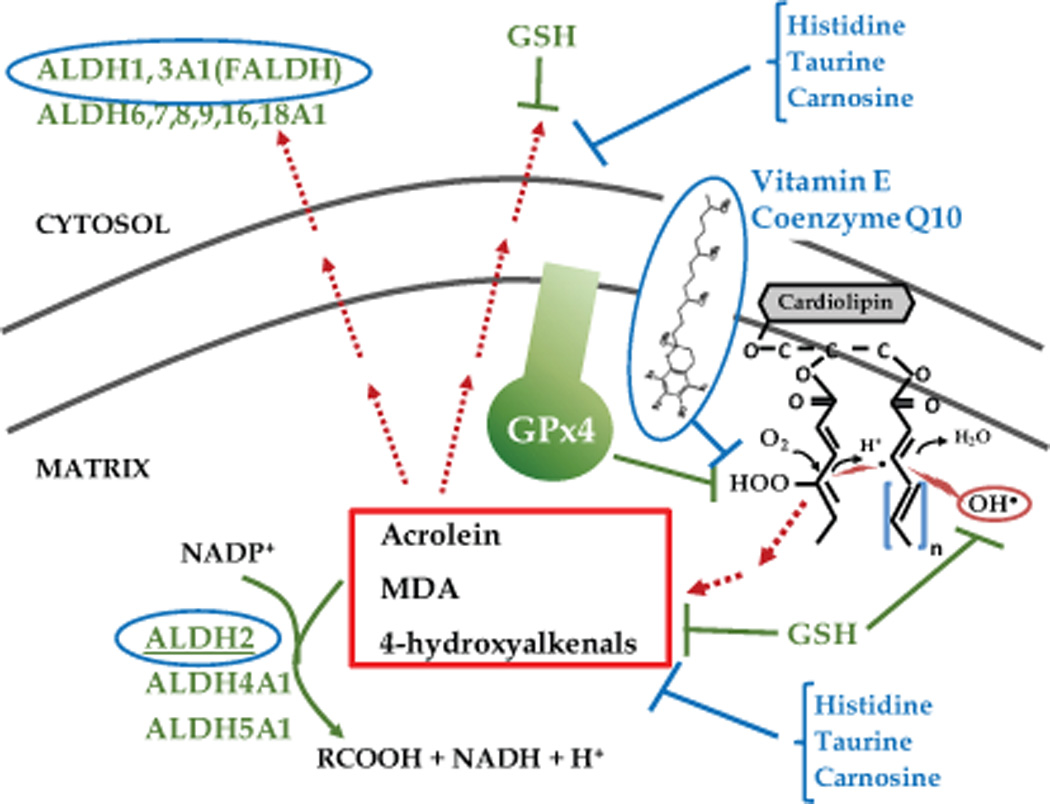
The major lipid peroxidation product detoxification systems present in the mitochondria and cytosol. Lipid peroxidation can be blunted directly by GSH removal of OH•− prior to imitation of lipid peroxidation. GSH is also used by the enzyme glutathione peroxidase 4 (GPx4), also known as phospholipid hydroperoxide glutathione peroxidase, to neutralize lipid peroxides as they are formed in mitochondrial membrane. Once aldehydes are formed (examples in red box), the most physiologically relevant enzyme in neutralizing these electrophiles is the aldehyde dehydrogenase (ALDH) family, with ALDH2 and ALDH3a1 as most likely candidate enzyme for removal of aldehydes in mitochondrial matrix and cytosol, respectively. Exogenous agents known to suppress lipid peroxidation and/or neutralize aldehydes are listed in blue text.
In addition to the endogenous systems in place to neutralize lipid peroxides, chemical agents have been identified that have the capacity to neutralize LPPs and aldehydes (see Figure 2). Lipophilic antioxidants such as vitamin E, α-lipoic acid, and co-enzyme Q10 have the capacity to inhibit lipid peroxidation and prevent adverse downstream effects34, 35. Agents that effectively neutralize and/or remove aldehydes in both experimental models of metabolic and cardiovascular diseases as well as clinical studies include taurine36, histidine analogs37, and carnosine38. These agents provide options for stand-alone or supplemental therapy in treating diseases where suppression or removal of LPPs is desired.
II. Mitochondria as a primary target of LPPs in heart
Studies of the effects of lipid peroxidation in cardiac tissue are sparse, even though this tissue may easily be considered to have the highest capacity for this process. Most research directed at identifying molecular targets of LPPs has been conducted in models of cancer, neuronal disease or in supra-physiological experimental conditions. This complicates extrapolation of these findings to other organs or to systems where lipid peroxidation may be a constitutive process that is fundamental to maintaining homeostasis, such as the heart39.
Cardiomyocytes possess the highest mitochondrial density of all tissues in the body, and therefore have a higher capacity for reactive oxygen species production. In addition to the high poly-unsaturated fatty acid (PUFA) content distributed across the mitochondrial membrane, there is also an omnipresent pool of free fatty acids, the preferred substrate of cardiac mitochondria. This fatty acid pool closely reflects a person’s dietary intake. According to some estimates the n-6:n-3 PUFA ratio approaches 10:1 in the archetypal Western diet40. Hence, HNE would be expected to constitute a much higher proportion of LPPs in individuals on such a diet. Approximately 2-8% of HNE produced is involved in largely irreversible cell conjugation reactions with both cellular and mitochondrial proteins41. The following sections discuss the resultant mitochondrial component damage and aberrations in homeostasis.
LPP effects on cellular membranes
The primary source of ROS in cardiomyocytes is the electron transport system located in the mitochondrial inner membrane42. Thus, the most immediate target of the 4-hydroxyalkenals are the lipids present in the phospholipid bilayer43, 44 and its associated proteins45. The reaction of 4-hydroxyalkenals with the mitochondrial membrane results in altered lipid-lipid and protein-lipid interactions. This occurs through a variety of mechanisms, including cross-linking of the lipid tails, which limits phospholipid mobility in the bilayer46. Of singular importance, the inner mitochondrial membrane mosaic contains cardiolipin, a highly unsaturated phospholipid specifically localized to this compartment, which is particularly prone to peroxidation. Its presence in the inner membrane, constituting ~20% of the lipids here, ensures the efficient function of a battery of mitochondrial components such as the electron transport chain complexes, adenine nucleotide transporter and the acylcarnitine carrier, among others47, 48. Cardiolipin levels diminish dramatically in robust lipid peroxidation conditions49, and diminished levels of cardiolipin have been observed in many pathological conditions including aging, Barth Syndrome, heart failure, ischemia/reperfusion injury, diabetes, and neurodegenerative diseases47. If 4-hydroxyalkenals migrate to the cell membrane, the high number of conjugated unsaturated carbon bonds in the cholesterol molecule make it an attractive target for attack50. Collectively, studies on the effects on lipid peroxidation on membrane physiology demonstrate a marked reduction in membrane fluidity46, 51, 52 and an increase in membrane permeability53, 54. Moreover, the production of HNE from lipid peroxidation gives rise to a chain of chemical reactions and products. The products formed from electrophilic attack by the 4-hydroxyalkenals degrade to generate more aldehydes, which serve to propagate the cycle55. Some of the more stable products include isoprostanes and neuroprostanes following cyclization of the fatty acid tails45.
LPP affects the Electron Transport Chain and disturbs intracellular Ca2+ in cardiomyocytes
4-hydroxyalkenals target enzymatic, structural, and signalling proteins present in the phospholipid bilayer and mitochondrial matrix. Like all electrophiles, 4-hydroxyalkenals have a characteristic affinity for sulfhydryl groups (e.g. cysteine, methionine), although lysine and histidine are also subject to covalent modification in their amide groups through Michael addition reactions23, 56–58. HNE augments the accumulation of damaged proteins by interfering with normal proteolysis pathways59,60. A recent proteomic study of the relative levels of adducted proteins in rat cardiac mitochondria treated with a spectrum of LPPs including 4-hydroxynonenal (HNE) and 4-hydroxyhexanal (HHE) revealed that proteins of the electron transport chain constituted the greatest percentage of altered proteins. Moreover, this group observed that cysteine constituted an overwhelmingly high percentage of total modified residues (85%), while histidine accounted for 12% and lysine less than 5%61. The concentration of 4-hydroxynonenal (HNE) and 4-hydroxyhexanal (HHE) adducts were relatively similar to one another. This study underscores the high propensity that LPPs exhibit towards attacking protein components involved in cellular respiration41, 62–64, expanding our current knowledge of the negative impact of the LPPS on mitochondrial homeostasis.
Prior to this point there have been multiple studies conducted to determine the effects of reactive aldehydes on mitochondrial components. Most of these studies to date, summarized in Table 1, are from experiments performed in a variety of oxidative tissues like brain, liver, kidney, as well as heart. Questions remain as to whether the observed effects of these LPPs seen in ‘other-than-cardiac’ tissues would also be observed in heart. In the heart, both 4-hydroxynonenal (HNE) and 4-hydroxyhexanal (HHE) have been shown to affect mitochondrial homeostasis by disturbing the balance of ions65, 66. 4-Hydroxynonenal attacks the sulfhydryl groups of the Na+/K+ ATPase67 resulting in diminished activity and elevated intracellular concentrations of free calcium. Both HNE and HHE have been implicated in inducing Ca2+ overload in cardiomyocytes68; activation of uncoupling proteins69 and premature opening of the mitochondrial permeability transition pore (MTP)70 thus increasing the risk of heart failure3, 71.
Table 1.
Summary of the of lipid peroxidation products on mitochondria.
| Mitochondrial Target |
LPP | Model | Effect |
|---|---|---|---|
| UCP | HNE | Heart-mouse | ↑ uncoupling of respiration at UCP1, UCP2 & UCP369, 151 |
| Kidney-mouse | |||
| Skeletal muscle-mouse | |||
| Complex I | MDA | Liver-rat | ↓ activity153 |
| HNE | Heart-rat | ↓ activity in model of diabetic cardiomyopathy154 | |
| Acrolein | Heart-rat | ↓ activity155 | |
| Brain-Guinea pig | ↓ activity156 | ||
| Brain & spinal cord-rat | ↓ activity157 | ||
| ↓ activity63 | |||
| Liver-rat | 80%↓ activity158 | ||
| Complex II | MDA | Liver | ↓ activity153 |
| HNE | Heart-rat | ↓ activity154 | |
| Acrolein | Heart-rat | ↓ activity155 | |
| Brain-Guinea pig | ↓ activity156 | ||
| Brain-rat | ↓ activity159 | ||
| Liver-rat | 80% ↓ activity158 | ||
| Complex III | HNE | Heart-Aging | protein modification, ↓activity especially in interfibrillar mitochondria160 |
| Complex IV | HNE | Heart-Aging | protein modification ↓ activity especially in interfibrillar mitochondria160 |
| MDA | Aging/Liver-rat | ↓ activity161 | |
| ATP synthase | MDA | Heart /Skeletal Muscle-Mouse | ↓ activity modification targeted at the F1 complex162 |
| Cytochrome c oxidase | HNE | Heart - rat | ~60%↓activity in rat reperfusion model163 |
| LPPs | Heart-rat | ↓activity in rat reperfusion model49 | |
| Adenine Nucleotide Translocase (ANT) | Acrolein | Brain-Guinea pig | ↓activity at concentrations higher than 5uM156 |
| HNE | Flight muscle/Aging-Housefly | ↓activity164 | |
| Heart/Kidney/Skeletal Muscle | ↑uncoupling of respiration at ANT69, 151 | ||
| Liver - rat | ↓ transportation capacity165 | ||
| α-Ketoglutarate dehydrogenase | Acrolein | Liver-rat | ↓activity158 |
| Enzyme assay | ↓activity166 | ||
| MDA | Liver-rat | ↓ activity153 | |
| Heart & Skeletal-Mouse | ↓ activity modification at E2 component of a-KD complex162 | ||
| HNE | Heart-rat | ↓activity, modifies E2 component of α-KD167 | |
| ↓ concentration, potent inhibitor of isolated subsarcolemmal mitochondria168 | |||
| ↓activity in reperfusion model169 | |||
| Succinate dehydrogenase | Acrolein | Liver-rat | ↓ activity170 |
| Brain-Guinea pig | ↓ activity156 | ||
| HNE | Heart-rat | 30% ↓oxygen consumption, modifies the FAD-containing subunit in diabetic cardiomyopathy154 | |
| Succinic semialdehyde dehydrogenase | HNE | Brain-rat | ↓activity at IC50 110uM171 |
| Acrolein | Brain-rat | ↓activity at 15uM respectively171 | |
| Pyruvate dehydrogenase complex | Acrolein | Liver-rat | ↓ activity158 |
| Brain-Guinea pig | ↓ activity166 | ||
| Brain & Spinal Cord-rat | ↓ activity157 | ||
| Enzyme assay | ↓ activity156 | ||
| MDA | Liver-rat | ↓ activity153 | |
| Isocitrate dehydrogenase complex | HNE | Heart-rat | 50 uM 85% ↓activity172 in spontaneously hypertensive rats |
| Alcohol dehydrogenase | Acrolein | Liver-rat | potent inhibitor of ADH at ↓ concentrations173 |
| Aconitase | Acrolein | Brain-Guinea pig | ↓activity156 |
| MDA | Heart& Skeletal Muscle-Mouse | ↓activity162 | |
| Heat shock protein 90 | HNE | Liver-rat | Inhibits refolding of HSP 90 by modification of cysteine174 |
| Mitochondrial permeability transition pore | Acrolein | Liver-rat | ↑MTP induction158 |
| HHE | Liver-rat | ↑MTP induction70 | |
| HNE | Liver-rat | ↑MTP induction70 | |
Abbreviations: ↓, decreased; ↑, increased; ADH; aldehyde dehydrogenase; HNE, 4-hydroxynonenal; HSP90, heat shock protein 90; LPP, lipid peroxidation products; MDA, malondialdehyde; MTP, mitochondrial transition pore; UCP1-3; uncoupling proteins 1–3
LPP effects on DNA and other cellular targets
The role of the 4-hydroxyalkenals in DNA adduct formation during mutagenesis is well documented with respect to HNE. HNE displays a differential affinity for the respective bases with the highest reactivity toward guanosine72, cytosine, adenine, and least toward thiamine73. Cellular machinery is capable of repairing damage to the DNA inflicted by LPPs by excising the damaged portion, although long term exposure to LPPs leads to an accumulation of damaged DNA. In addition to its well-documented reactivity with DNA, HNE is both directly and indirectly involved in the upregulation of several cell signalling pathways including protein kinase C, mitogen activated protein kinases, IκB Kinase Complex, tyrosine receptor kinases, serine/threonine protein kinases, and C-Jun H-Terminal kinases57, 71, 74–78.
III. The effects of clinically relevant LPPs on cardiac function
When LPPs are increased in the heart, their effect on cardiac function is uniformly detrimental. However, with respect to the effects of individual LPPs on cardiac function at the molecular level, either as protein adducts or free LPPs, very little is known. The LPPs acrolein, trans-2-hexanal, 4-hydroxynonenal, and acetaldehyde all affect cardiac function and cardiomyocyte viability, each contributing synergistically to adversely affect the heart during times of increased oxidative stress and lipid peroxidation. Since other aldehydes described in previous sections react similar to the LPPs described here, the following findings may act as general examples by which LPPs might affect cardiac function and viability.
Acrolein
Acrolein, the simplest unsaturated aldehyde, is produced widely in different tissues and is found ubiquitously in the environment as a pollutant. It has also recently been identified as a product of lipid peroxidation formed in response to increased oxidative stress. Acrolein binds to cysteine and lysine residues on proteins and has been suggested to be a mediator of oxidative damage in a variety of human diseases79–81. Despite the well-known reactivity of unsaturated aldehydes, their effects on cardiac function were only recently described. Mice challenged with intravenous acrolein (0.5 mg/kg) responded with a rapid and reversible left ventricular dilation and dysfunction82. In isolated cardiomyocytes, this same study identified that micromolar concentrations of acrolein acutely decreased contractile responses to Ca2+, but did not alter catecholamine sensitivity, paralleling what is found in the stunned myocardium82. Attenuation of acrolein’s effects on cardiac function and protein adduct formation could be achieved by pre-treatment with N-acetylcysteine, indicating that ROS mediates some of the effects of acrolein on the heart82. Mass spec analysis identified that acrolein formed protein adducts on both sarcomeric proteins (cardiac α-actin, desmin) and proteins involved in energy metabolism, including creatine kinase-2 and ATP synthase82. Experimental acrolein therefore induces cardiac dysfunction, selective myofilament impairment, and modifies proteins involved in energy metabolism, which possibly affects their function. Cardiac pathologies that produce acrolein in response to oxidative stress may therefore have some or all of these mechanisms at work to inhibit cardiac function.
Trans-2-hexanal
Like acrolein, trans-2-hexanal is a ubiquitous pollutant as well as an endogenous product of lipid peroxidation83, which similarly has potent effects on cardiac function. When 8 week old ICR mice were fed trans-2-hexanal for 4 weeks, significant impairment in cardiac function ensued84. Echocardiographic studies identified impaired contractile function within 4 weeks of trans-2-hexanal feeding and histological analysis of the heart identified myofibril disarray. Trans-2-hexanal feeding also resulted in increased cardiomyocyte apoptosis and aldehyde-protein adduct formation84. These findings demonstrate that trans-2-hexanal exposure can lead to cardiac dysfunction by its effects on sarcomere stability and apoptosis, resulting in significant functional deficits84. The specific mechanisms by which these effects are mediated, either through trans-2-hexanal adducts or through direct aldehyde effects on signalling, have not yet been determined.
4-hydroxynonenal
In cardiac ischemia reperfusion injury, the production of excess reactive oxygen species trigger increases in lipid peroxidation, resulting in the generation of α,β-unsaturated aldehydes, including 4-hydroxynonenal (HNE). Recent studies have started to delineate the role of HNE on cardiac function. While lower concentrations of HNE are not toxic to cultured cardiomyocytes, higher levels of HNE (≥ 20 µM) are85. Interestingly, priming cardiomyocytes with sub-lethal concentrations of HNE (5 µM) results in a “pre-conditioning” that protects cardiomyocytes from subsequent cytotoxic concentrations of HNE85. This is due, in part, to HNE-induced nuclear translocation of NF-E2 p45 related factor 2 (Nrf2), which increases intracellular glutathione (GSH) levels through its transcriptional upregulation of glutathione-S-transferase (GST)85. Nrf2 is a transcription factor which acts as a master regulator of the antioxidant response, transcriptionally inducing antioxidant systems such as glutathione S-transferase (GST)86. Therefore, these studies demonstrate that ≥ 20 µM of HNE is cardio-toxic, while levels around 5 µM of HNE induce endogenous anti-oxidant systems through Nrf2 activation. The mechanisms by which HNE induces nuclear translocation of Nrf2, and subsequent cardioprotection, have not yet been completely delineated. The implications of these findings in the context of ischemia-reperfusion injury are discussed in the next section.
Acetaldehyde
Acetaldehyde is formed in cardiac ischemia/reperfusion (I/R) injury and during the metabolism of ethanol in the heart. The direct effects of acute acetaldehyde on the heart after 5-10 minutes of exposure have been reported at least 8 times87–92. Acetaldehyde causes vasoconstriction and positive inotropic and chronotropic responses at concentrations ≤ 3 mM. Levels >3 mM induce cardiac dysfunction, vasodilation, and hypotension93, 94. The negative inotropy seen with these higher levels of acetaldehyde is associated with decreased release of SR Ca2+ 87, 89 and inhibition of voltage-dependent Ca2+ channels95. Acetaldehyde depresses myocardial contraction, cardiac myocyte shortening and intracellular Ca2+ transients by increasing β-adrenergic activity at low doses, and reducing Ca2+ entry and/or release at high doses90. The decrease in myocardial function by acute acetaldehyde exposure has been reported to be enhanced in the diabetic state91 and in the presence of nicotine96. Acetaldehyde may inhibit cardiac contractility by other mechanisms, including its effects on cytochrome P450 oxidase, xanthine oxidase, and lipid peroxidation97. The implications of these findings in the context of ischemia reperfusion injury and ethanol exposure is discussed in the next section.
IV. LPPs in Diseased Heart
Even though the functional consequences of LPPs in the heart have not been studied extensively, their presence in the heart has been widely reported. What has been most extensively studied is how anti-oxidants are cardioprotective in disease98–100, although the disappointing clinical utility of these therapies illustrates our lack of knowledge of the pathophysiology of oxidative stress, and the limited utility of studies using anti-oxidants101, 102. In this section, we focus on LPPs that have been identified in cardiac disease. LPPs have been reported in heart failure and myocardial infarction, in addition to the cardiomyopathies associated with diabetes and ethanol use. While these observations do not constitute direct evidence that LPPs are involved in the pathogenesis of these diseases, they do suggest clinical scenarios by which LPPs should be investigated for evidence that they directly mediate the pathophysiology of disease.
Clinical studies implicating LPPs in cardiovascular disease
Probably the best described role for LPPs in human heart disease is in the etiology of atherosclerosis, where LPPs initiate and sustain inflammatory reactions along the arterial wall103. Oxidized phospholipids have a high affinity for lipoproteins (oxLDL), and increased levels of these adducts are seen in serum of patients with cardiovascular disease104, as were antibodies to them105. The correlation between LPPs in serum and cardiovascular diseases is now so strong that they are frequently used as biomarkers of prognosis for patients with acute coronary syndrome106, or for atherosclerotic development107.
In addition to the vascular effects of LPPs, a number of studies have shown increased levels of MDA-adducts in myocardial samples of patients with heart failure108, 109. Heart failure is associated with numerous metabolic, functional, and structural defects. The role of anti-oxidant stress in heart failure is just beginning to be appreciated. When heart biopsies from dilated cardiomyopathy (DCM) patients were compared to donor controls, increased malondialdehyde (MDA) was found regionally in hearts of DCM patients110. MDA concentrations were generally higher in the DCM group compared to controls, but were decreased in the endocardium and aortic samples in the DCM group110. These studies indicate not only that in DCM-mediated heart failure ventricular MDA is increased, but also that this increase is not uniform throughout the heart110. In a model of chronic heart failure in hamsters (TO-2 strain), recent studies have identified HNE adducts both histologically and by ELISA, paralleling increases in other markers of oxidative stress, such as HSP27, HSP32, and manganese superoxide dismutase111. Persistent oxidative stress has been shown to be present in serum of patients with congestive heart failure as well112, indicating that a more ‘global’ cardiovascular oxidative stress is likely predominant in heart failure. The recent success of the non-selective β-blocker Carvedilol in demonstrating marked improvements in suppressing markers of oxidative stress by reducing the levels of HNE-adducts113 in hearts of treated patients, highlights the importance of this characteristic in the pathophysiology of heart failure. While the lipid peroxidation products MDA and HNE have both been reported elevated in models of heart failure, their role in the pathophysiology of heart failure has not been elucidated.
Cardiac ischemia reperfusion injury generates HNE, H2O2, and MDA
In addition to formation of HNE (described above), cardiac ischemia reperfusion injury also generates hydrogen peroxide (H2O2) and MDA. When isolated rat hearts are challenged with I/R injury, they generate elevated concentrations of hydrogen peroxide, MDA, and calcium114. The formation of hydrogen peroxide appears to lead to increased lipid peroxidation, evidenced by increased levels of MDA. Pre-treating hearts with scavenger oxyradicals (superoxide dismutase and catalase) or antioxidants attenuate I/R damage, also implicating ROS in I/R induced damage115. The specific mechanisms in cardiac I/R by which LPPs induces membrane defects to affect calcium overload and contractile dysfunction have yet to be elucidated, but may be related to ROS and LPP production.
Diabetes generates glycoaldehyde
While the cardiovascular complications of diabetes account for more than 80% of patient mortality, mechanisms by which glucose intermediates induce diabetic cardiomyopathy have not been fully elucidated. Recent studies suggest that the glycoaldehyde may mediate some of the cardiac defects. Hyperglycemia increases oxidative stress in the diabetic state, which leads to increases in advanced glycation endproducts (AGE) formed from glucose itself. Glucose itself is an aldehyde, which can react non-enzymatically with proteins (via lysine) to make AGE products (adducts) when glucose is elevated chronically. One of the early reactive intermediates formed during the formation of AGE products is glycoaldehyde (GA). Recent studies have identified that glycoaldehyde induces oxidative stress in the heart. When male Wistar rats are challenged with a single injection of GA, decreased catalase and glyoxalase I activities are identified, resulting in increased protein carbonylation and lipid peroxidation116. These studies suggest that the short chain glycoaldehyde (GA) produced during the formation of AGE products may play a significant role in diabetic cardiomyopathy. The direct effects of AGE products or the GA intermediate on the heart to mediate diabetic cardiomyopathy has yet to be determined. Hyperglycemia also induces the production of cardiac MDA in vivo. Using streptozotocin (STZ)-induced diabetic male Wistar rats, cardiac tissue was assayed after 4 weeks of hyperglycemia. Hyperglycemia in this model resulted in significant increases in cardiac MDA117. However, the role of MDA on cardiac function in this model remains to be determined.
Cardiac Acetaldehyde and Oxysterols are found with chronic ethanol feeding and in ischemia reperfusion injury
The relevance of acetaldehyde in cardiac disease comes from recent studies demonstrating that acetaldehyde is produced in states in which lipid peroxidation is increased, including I/R injury, similar to other reactive aldehydes such as HNE and MDA80, 118. Acetaldehyde is also formed in the heart as well as in the liver during ethanol exposure119. It is the direct effects of acetaldehyde on the heart, discussed in the previous section, which have been implicated in the pathogenesis of cardiac I/R injury33, 120–127 and alcoholic cardiomyopathy90, 91, 96, 97, 128–130.
Acetaldehyde formed during ethanol consumption may also mediate cardiac dysfunction due to its effects on mitochondrial damage and the regulation of the mitochondrial death pathway129, 130. This was determined in experiments using mice with cardiac-specific over-expression of alcohol dehydrogenase (ADH), which leads to an increase in the formation of acetaldehyde. When these ADH transgenic mice are challenged with ethanol (3/g/kg/d) for 3 days, myocardial mitochondrial damage and apoptosis occurred, leading to reduced contractility, and enlarged cardiomyocytes130. Additional studies using these ADH transgenic mice have also found dysregulation in ER stress and insulin sensitivity. In response to ethanol challenge, ADH transgenic mice develop cardiac hypertrophy and dilated cardiomyopathy, suggesting a role of acetaldehyde in this process131, 132. While acetaldehyde is significantly increased in this transgenic mouse model fed ethanol, its direct role in the development of cardiac hypertrophy and dilated cardiomyopathy has yet to be determined.
A role for acetaldehyde in ischemia reperfusion injury has been determined using genetic mouse models33, 120–127. Recent studies have found that I/R injury is exacerbated when aldehyde dehydrogenase 2 (ALDH2) is missing in ALDH2 −/− mice133. One of the primary functions of ALDH2 is to metabolize acetaldehyde (among other aldehydes). By decreasing toxic aldehydes, ALDH2 inhibits the production of free radicals, preventing mitochondrial dysfunction33, 121, 134. The ALDH2 −/− mice, therefore, have an exaggerated I/R injury due to increased aldehydes and ROS, both of which lead to impairment of the mitochondria. However, ALDH2 has additional beneficial mechanisms in the heart unrelated to its ability to clear aldehydes, including its role in regulating nitro-glycerine bioactivation, mTOR-STAT3-notch signalling, and autophagy33, 121, 134. So the cardioprotective effects of ALDH2 in I/R injury may be due to either direct clearance of acetaldehyde or through indirect regulation of other biologies. The exact mechanisms have not yet been delineated.
V. The transcriptional regulation of endogenous anti-oxidant systems by LPPs and their role in Hormesis and Cardioprotection
We previously introduced the endogenous antioxidant systems that are induced in response to LPPs. Here we introduce the underlying mechanisms by which LPPs induce antioxidant system by their induction of the Nrf2 transcription factor activity. One of the prominent sensors of oxidative stress involves the Keap1/Nrf2/ARE signalling network (see recent reviews for details135–139). Briefly, under normal conditions, the Nrf2 (NF-E2-related factors 2) transcription factor associates with the suppressor protein Keap1 (Kelchlike ECH-associated protein 1) in the cytosol (34). When Nrf2 is bound to Keap1, it is ubiquitinated by the Cullen3-Rbx1 ubiquitin ligase, which targets this complex for degradation by the 26S proteasome140. Keap1 has many cysteines, 27 in all, which can be modified in the presence of oxidative stress, making it an ideal redox sensor. In this way, free aldehydes and other electrophilic LPPs formed in oxidative conditions can attack and form adducts on these cysteines. Chemical modification of Keap1 with free aldehydes leads to its disassociation with Nrf2, allowing Nrf2 to move into the nucleus139. Cysteines in the 273, 288, and 151 positions of Keap1 have been reported as particularly sensitive to chemical modification and play a key role in its interaction with Nrf2141, 142. Upon entering the nucleus, Nrf2 binds to the antioxidant response element (ARE) found in the promoter regions of key antioxidant enzymes. These include glutathione S-transferase, UDP-glucuronosyl transferase (UGT), glutathione peroxidase (GPx), superoxide dismutase (SOD), and peroxiredoxin139. Additionally, Nrf2 activates genes involved in the cellular redox regulation including glutathione synthetase, thioredoxin, thioredoxin reductase, NAD(P)H: quinone oxidoreductase 1 (NQO1), among others139. Nrf2 is also regulated by phosphorylation by protein kinases involved in the stress response, such as ERK1/2, PKC, JNK, which enable its disassociation from its repressor Keap1137.
Understanding how exposure to LPPs initiate the antioxidant response via the Keap1/Nrf2/ARE signalling pathway allows a mechanistic discussion of how exposure to LPPs may initiate a series of protective actions in the heart. This concept has been previously described as Hormesis. Hormesis is the cellular response by a low-dosed sub-lethal stressor, which results in a cellular reaction that over-compensates, allowing the resistance of subsequent lethal doses of the stressor139, 143. There are many examples of this concept in biology, including exercise. The “exercise paradox”, which is the observation that exercise increases oxygen consumption and the generation of ROS, also leads to increases in cyto-protective elements, including glutathione as discussed in the previous paragraph144, 145. This finding seems to contradict the paradigm that oxidative stress is unconditionally detrimental in physiological systems, and responsible for age-related decline. Perhaps the words of the early Swiss intellectual Paracelsus provide a simple but insightful answer “Poison is in everything, and nothing is without poison. The dosage makes it either a poison or remedy”. This concept underscores a growing consensus that the relationship between oxidative stress and aging may not be as clear-cut and unidirectional after all, as highlighted in a recent review by Ristow and Schmeisser146. As our understanding of redox signalling in homeostasis grows, it is becoming increasing evident that the body needs small doses of ‘poison’ to mount and maintain adequate defences to blunt the effects of a full-on oxidative onslaught. This precarious balance between poison and remedy is a familiar feature of bi-phasic dose-response curves147 and is called hormesis. The direct relevance of inducing this pre-emptive cytoprotective response specifically in mitochondria has led to the term ‘mitohormesis’148–151.
The concept of mitohormesis causing cardioprotection via Nrf2 (e.g. through induction of anti-oxidant networks in the mitochondria) is new and uncharted ground, yet pioneering studies are very promising. The role of Nrf2 in cardioprotection is, at this point, unequivocal, and has been demonstrated in multiple transgenic and Nrf2−/− mouse models. In an experimental model of pressure overload induced by transverse aortic constriction, overexpression of Nrf2 proved to be protective against cardiac hypertrophy152. Furthermore, in the previous section we described how pre-treatment with non-lethal doses of HNE (5 µM) could induce cardioprotection to larger, lethal doses (≥20 µM) of HNE85. The role of this pre-conditioning has been investigated in cardiac ischemia in vivo. When mice are pre-treated with HNE (4 mg/kg) prior to an I/R challenge, their heart is protected against injury compared to vehicle-treated mice85. The mechanism of this protection paralleled in vitro findings described in the previous section. HNE activates the transcription factor Nrf2, which in turn up-regulates endogenous anti-oxidant systems, including GST. This in turn appears to increase intramyocardial GSH content and decrease ROS, resulting in a better functional recovery of the LV following I/R injury. The molecular mechanisms by which HNE regulates Nrf2 have yet to be elucidated, but the importance of Nrf2 in cardioprotection has been determined using Nrf2 −/− mice. The HNE induced “pre-conditioning” cardioprotection in I/R injury is absent in Nrf2 −/− mice, implicating Nrf2’s role in mediating HNE induced GSH biosynthesis in the heart85.
VI. Concluding Perspectives
The importance of LPPs in the pathogenesis, and paradoxically, prevention, of cardiac disease remains an area of research that demands renewed attention. Clearly lipid peroxidation and the reactive lipid peroxidation products have context-specific effects on physiology, with differential effects being driven by organ-specific localization, structure and reactivity of LPP in question, dosage and temporal parameters. An increased understanding of these complex questions may lead to better understanding and improved therapies of cardiovascular diseases associated with redox imbalances.
Acknowledgements
The authors’ laboratories are supported by the National Institutes of Health R21HL098780 (to E.A.) and R01HL104129 (to M.W.).
Non-standard abbreviations
- ADH
alcohol dehydrogenase
- AGE
advanced glycation endproducts
- ALDH
aldehyde dehydrogenase
- ARE
Antioxidant Response Element
- Cu/ZnSOD
copper/zinc super oxide dismutase
- GSH
glutathione
- GST
glutathione-S-transferase
- HHE
4-hydroxyhexenal
- HNE
4-hydroxyhexenal
- H2O2
hydrogen peroxide
- I/R
ischemia reperfusion
- Keap1
Kelchlike ECH-associated protein 1
- LPP
lipid peroxidation products
- MTP
mitochondria transition pore
- MDA
malondialdehyde
- MnSOD
Manganese Superoxide Dismutase
- NO
nitric oxide
- Nrf2
NF-E2 p45 related factor 2
- O2•−
superoxide
- ONOO•−
peroxynitrite
- PUFA
poly-unsaturated fatty acids
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
Footnotes
Disclosures
None of the authors have any conflict of interest to declare.
References
- 1.Negre-Salvayre A, Auge N, Ayala V, Basaga H, Boada J, Brenke R, Chapple S, Cohen G, Feher J, Grune T, Lengyel G, Mann GE, Pamplona R, Poli G, Portero-Otin M, Riahi Y, Salvayre R, Sasson S, Serrano J, Shamni O, Siems W, Siow RC, Wiswedel I, Zarkovic K, Zarkovic N. Pathological aspects of lipid peroxidation. Free Radic Res. 2010;44:1125–1171. doi: 10.3109/10715762.2010.498478. [DOI] [PubMed] [Google Scholar]
- 2.Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-Hydroxynonenal: a Membrane Lipid Oxidation Product of Medicinal Interest. Medicinal research reviews. 2008;28:569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 3.Negre-Salvayre A, Auge N, Ayala V, Basaga H, Boada J, Brenke R, Chapple S, Cohen G, Feher J, Grune T, Lengyel G, Mann GE, Pamplona R, Poli G, Portero-Otin M, Riahi Y, Salvayre R, Sasson S, Serrano J, Shamni O, Siems W, Siow RC, Wiswedel I, Zarkovic K, Zarkovic N. Pathological aspects of lipid peroxidation. Free radical research. 44:1125–1171. doi: 10.3109/10715762.2010.498478. [DOI] [PubMed] [Google Scholar]
- 4.Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Experimental gerontology. 2009;44:625–633. doi: 10.1016/j.exger.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riahi Y, Cohen G, Shamni O, Sasson S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am J Physiol Endocrinol Metab. 2010;299:E879–E886. doi: 10.1152/ajpendo.00508.2010. [DOI] [PubMed] [Google Scholar]
- 6.Jezek P, Hlavata L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol. 2005;37:2478–2503. doi: 10.1016/j.biocel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 7.Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free radical biology & medicine. 2010;48:1286–1295. doi: 10.1016/j.freeradbiomed.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 8.Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium. 2009;45:643–650. doi: 10.1016/j.ceca.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loschen G, Azzi A. On the formation of hydrogen peroxide and oxygen radicals in heart mitochondria. Recent Adv Stud Cardiac Struct Metab. 1975;7:3–12. [PubMed] [Google Scholar]
- 11.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 12.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free radical biology & medicine. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 13.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hauptmann N, Grimsby J, Shih JC, Cadenas E. The metabolism of tyramine by monoamine oxidase A/B causes oxidative damage to mitochondrial DNA. Arch Biochem Biophys. 1996;335:295–304. doi: 10.1006/abbi.1996.0510. [DOI] [PubMed] [Google Scholar]
- 15.Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, Chen K, Gabrielson KL, Blakely RD, Shih JC, Pacak K, Kass DA, Di Lisa F, Paolocci N. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res. 2010;106:193–202. doi: 10.1161/CIRCRESAHA.109.198366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ascenzi P, di Masi A, Sciorati C, Clementi E. Peroxynitrite-An ugly biofactor? Biofactors. 2010;36:264–273. doi: 10.1002/biof.103. [DOI] [PubMed] [Google Scholar]
- 17.Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26:190–195. doi: 10.1016/j.tips.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Lacza Z, Pankotai E, Csordas A, Gero D, Kiss L, Horvath EM, Kollai M, Busija DW, Szabo C. Mitochondrial NO and reactive nitrogen species production: does mtNOS exist? Nitric Oxide. 2006;14:162–168. doi: 10.1016/j.niox.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 19.Lesnefsky EJ, Hoppel CL. Cardiolipin as an oxidative target in cardiac mitochondria in the aged rat. Biochimica et biophysica acta. 2008;1777:1020–1027. doi: 10.1016/j.bbabio.2008.05.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montero J, Mari M, Colell A, Morales A, Basanez G, Garcia-Ruiz C, Fernandez-Checa JC. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochimica et biophysica acta. 2010;1797:1217–1224. doi: 10.1016/j.bbabio.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kagan VE, Bayir A, Bayir H, Stoyanovsky D, Borisenko GG, Tyurina YY, Wipf P, Atkinson J, Greenberger JS, Chapkin RS, Belikova NA. Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: a new strategy in anti-apoptotic drug discovery. Mol Nutr Food Res. 2009;53:104–114. doi: 10.1002/mnfr.200700402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musatov A. Contribution of peroxidized cardiolipin to inactivation of bovine heart cytochrome c oxidase. Free radical biology & medicine. 2006;41:238–246. doi: 10.1016/j.freeradbiomed.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 23.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free radical biology & medicine. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 24.Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. American journal of physiology. Heart and circulatory physiology. 2007;292:H1227–H1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 27.Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free radical biology & medicine. 2003;34:496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 28.Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free radical biology & medicine. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 29.Franco R, Schoneveld OJ, Pappa A, Panayiotidis MI. The central role of glutathione in the pathophysiology of human diseases. Arch Physiol Biochem. 2007;113:234–258. doi: 10.1080/13813450701661198. [DOI] [PubMed] [Google Scholar]
- 30.Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishikawa T, Esterbauer H, Sies H. Role of cardiac glutathione transferase and of the glutathione S-conjugate export system in biotransformation of 4-hydroxynonenal in the heart. The Journal of biological chemistry. 1986;261:1576–1581. [PubMed] [Google Scholar]
- 32.Coleman JD, Prabhu KS, Thompson JT, Reddy PS, Peters JM, Peterson BR, Reddy CC, Vanden Heuvel JP. The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) Free radical biology & medicine. 2007;42:1155–1164. doi: 10.1016/j.freeradbiomed.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen CH, Sun L, Mochly-Rosen D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovascular research. 2010;88:51–57. doi: 10.1093/cvr/cvq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sena CM, Nunes E, Gomes A, Santos MS, Proenca T, Martins MI, Seica RM. Supplementation of coenzyme Q10 and alpha-tocopherol lowers glycated hemoglobin level and lipid peroxidation in pancreas of diabetic rats. Nutr Res. 2008;28:113–121. doi: 10.1016/j.nutres.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Baydas G, Donder E, Kiliboz M, Sonkaya E, Tuzcu M, Yasar A, Nedzvetskii VS. Neuroprotection by alpha-lipoic acid in streptozotocin-induced diabetes. Biochemistry (Mosc) 2004;69:1001–1005. doi: 10.1023/b:biry.0000043542.39691.95. [DOI] [PubMed] [Google Scholar]
- 36.Xiao C, Giacca A, Lewis GF. Oral taurine but not N-acetylcysteine ameliorates NEFA-induced impairment in insulin sensitivity and beta cell function in obese and overweight, non-diabetic men. Diabetologia. 2008;51:139–146. doi: 10.1007/s00125-007-0859-x. [DOI] [PubMed] [Google Scholar]
- 37.Tang SC, Arumugam TV, Cutler RG, Jo DG, Magnus T, Chan SL, Mughal MR, Telljohann RS, Nassar M, Ouyang X, Calderan A, Ruzza P, Guiotto A, Mattson MP. Neuroprotective actions of a histidine analogue in models of ischemic stroke. J Neurochem. 2007;101:729–736. doi: 10.1111/j.1471-4159.2006.04412.x. [DOI] [PubMed] [Google Scholar]
- 38.Guiotto A, Calderan A, Ruzza P, Borin G. Carnosine and carnosine-related antioxidants: a review. Curr Med Chem. 2005;12:2293–2315. doi: 10.2174/0929867054864796. [DOI] [PubMed] [Google Scholar]
- 39.Koenitzer JR, Freeman BA. Redox signaling in inflammation: interactions of endogenous electrophiles and mitochondria in cardiovascular disease. Annals of the New York Academy of Sciences. 1203:45–52. doi: 10.1111/j.1749-6632.2010.05559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, O'Keefe JH, Brand-Miller J. Origins and evolution of the Western diet: health implications for the 21st century. The American Journal of Clinical Nutrition. 2005;81:341–354. doi: 10.1093/ajcn.81.2.341. [DOI] [PubMed] [Google Scholar]
- 41.Siems W, Grune T. Intracellular metabolism of 4-hydroxynonenal. Molecular aspects of medicine. 2003;24:167–175. doi: 10.1016/s0098-2997(03)00011-6. [DOI] [PubMed] [Google Scholar]
- 42.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free radical biology & medicine. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 43.Bridges JW, Benford DJ, Hubbard SA. Mechanisms of toxic injury. Annals of the New York Academy of Sciences. 1983;407:42–63. doi: 10.1111/j.1749-6632.1983.tb47813.x. [DOI] [PubMed] [Google Scholar]
- 44.Bacot S, Bernoud-Hubac N, Baddas N, Chantegrel B, Deshayes C, Doutheau A, Lagarde M, Guichardant M. Covalent binding of hydroxy-alkenals 4-HDDE, 4-HHE, and 4-HNE to ethanolamine phospholipid subclasses. Journal of lipid research. 2003;44:917–926. doi: 10.1194/jlr.M200450-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Catala A. Lipid peroxidation of membrane phospholipids generates hydroxyalkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chemistry and physics of lipids. 2009;157:1–11. doi: 10.1016/j.chemphyslip.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 46.Chen JJ, Yu BP. Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free radical biology & medicine. 1994;17:411–418. doi: 10.1016/0891-5849(94)90167-8. [DOI] [PubMed] [Google Scholar]
- 47.Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. American journal of physiology.Cell physiology. 2007;292:C33–C44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- 48.Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. The Biochemical journal. 2000;351:183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Serena D, Ruggiero FM. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free radical biology & medicine. 1999;27:42–50. doi: 10.1016/s0891-5849(99)00032-5. [DOI] [PubMed] [Google Scholar]
- 50.Niki E. Lipid peroxidation: physiological levels and dual biological effects. Free radical biology & medicine. 2009;47:469–484. doi: 10.1016/j.freeradbiomed.2009.05.032. [DOI] [PubMed] [Google Scholar]
- 51.Chatterjee SN, Agarwal S, Kumarjana A, Bose B. Membrane lipid peroxidation and its pathological consequences. Indian journal of biochemistry & biophysics. 1988;25:25–31. [PubMed] [Google Scholar]
- 52.Borchman D, Lamba OP, Salmassi S, Lou M, Yappert MC. The dual effect of oxidation on lipid bilayer structure. Lipids. 1992;27:261–265. doi: 10.1007/BF02536472. [DOI] [PubMed] [Google Scholar]
- 53.Goldstein RM, Weissmann G. Effects of the generation of superoxide anion on permeability of liposomes. Biochemical and biophysical research communications. 1977;75:604–609. doi: 10.1016/0006-291x(77)91515-7. [DOI] [PubMed] [Google Scholar]
- 54.Kunimoto M, Inoue K, Nojima S. Effect of ferrous ion and ascorbate-induced lipid peroxidation on liposomal membranes. Biochimica et biophysica acta. 1981;646:169–178. doi: 10.1016/0005-2736(81)90284-4. [DOI] [PubMed] [Google Scholar]
- 55.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free radical biology & medicine. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 56.LoPachin RM, Gavin T, Petersen DR, Barber DS. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chemical research in toxicology. 2009;22:1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free radical biology & medicine. 2004;37:937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 58.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Progress in lipid research. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 59.Nakashima I, Liu W, Akhand AA, Takeda K, Kawamoto Y, Kato M, Suzuki H. 4-Hydroxynonenal Triggers Multistep Signal Transduction Cascades for Suppression of Cellular Functions. Molecular aspects of medicine. 2003;24:231–238. doi: 10.1016/s0098-2997(03)00018-9. [DOI] [PubMed] [Google Scholar]
- 60.Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. The Journal of biological chemistry. 1999;274:23787–23793. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- 61.Chavez JD, Wu J, Bisson W, Maier CS. Site-specific proteomic analysis of lipoxidation adducts in cardiac mitochondria reveals chemical diversity of 2-alkenal adduction. Journal of proteomics. doi: 10.1016/j.jprot.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lucas DT, Szweda LI. Cardiac reperfusion injury: aging, lipid peroxidation, and mitochondrial dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:510–514. doi: 10.1073/pnas.95.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Picklo MJ, Amarnath V, McIntyre JO, Graham DG, Montine TJ. 4-Hydroxy-2(E)-nonenal inhibits CNS mitochondrial respiration at multiple sites. Journal of neurochemistry. 1999;72:1617–1624. doi: 10.1046/j.1471-4159.1999.721617.x. [DOI] [PubMed] [Google Scholar]
- 64.Choksi KB, Boylston WH, Rabek JP, Widger WR, Papaconstantinou J. Oxidatively damaged proteins of heart mitochondrial electron transport complexes. Biochimica et biophysica acta. 2004;1688:95–101. doi: 10.1016/j.bbadis.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 65.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. Journal of neurochemistry. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 66.Mattson MP. Modification of ion homeostasis by lipid peroxidation: roles in neuronal degeneration and adaptive plasticity. Trends in neurosciences. 1998;21:53–57. doi: 10.1016/s0166-2236(97)01188-0. [DOI] [PubMed] [Google Scholar]
- 67.Siems WG, Hapner SJ, van Kuijk FJ. 4-hydroxynonenal inhibits Na(+)-K(+)-ATPase. Free radical biology & medicine. 1996;20:215–223. doi: 10.1016/0891-5849(95)02041-1. [DOI] [PubMed] [Google Scholar]
- 68.Nakamura K, Miura D, Kusano KF, Fujimoto Y, Sumita-Yoshikawa W, Fuke S, Nishii N, Nagase S, Hata Y, Morita H, Matsubara H, Ohe T, Ito H. 4-Hydroxy-2-nonenal induces calcium overload via the generation of reactive oxygen species in isolated rat cardiac myocytes. Journal of cardiac failure. 2009;15:709–716. doi: 10.1016/j.cardfail.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 69.Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otin M, Pamplona R, Vidal-Puig AJ, Wang S, Roebuck SJ, Brand MD. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. The EMBO journal. 2003;22:4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kristal BS, Park BK, Yu BP. 4-Hydroxyhexenal is a potent inducer of the mitochondrial permeability transition. The Journal of biological chemistry. 1996;271:6033–6038. doi: 10.1074/jbc.271.11.6033. [DOI] [PubMed] [Google Scholar]
- 71.Uchida K. Role of reactive aldehyde in cardiovascular diseases. Free radical biology & medicine. 2000;28:1685–1696. doi: 10.1016/s0891-5849(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 72.Minko IG, Kozekov ID, Harris TM, Rizzo CJ, Lloyd RS, Stone MP. Chemistry and biology of DNA containing 1,N(2)-deoxyguanosine adducts of the alpha,beta-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal. Chemical research in toxicology. 2009;22:759–778. doi: 10.1021/tx9000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blair IA. DNA adducts with lipid peroxidation products. The Journal of biological chemistry. 2008;283:15545–15549. doi: 10.1074/jbc.R700051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ji C, Kozak KR, Marnett LJ. IkappaB kinase, a molecular target for inhibition by 4-hydroxy-2-nonenal. The Journal of biological chemistry. 2001;276:18223–18228. doi: 10.1074/jbc.M101266200. [DOI] [PubMed] [Google Scholar]
- 75.Rudolph TK, Freeman BA. Transduction of redox signaling by electrophile-protein reactions. Science signaling. 2009;2:re7. doi: 10.1126/scisignal.290re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leonarduzzi G, Robbesyn F, Poli G. Signaling kinases modulated by 4-hydroxynonenal. Free radical biology & medicine. 2004;37:1694–1702. doi: 10.1016/j.freeradbiomed.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 77.Murphy MP. Induction of mitochondrial ROS production by electrophilic lipids: a new pathway of redox signaling? American journal of physiology.Heart and circulatory physiology. 2006;290:H1754–H1755. doi: 10.1152/ajpheart.00040.2006. [DOI] [PubMed] [Google Scholar]
- 78.Uchida K. Lipid peroxidation and redox-sensitive signaling pathways. Current atherosclerosis reports. 2007;9:216–221. doi: 10.1007/s11883-007-0022-7. [DOI] [PubMed] [Google Scholar]
- 79.Uchida K, Kanematsu M, Sakai K, Matsuda T, Hattori N, Mizuno Y, Suzuki D, Miyata T, Noguchi N, Niki E, Osawa T. Protein-bound acrolein: potential markers for oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4882–4887. doi: 10.1073/pnas.95.9.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Uchida K. Current status of acrolein as a lipid peroxidation product. Trends in cardiovascular medicine. 1999;9:109–113. doi: 10.1016/s1050-1738(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 81.Voulgaridou GP, Anestopoulos I, Franco R, Panayiotidis MI, Pappa A. DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutation research. 2011;711:13–27. doi: 10.1016/j.mrfmmm.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 82.Luo J, Hill BG, Gu Y, Cai J, Srivastava S, Bhatnagar A, Prabhu SD. Mechanisms of acrolein-induced myocardial dysfunction: implications for environmental and endogenous aldehyde exposure. American journal of physiology. Heart and circulatory physiology. 2007;293:H3673–H3684. doi: 10.1152/ajpheart.00284.2007. [DOI] [PubMed] [Google Scholar]
- 83.Jaar V, Ste-Marie L, Montgomery JA. Striatal metabolism of hexanal, a lipid peroxidation product, in the rat. Metabolic brain disease. 1999;14:71–82. doi: 10.1023/a:1020701612639. [DOI] [PubMed] [Google Scholar]
- 84.Ping P, Baines CP, Gu Y, Prabhu SD, Zhang J, Tsai LL, Cardwell E, Zong NC, Vondriska TM, Korge P, Bhatnagar A, Wang GW. Cardiac toxic effects of trans-2-hexenal are mediated by induction of cardiomyocyte apoptotic pathways. Cardiovascular toxicology. 2003;3:341–351. doi: 10.1385/ct:3:4:341. [DOI] [PubMed] [Google Scholar]
- 85.Zhang Y, Sano M, Shinmura K, Tamaki K, Katsumata Y, Matsuhashi T, Morizane S, Ito H, Hishiki T, Endo J, Zhou H, Yuasa S, Kaneda R, Suematsu M, Fukuda K. 4-hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. Journal of molecular and cellular cardiology. 2010;49:576–586. doi: 10.1016/j.yjmcc.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 86.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. The Journal of biological chemistry. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Savage AO, Dunbar JC, Brown RA. Effects of acetaldehyde on the isolated papillary muscle of diabetic rats. Journal of cardiovascular pharmacology. 1995;26:251–258. [PubMed] [Google Scholar]
- 88.Brown RA, Savage AO. Effects of acute acetaldehyde, chronic ethanol, and pargyline treatment on agonist responses of the rat aorta. Toxicology and applied pharmacology. 1996;136:170–178. doi: 10.1006/taap.1996.0021. [DOI] [PubMed] [Google Scholar]
- 89.Ren J, Davidoff AJ, Brown RA. Acetaldehyde depresses shortening and intracellular Ca2+ transients in adult rat ventricular myocytes. Cell Mol Biol (Noisy-le-grand) 1997;43:825–834. [PubMed] [Google Scholar]
- 90.Brown RA, Jefferson L, Sudan N, Lloyd TC, Ren J. Acetaldehyde depresses myocardial contraction and cardiac myocyte shortening in spontaneously hypertensive rats: role of intracellular Ca2+ Cell Mol Biol (Noisy-le-grand) 1999;45:453–465. [PubMed] [Google Scholar]
- 91.Brown RA, Anthony MJ, Petrovski P, Ren J. The influence of gender, diabetes, and acetaldehyde on the intrinsic contractile properties of isolated rat myocardium. Cardiovascular toxicology. 2001;1:35–42. doi: 10.1385/ct:1:1:35. [DOI] [PubMed] [Google Scholar]
- 92.Aistrup GL, Kelly JE, Piano MR, Wasserstrom JA. Biphasic changes in cardiac excitation-contraction coupling early in chronic alcohol exposure. American journal of physiology. Heart and circulatory physiology. 2006;291:H1047–H1057. doi: 10.1152/ajpheart.00214.2006. [DOI] [PubMed] [Google Scholar]
- 93.Brown RA, Carpentier RG. Effects of acetaldehyde on the automaticity of the guinea pig sinus node. Alcohol. 1989;6:103–107. doi: 10.1016/0741-8329(89)90033-5. [DOI] [PubMed] [Google Scholar]
- 94.Brown RA, Carpentier RG. Effects of acetaldehyde on membrane potentials of sinus node pacemaker fibers. Alcohol. 1990;7:33–36. doi: 10.1016/0741-8329(90)90057-j. [DOI] [PubMed] [Google Scholar]
- 95.Morales JA, Ram JL, Song J, Brown RA. Acetaldehyde inhibits current through voltage-dependent calcium channels. Toxicology and applied pharmacology. 1997;143:70–74. doi: 10.1006/taap.1996.8072. [DOI] [PubMed] [Google Scholar]
- 96.Aberle NS, 2nd, Privratsky JR, Burd L, Ren J. Combined acetaldehyde and nicotine exposure depresses cardiac contraction in ventricular myocytes: prevention by folic acid. Neurotoxicology and teratology. 2003;25:731–736. doi: 10.1016/j.ntt.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 97.Aberle NS, 2nd, Ren J. Short-term acetaldehyde exposure depresses ventricular myocyte contraction: role of cytochrome P450 oxidase, xanthine oxidase, and lipid peroxidation. Alcoholism, clinical and experimental research. 2003;27:577–583. doi: 10.1097/01.ALC.0000060522.40447.8E. [DOI] [PubMed] [Google Scholar]
- 98.Nagoor Meeran MF, Mainzen Prince PS. Protective effects of N-acetyl cysteine on lipid peroxide metabolism on isoproterenol-induced myocardial infarcted rats. Journal of biochemical and molecular toxicology. 2011;25:151–157. doi: 10.1002/jbt.20371. [DOI] [PubMed] [Google Scholar]
- 99.Basha RH, Priscilla DH. An in vivo and in vitro study on the protective effects of N-acetylcysteine on mitochondrial dysfunction in isoproterenol treated myocardial infarcted rats. Experimental and toxicologic pathology : official journal of the Gesellschaft fur Toxikologische Pathologie. 2011 doi: 10.1016/j.etp.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 100.Nagoor Meeran MF, Mainzen Prince PS. Protective effects of N-acetyl cysteine on lipid peroxide metabolism on isoproterenol-induced myocardial infarcted rats. Journal of biochemical and molecular toxicology. 2010 doi: 10.1002/jbt.20371. [DOI] [PubMed] [Google Scholar]
- 101.Sawyer DB. Oxidative Stress in Heart Failure: What Are We Missing? The American journal of the medical sciences. 2011 doi: 10.1097/MAJ.0b013e3182249fcd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sugamura K, Keaney JF., Jr Reactive oxygen species in cardiovascular disease. Free radical biology & medicine. 2011 doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stocker R, Keaney JF., Jr New insights on oxidative stress in the artery wall. J Thromb Haemost. 2005;3:1825–1834. doi: 10.1111/j.1538-7836.2005.01370.x. [DOI] [PubMed] [Google Scholar]
- 104.Tsimikas S, Witztum JL. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Curr Opin Lipidol. 2008;19:369–377. doi: 10.1097/MOL.0b013e328308b622. [DOI] [PubMed] [Google Scholar]
- 105.Frostegard J. Low level natural antibodies against phosphorylcholine: a novel risk marker and potential mechanism in atherosclerosis and cardiovascular disease. Clin Immunol. 2010;134:47–54. doi: 10.1016/j.clim.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 106.Tsimikas S. Oxidized low-density lipoprotein biomarkers in atherosclerosis. Curr Atheroscler Rep. 2006;8:55–61. doi: 10.1007/s11883-006-0065-1. [DOI] [PubMed] [Google Scholar]
- 107.Fredrikson GN, Berglund G, Alm R, Nilsson JA, Shah PK, Nilsson J. Identification of autoantibodies in human plasma recognizing an apoB-100 LDL receptor binding site peptide. J Lipid Res. 2006;47:2049–2054. doi: 10.1194/jlr.M600217-JLR200. [DOI] [PubMed] [Google Scholar]
- 108.Campolo J, De Maria R, Caruso R, Accinni R, Turazza F, Parolini M, Roubina E, De Chiara B, Cighetti G, Frigerio M, Vitali E, Parodi O. Blood glutathione as independent marker of lipid peroxidation in heart failure. Int J Cardiol. 2007;117:45–50. doi: 10.1016/j.ijcard.2006.04.065. [DOI] [PubMed] [Google Scholar]
- 109.Nakamura K, Kusano KF, Matsubara H, Nakamura Y, Miura A, Nishii N, Banba K, Nagase S, Miyaji K, Morita H, Saito H, Emori T, Ohe T. Relationship between oxidative stress and systolic dysfunction in patients with hypertrophic cardiomyopathy. J Card Fail. 2005;11:117–123. doi: 10.1016/j.cardfail.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 110.Rochette L, Tatou E, Maupoil V, Zeller M, Cottin Y, Jazayeri S, Brenot R, Girard C, David M, Vergely C. Atrial and vascular oxidative stress in patients with heart failure. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2011;27:497–502. doi: 10.1159/000329951. [DOI] [PubMed] [Google Scholar]
- 111.Fujita S, Ikeda Y, Miyata M, Shinsato T, Kubozono T, Kuwahata S, Hamada N, Miyauchi T, Yamaguchi T, Torii H, Hamasaki S, Tei C. Effect of Waon therapy on oxidative stress in chronic heart failure. Circulation journal : official journal of the Japanese Circulation Society. 2011;75:348–356. doi: 10.1253/circj.cj-10-0630. [DOI] [PubMed] [Google Scholar]
- 112.Keith M, Geranmayegan A, Sole MJ, Kurian R, Robinson A, Omran AS, Jeejeebhoy KN. Increased oxidative stress in patients with congestive heart failure. Journal of the American College of Cardiology. 1998;31:1352–1356. doi: 10.1016/s0735-1097(98)00101-6. [DOI] [PubMed] [Google Scholar]
- 113.Nakamura K, Kusano K, Nakamura Y, Kakishita M, Ohta K, Nagase S, Yamamoto M, Miyaji K, Saito H, Morita H, Emori T, Matsubara H, Toyokuni S, Ohe T. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation. 2002;105:2867–2871. doi: 10.1161/01.cir.0000018605.14470.dd. [DOI] [PubMed] [Google Scholar]
- 114.Dhalla NS, Golfman L, Takeda S, Takeda N, Nagano M. Evidence for the role of oxidative stress in acute ischemic heart disease: a brief review. The Canadian journal of cardiology. 1999;15:587–593. [PubMed] [Google Scholar]
- 115.Makazan Z, Saini HK, Dhalla NS. Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts. American journal of physiology. Heart and circulatory physiology. 2007;292:H1986–H1994. doi: 10.1152/ajpheart.01214.2006. [DOI] [PubMed] [Google Scholar]
- 116.Lorenzi R, Andrades ME, Bortolin RC, Nagai R, Dal-Pizzol F, Moreira JC. Glycolaldehyde induces oxidative stress in the heart: a clue to diabetic cardiomyopathy? Cardiovascular toxicology. 2010;10:244–249. doi: 10.1007/s12012-010-9083-x. [DOI] [PubMed] [Google Scholar]
- 117.Saddala RR, Thopireddy L, Ganapathi N, Kesireddy SR. Regulation of cardiac oxidative stress and lipid peroxidation in streptozotocin-induced diabetic rats treated with aqueous extract of Pimpinella tirupatiensis tuberous root. Experimental and toxicologic pathology : official journal of the Gesellschaft fur Toxikologische Pathologie. 2011 doi: 10.1016/j.etp.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 118.Wang G, Konig R, Ansari GA, Khan MF. Lipid peroxidation-derived aldehyde-protein adducts contribute to trichloroethene-mediated autoimmunity via activation of CD4+ T cells. Free radical biology & medicine. 2008;44:1475–1482. doi: 10.1016/j.freeradbiomed.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang Y, Ren J. ALDH2 in alcoholic heart diseases: Molecular mechanism and clinical implications. Pharmacology & therapeutics. 2011 doi: 10.1016/j.pharmthera.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Budas GR, Disatnik MH, Mochly-Rosen D. Aldehyde dehydrogenase 2 in cardiac protection: a new therapeutic target? Trends in cardiovascular medicine. 2009;19:158–164. doi: 10.1016/j.tcm.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Doser TA, Turdi S, Thomas DP, Epstein PN, Li SY, Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;119:1941–1949. doi: 10.1161/CIRCULATIONAHA.108.823799. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 123.Ma H, Li J, Gao F, Ren J. Aldehyde dehydrogenase 2 ameliorates acute cardiac toxicity of ethanol: role of protein phosphatase and forkhead transcription factor. Journal of the American College of Cardiology. 2009;54:2187–2196. doi: 10.1016/j.jacc.2009.04.100. [DOI] [PubMed] [Google Scholar]
- 124.Ren J, Babcock SA, Li Q, Huff AF, Li SY, Doser TA. Aldehyde dehydrogenase-2 transgene ameliorates chronic alcohol ingestion-induced apoptosis in cerebral cortex. Toxicology letters. 2009;187:149–156. doi: 10.1016/j.toxlet.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ma J, Meng Y, Kwiatkowski DJ, Chen X, Peng H, Sun Q, Zha X, Wang F, Wang Y, Jing Y, Zhang S, Chen R, Wang L, Wu E, Cai G, Malinowska-Kolodziej I, Liao Q, Liu Y, Zhao Y, Xu K, Dai J, Han J, Wu L, Zhao RC, Shen H, Zhang H. Mammalian target of rapamycin regulates murine and human cell differentiation through STAT3/p63/Jagged/Notch cascade. The Journal of clinical investigation. 2010;120:103–114. doi: 10.1172/JCI37964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ma H, Yu L, Byra EA, Hu N, Kitagawa K, Nakayama KI, Kawamoto T, Ren J. Aldehyde dehydrogenase 2 knockout accentuates ethanol-induced cardiac depression: role of protein phosphatases. Journal of molecular and cellular cardiology. 2010;49:322–329. doi: 10.1016/j.yjmcc.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Koda K, Salazar-Rodriguez M, Corti F, Chan NY, Estephan R, Silver RB, Mochly-Rosen D, Levi R. Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation. 2010;122:771–781. doi: 10.1161/CIRCULATIONAHA.110.952481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cai L. Alcoholic cardiomyopathy: acetaldehyde, insulin insensitization and ER stress. Journal of molecular and cellular cardiology. 2008;44:979–982. doi: 10.1016/j.yjmcc.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 129.Guo R, Ren J. Alcohol and acetaldehyde in public health: from marvel to menace. International journal of environmental research and public health. 2010;7:1285–1301. doi: 10.3390/ijerph7041285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Guo R, Ren J. Alcohol dehydrogenase accentuates ethanol-induced myocardial dysfunction and mitochondrial damage in mice: role of mitochondrial death pathway. PloS one. 2010;5:e8757. doi: 10.1371/journal.pone.0008757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liang Q, Carlson EC, Borgerding AJ, Epstein PN. A transgenic model of acetaldehyde overproduction accelerates alcohol cardiomyopathy. The Journal of pharmacology and experimental therapeutics. 1999;291:766–772. [PubMed] [Google Scholar]
- 132.Li SY, Ren J. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: role of insulin signaling and ER stress. Journal of molecular and cellular cardiology. 2008;44:992–1001. doi: 10.1016/j.yjmcc.2008.02.276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 133.Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. European heart journal. 2011;32:1025–1038. doi: 10.1093/eurheartj/ehq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ge W, Ren J. mTOR-STAT3-notch signaling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. Journal of cellular and molecular medicine. 2011 doi: 10.1111/j.1582-4934.2011.01347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Brigelius-Flohe R, Flohe L. Basic Principles and Emerging Concepts in the Redox Control of Transcription Factors. Antioxidants & redox signaling. 2011 doi: 10.1089/ars.2010.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jung KA, Kwak MK. The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules. 2010;15:7266–7291. doi: 10.3390/molecules15107266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta medica. 2008;74:1526–1539. doi: 10.1055/s-0028-1088302. [DOI] [PubMed] [Google Scholar]
- 138.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–99. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Birringer M. Hormetics: Dietary Triggers of an Adaptive Stress Response. Pharmaceutical research. 2011 doi: 10.1007/s11095-011-0551-1. [DOI] [PubMed] [Google Scholar]
- 140.Kaspar JW, Jaiswal AK. An autoregulatory loop between Nrf2 and Cul3-Rbx1 controls their cellular abundance. The Journal of biological chemistry. 2010;285:21349–21358. doi: 10.1074/jbc.M110.121863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Moshev B. Severe combined trauma and the autotransplantation of splenic tissue. Khirurgiia. 1990;43:108–110. [PubMed] [Google Scholar]
- 144.Goto S, Naito H, Kaneko T, Chung HY, Radak Z. Hormetic effects of regular exercise in aging: correlation with oxidative stress. Applied physiology, nutrition, and metabolism = Physiologie appliquee, nutrition et metabolisme. 2007;32:948–953. doi: 10.1139/H07-092. [DOI] [PubMed] [Google Scholar]
- 145.Radak Z, Chung HY, Koltai E, Taylor AW, Goto S. Exercise, oxidative stress and hormesis. Ageing research reviews. 2008;7:34–42. doi: 10.1016/j.arr.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 146.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free radical biology & medicine. 2011;51:327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 147.McCord JM. Superoxide dismutase, lipid peroxidation, and bell-shaped dose response curves. Dose-response : a publication of International Hormesis Society. 2008;6:223–238. doi: 10.2203/dose-response.08-012.McCord. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis) Experimental gerontology. 45:410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 149.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free radical biology & medicine. 51:327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 150.Sano M, Fukuda K. Activation of mitochondrial biogenesis by hormesis. Circulation research. 2008;103:1191–1193. doi: 10.1161/CIRCRESAHA.108.189092. [DOI] [PubMed] [Google Scholar]
- 151.Echtay KS, Pakay JL, Esteves TC, Brand MD. Hydroxynonenal and uncoupling proteins: a model for protection against oxidative damage. BioFactors (Oxford, England) 2005;24:119–130. doi: 10.1002/biof.5520240114. [DOI] [PubMed] [Google Scholar]
- 152.Li J, Ichikawa T, Villacorta L, Janicki JS, Brower GL, Yamamoto M, Cui T. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29:1843–1850. doi: 10.1161/ATVBAHA.109.189480. [DOI] [PubMed] [Google Scholar]
- 153.Long J, Wang X, Gao H, Liu Z, Liu C, Miao M, Liu J. Malonaldehyde acts as a mitochondrial toxin: Inhibitory effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Life Sciences. 2006;79:1466–1472. doi: 10.1016/j.lfs.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 154.Lashin OM, Szweda PA, Szweda LI, Romani AM. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free radical biology & medicine. 2006;40:886–896. doi: 10.1016/j.freeradbiomed.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 155.Biagini RE, Toraason MA, Lynch DW, Winston GW. Inhibition of rat heart mitochondrial electron transport in vitro: implications for the cardiotoxic action of allylamine or its primary metabolite, acrolein. Toxicology. 1990;62:95–106. doi: 10.1016/0300-483x(90)90034-e. [DOI] [PubMed] [Google Scholar]
- 156.Luo J, Shi R. Acrolein induces oxidative stress in brain mitochondria. Neurochemistry international. 2005;46:243–252. doi: 10.1016/j.neuint.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 157.Vaishnav RA, Singh IN, Miller DM, Hall ED. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. Journal of neurotrauma. 27:1311–1320. doi: 10.1089/neu.2009.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sun L, Luo C, Long J, Wei D, Liu J. Acrolein is a mitochondrial toxin: effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Mitochondrion. 2006;6:136–142. doi: 10.1016/j.mito.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 159.Picklo MJ, Montine TJ. Acrolein inhibits respiration in isolated brain mitochondria. Biochimica et biophysica acta. 2001;1535:145–152. doi: 10.1016/s0925-4439(00)00093-4. [DOI] [PubMed] [Google Scholar]
- 160.Suh JH, Heath SH, Hagen TM. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free radical biology & medicine. 2003;35:1064–1072. doi: 10.1016/s0891-5849(03)00468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bacon BR, O'Neill R, Park CH. Iron-induced peroxidative injury to isolated rat hepatic mitochondria. Journal of free radicals in biology & medicine. 1986;2:339–347. doi: 10.1016/s0748-5514(86)80034-4. [DOI] [PubMed] [Google Scholar]
- 162.Yarian CS, Rebrin I, Sohal RS. Aconitase and ATP synthase are targets of malondialdehyde modification and undergo an age-related decrease in activity in mouse heart mitochondria. Biochemical and biophysical research communications. 2005;330:151–156. doi: 10.1016/j.bbrc.2005.02.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen J, Henderson GI, Freeman GL. Role of 4-hydroxynonenal in modification of cytochrome c oxidase in ischemia/reperfused rat heart. Journal of molecular and cellular cardiology. 2001;33:1919–1927. doi: 10.1006/jmcc.2001.1454. [DOI] [PubMed] [Google Scholar]
- 164.Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12896–12901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen JJ, Bertrand H, Yu BP. Inhibition of adenine nucleotide translocator by lipid peroxidation products. Free radical biology & medicine. 1995;19:583–590. doi: 10.1016/0891-5849(95)00066-7. [DOI] [PubMed] [Google Scholar]
- 166.Pocernich CB, Butterfield DA. Acrolein inhibits NADH-linked mitochondrial enzyme activity: implications for Alzheimer's disease. Neurotoxicity research. 2003;5:515–520. doi: 10.1007/BF03033161. [DOI] [PubMed] [Google Scholar]
- 167.Moreau R, Nguyen BT, Doneanu CE, Hagen TM. Reversal by aminoguanidine of the age-related increase in glycoxidation and lipoxidation in the cardiovascular system of Fischer 344 rats. Biochemical pharmacology. 2005;69:29–40. doi: 10.1016/j.bcp.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 168.Humphries KM, Szweda LI. Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry. 1998;37:15835–15841. doi: 10.1021/bi981512h. [DOI] [PubMed] [Google Scholar]
- 169.Lucas DT, Szweda LI. Declines in mitochondrial respiration during cardiac reperfusion: age-dependent inactivation of alpha-ketoglutarate dehydrogenase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6689–6693. doi: 10.1073/pnas.96.12.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zollner H. Inhibition of some mitochondrial functions by acrolein and methylvinylketone. Biochemical pharmacology. 1973;22:1171–1178. doi: 10.1016/0006-2952(73)90234-7. [DOI] [PubMed] [Google Scholar]
- 171.Nguyen E, Picklo MJ., Sr Inhibition of succinic semialdehyde dehydrogenase activity by alkenal products of lipid peroxidation. Biochimica et biophysica acta. 2003;1637:107–112. doi: 10.1016/s0925-4439(02)00220-x. [DOI] [PubMed] [Google Scholar]
- 172.Benderdour M, Charron G, DeBlois D, Comte B, Des Rosiers C. Cardiac mitochondrial NADP+-isocitrate dehydrogenase is inactivated through 4-hydroxynonenal adduct formation: an event that precedes hypertrophy development. The Journal of biological chemistry. 2003;278:45154–45159. doi: 10.1074/jbc.M306285200. [DOI] [PubMed] [Google Scholar]
- 173.Mitchell DY, Petersen DR. Inhibition of rat liver aldehyde dehydrogenases by acrolein. Drug metabolism and disposition: the biological fate of chemicals. 1988;16:37–42. [PubMed] [Google Scholar]
- 174.Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. The Journal of pharmacology and experimental therapeutics. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]