

Abstract
Genome duplication requires that replication forks track the entire length of every chromosome. When complications occur, homologous recombination-mediated repair supports replication fork movement and recovery. This leads to physical connections between the nascent sister chromatids in the form of Holliday junctions and other branched DNA intermediates. A key role in the removal of these recombination intermediates falls to structure-specific nucleases such as the Holliday junction resolvase RuvC in Escherichia coli. RuvC is also known to cut branched DNA intermediates that originate directly from blocked replication forks, targeting them for origin-independent replication restart. In eukaryotes, multiple structure-specific nucleases, including Mus81–Mms4/MUS81–EME1, Yen1/GEN1, and Slx1–Slx4/SLX1–SLX4 (FANCP) have been implicated in the resolution of branched DNA intermediates. It is becoming increasingly clear that, as a group, they reflect the dual function of RuvC in cleaving recombination intermediates and failing replication forks to assist the DNA replication process.
Introduction
Faithful duplication of the eukaryotic genome is a formidable challenge and requires that every segment of DNA be replicated once and only once. Origins of replication are distributed along the chromosomes and fire throughout synthesis phase (S phase) in a specific temporal pattern. Origins are licensed prior to S phase by a system that allows them to send out bidirectional replication forks (RFs) precisely once per cell cycle (Diffley 2011). A given segment of DNA is therefore replicated by the combined efforts of two converging RFs that emanate from adjacent origins. Key in completing the replication of an inter-origin space is that the converging RFs meet at some point on their traverse.
Complications in the replication process can arise from a number of different stressors, which impede RF progression and may preclude RF convergence. Firstly, replication occurs in the presence of lesions within the DNA template. This is due to the susceptibility of DNA to hydrolysis and oxidation in the cellular milieu and its constant exposure to reactive cellular metabolites and various exogenous DNA damaging agents (Hoeijmakers 2001; Lindahl 1993). The consequence is a high steady-state background level of DNA damage, making collisions between the replication machinery and lesions in the template inevitable. Secondly, RFs have to negotiate obstructions arising from proteins tightly bound to the DNA template, DNA secondary structures, ongoing transcription in their path, and buildup of torsional stress in the DNA (Aguilera and Gómez-González 2008). Thirdly, low supplies of deoxyribonucleoside triphosphates (dNTPs) during S phase, a condition that may arise from untimely S phase entry and altered replication dynamics, can severely retard RF progression (Alvino et al. 2007; Beck et al. 2010; Bester et al. 2011). Stalled RFs are sensed by the S phase checkpoint that inhibits cell cycle progression and late origin firing (Errico and Costanzo 2012). Activated checkpoint proteins also preserve the integrity of stalling RFs, allowing them to resume DNA synthesis once the replication block is removed. RF restart is further facilitated by homologous recombination (HR) reactions, which entail the formation of branched intermediates that physically link sister chromatids. These must be resolved to restore discrete chromosome entities that can be segregated during mitosis. If RF restart is delayed, RFs become prone to breakage (Petermann et al. 2010; Saintigny et al. 2001), perhaps reflecting the need to remove idling RFs that have become a hindrance to—rather than a facilitator of—genome duplication. In Escherichia coli, the structure-specific nuclease RuvC serves both of these purposes by processing HR repair intermediates and by mediating RF cleavage so that a new RF may be assembled to complete the replication process. In eukaryotes, three conserved nucleases have been found to have properties similar to those of RuvC: Mus81–Mms4/MUS81–EME1 (budding yeast/human), and, more recently, Yen1/GEN1 and Slx1–Slx4/SLX1–SLX4 (SLX4 is also known as BTBD12 and FANCP). The purpose of this review is to highlight how side-by-side analysis of these nucleases is revealing that they too play a multifaceted role by resolving branched recombination intermediates as well as replication structures that arise in S phase (Fig. 1).
Fig. 1.
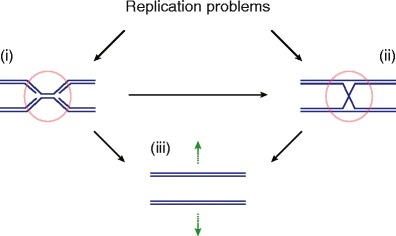
DNA replication problems can lead to replication fork stalling and unreplicated chromosomal areas (i), and to the formation of branched repair intermediates from homologous recombination-dependent RF recovery pathways (ii). The resulting physical links between sister chromatids can be targeted by structure-specific nucleases to facilitate the completion of S phase and chromosome segregation (iii)
Homologous recombination underpins the replication process
HR is an ancient process that is conserved in all domains of life. In eukaryotes, HR plays important roles in meiosis, mitosis, and DNA repair. Disruption of key recombination genes of the RAD52 epistasis group in yeast and vertebrate cells causes sensitivity to a broad spectrum of replication blocking agents (Chang et al. 2002; Essers et al. 1997; Parsons et al. 2004; Takata et al. 2001). These include interstrand DNA cross-linkers such as mitomycin C (MMC), DNA alkylating agents such as methyl methanesulfonate (MMS), the topoisomerase I (TOP1) poison camptothecin (CPT) that interferes with the release of torsional stress from replicating DNA, and the ribonucleotide reductase inhibitor hydroxyurea (HU) whose presence causes dNTP depletion. Vertebrate cells are unable to complete DNA replication in the absence of the central recombinase RAD51 (Sonoda et al. 1998; Su et al. 2008), while rad51 mutant yeast cells exhibit growth defects (Fingerhut et al. 1984) and accumulate chromosomes with unreplicated areas in the presence of DNA damage (Alabert et al. 2009). These observations underline the critically important role of HR to lend support to troubled RFs.
Molecular aspects of HR
HR is part of the meiotic program in eukaryotes, allowing for reciprocal genetic exchange (crossover) between maternal and paternal homologous chromosomes, which is required for their accurate segregation. Careful analysis of the meiotic products in fungi has provided early insights into the mechanism of HR (Holliday 1964), providing the groundwork for the current DNA double-strand break (DSB) repair model of HR (Szostak et al. 1983). The key steps are illustrated in Fig. 2 (steps 1–6). The signature reaction is strand exchange (mediated by Rad51/RAD51) that occurs between the damaged molecule and an intact donor duplex of homologous sequence. In the context of DSB repair, the donor serves as a template for repair synthesis to retrieve all sequence information lost at the break. The recombining DNA molecules may ultimately become covalently attached to one another at DNA four-way junctions known as Holliday junctions (HJs) (Holliday 1964; Liu and West 2004). These late recombination structures must be removed prior to chromosome segregation. Specialized structure-specific nucleases, so-called HJ resolvases, cleave HJs by the introduction of two symmetrically related nicks (Fig. 2, step 5). Depending on the orientation of the nicks, crossover (associated with the reciprocal exchange of flanking markers) or non-crossover duplex products are generated. Other HR subpathways have been described, and a growing number of proteins are known to be involved in HR-mediated DSB repair (Mazón et al. 2010). The RecQ helicase Sgs1-type IA topoisomerase Top3–Rmi1 protein complex (BLM–TOPOIIIα–RMI1–RMI2 in humans) catalyzes convergent branch migration and DNA decatenation to separate recombining molecules along the nuclease-independent non-crossover pathway of double HJ dissolution (Cejka et al. 2010; Ira et al. 2003; Wu and Hickson 2003) (Fig. 2, steps 7 and 8). The early disassembly of recombination intermediates sidesteps the formation of HJs on a pathway known as synthesis-dependent strand annealing (SDSA) (Pâques and Haber 1999) (Fig. 2, step 9).
Fig. 2.
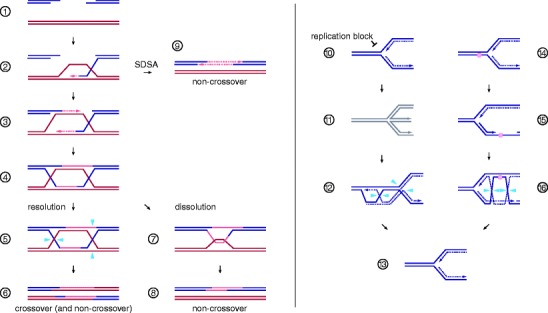
DNA double-strand break repair and replication fork support mediated by homologous recombination. Steps 1–6 describe the canonical DSB repair model of HR. 1, DNA end resection produces 3′-single-stranded overhangs which are bound by strand exchange protein Rad51/RAD51; 2, the resulting nucleoprotein-filament is capable of identifying and invading a homologous donor duplex, thereby creating a displacement-loop (D-loop) and initiating repair synthesis; 3, second-end capture, the association of the D-loop with the non-invading break end, also initiates repair synthesis; 4, after repair and nick ligation, the recombining molecules are joined together at a double HJ intermediate; 5, HJ resolvases introduce symmetrically related nicks (blue arrowheads) to produce discrete duplex products; 6, resolution of a pair of HJs along the same axis produces non-crossover products, while the use of different axes leads to crossover products characterized by reciprocal genetic exchange of flanking markers (as shown). Double HJ dissolution offers an alternative to nucleolytic cleavage: 7, the Sgs1–Top3–Rmi1/BLM–TOPOIIIα–RMI1–RMI2 complex drives HJ convergence by branch migration and removal of the resulting hemi-catenate; 8, dissolution results exclusively in non-crossover products, which differ from the DNA molecules at outset only by the presence of a repair patch. 9, Synthesis-dependent strand annealing (SDSA). Disassembly of the D-loop by expulsion of the invading DNA single-strand frees up a repair template for the second break end. 10–16, HR facilitates DNA replication: 10, a range of replication blocks may stall RF progression (see text); 11, a blocked RF (gray) can regress to form a HJ-like structure with a recombinogenic DSB end homologous to the DNA ahead of the four-way DNA junction; 12, HR-mediated strand-invasion forms a D-loop at which DNA synthesis may be re-initiated. HJs that arise during the process can be removed by nucleolytic cleavage (blue arrowheads) to reestablish a processive RF (13). 14, A lesion in the lagging strand template (pink sphere) can be bypassed without RF arrest; 15, the lesion is tolerated at the cost of a single-stranded DNA gap known as daughter strand gap; 16, HR-mediated gap repair and HJ formation. HJ resolution (blue arrowheads) or dissolution can restore a normal RF (13)
Pathways of HR-mediated RF repair
Models of the ways in which HR repair reactions facilitate the DNA replication process have been derived from extensive studies in E. coli (Cox et al. 2000; McGlynn and Lloyd 2002; Michel et al. 2007). The strategies found in prokaryotes are thought to be broadly conserved in eukaryotes (Lambert et al. 2007; Petermann and Helleday 2010). In this context, the recombination substrates comprise double-stranded DNA ends/single-ended DSBs and DNA gaps rather than canonical two-ended DSBs. For example, blocked RFs have been shown to regress by removal of the nascent leading and lagging strands from the template and their annealing with one another. This generates an HJ-like structure with a recombinogenic double-stranded DNA end homologous to the replication template upstream of the RF. Thus, Rad51/RAD51 may catalyze strand exchange to rebuild a RF in an origin-independent manner (Fig. 2, steps 10–13). HR is also useful for the repair of single-stranded DNA gaps that are left behind the RF when the replicative DNA polymerase skips over a lesion and reinitiates DNA synthesis downstream of it. Strand exchange between the sister chromatids can provide an intact template for gap repair without the need for immediate lesion repair (lesion bypass) (Fig. 2, steps 14–16). Finally, if a RF collapses into a single-ended DSB, for example by replication run-off at a preexisting nick in the template, HR can mediate the reestablishment of a RF. Such a single-ended break may contain single-stranded DNA or be processed to expose a 3′-single-stranded overhang for Rad51/RAD51 to polymerize on, which is followed by strand invasion at the intact sister chromatid and assembly of a processive RF (a reaction depicted in Fig. 3, steps 7–10). In all these cases, and similar to the situation in HR-mediated DSB repair, HJ-containing intermediates are generated (Fig. 2, steps 12 and 16 and Fig. 3, step 9), which require the attention of resolution and/or dissolution factors.
Fig. 3.
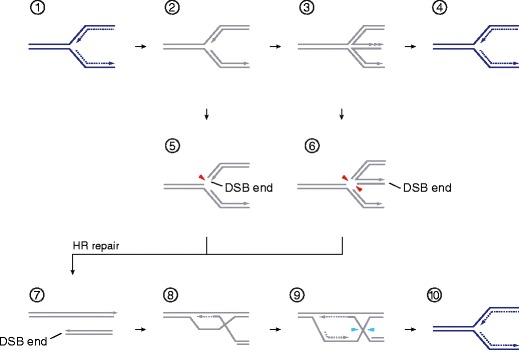
Replication fork recovery by simple resetting or deliberate breakage. 1, RF movement can be impeded by a range of factors (see text); 2, potentially leading to RF arrest (inactive fork, gray); 3, RF regression, by annealing of the nascent leading and lagging strands with one another, leads to the formation of a HJ-like structure. Template switching allows leading strand extension (indicated by the triple arrowhead) if the lagging strand had been extended further than the leading stand prior to regression. This might occur via the RAD6–RAD18–RAD5 DNA damage tolerance pathway when a RF is blocked at a lesion in the leading strand template (Ghosal and Chen 2013). Regression also ensures that any blocking lesion is moved away from the fork and placed back into a duplex DNA context enabling excision repair pathways. 4, If the cause for regression can be removed (or if template switch enables bypass/DNA damage tolerance), HJ branch migration may reset an active fork (blue). Alternatively, deliberate fork cleavage might occur: 5, the substrate spectra of eukaryotic structure-specific nucleases such as Mus81–Mms4/MUS81–EME1 may allow them to act directly on RFs (red arrowhead); 6, RF cleavage may also occur after conversion of the three-way junction into a four-way HJ intermediate. 7–10, Recombination-dependent replication/break-induced replication (BIR) pathways can reestablish a processive RF from a single-ended DSB after RF breakage (or spontaneous collapse at a preexisting nick in the template). Note that this pathway entails the formation of a single HJ (step 9), which requires the attention of a structure-specific nuclease (blue arrowheads)
Structure-specific nucleases and the cleavage of recombination and replication intermediates
The RuvC paradigm
The RuvC HJ resolvase is highly selective for fully double-stranded DNA four-way junctions (Benson and West 1994; Takahagi et al. 1994). Being a constitutive dimer, the enzyme uses two active sites to resolve HJs by double-sided incision (West 1997). It has long been known that loss of RuvC function in bacteria causes recombination defects and DNA damage sensitivity (Benson et al. 1991; Lloyd 1991; Lloyd et al. 1984; Sharples et al. 1990), indicating a key role in the removal of HJ intermediates during recombination-dependent DNA repair (Michel et al. 2007). Interestingly, RuvC also attacks HJs formed by RF regression, which leads to DSB formation (Seigneur et al. 1998, 2000) (Fig. 3, steps 1–3 and 6). This offers the possibility for origin-independent RF establishment by recombination-dependent replication (Asai et al. 1994; Heller and Marians 2006; Kogoma 1997) (Fig. 3, steps 7–10), providing a survival strategy in situations of DNA damage-induced RF blockage (Khan and Kuzminov 2012). RuvC is not conserved in eukaryotes, but structure-specific nucleases, which have the ability to resolve HJs and may function analogously, have been identified.
HJ-resolving nucleases in eukaryotes
Three HJ-resolving enzymes found in eukaryotes are Mus81–Mms4 (Mus81–Eme1 in fission yeast)/MUS81–EME1, Yen1/GEN1, and Slx1–Slx4/SLX1–SLX4 (Fig. 4). Mus81–Mms4/MUS81–EME1 belongs to the XPF family of nucleases (Ciccia et al. 2008). Both subunits contain ERCC4 endonuclease domains, but while the one found in the catalytic subunit Mus81/MUS81 is active, the one in Mms4/EME1 has diverged and lost its activity. The substrate spectrum of Mus81–Mms4/MUS81–EME1 comprises 3′-flaps (duplex DNA with a 3′-single-stranded flap), double-stranded three-way junctions that resemble RFs, HJ precursors, and fully ligated HJs (Boddy et al. 2001; Chen et al. 2001; Ciccia et al. 2003; Constantinou et al. 2002; Doe et al. 2002; Kaliraman et al. 2001). The activity of purified Mus81–Mms4/MUS81–EME1 complexes towards fully fledged HJs is relatively weak, but nicked HJs are efficiently resolved by a counter-nicking mechanism (Ehmsen and Heyer 2008; 2009; Fricke et al. 2005; Osman et al. 2003). This is consistent with budding yeast Mus81–Mms4 operating as a single heterodimer with only one active site (Schwartz et al. 2012). Therefore, Mus81–Mms4/MUS81–EME1 resembles the propensity of the bacterial HJ resolvase RuvC to target branched DNA structures but is not biochemically equivalent.
Fig. 4.
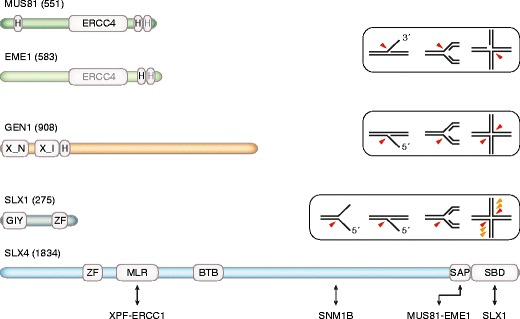
Domain structure and in vitro DNA substrate specificities of the eukaryotic HJ-resolving enzymes. These structure-specific nucleases are largely conserved from yeast to human (Schwartz and Heyer 2011), with the human proteins depicted (number of amino acid residues in brackets). MUS81 and EME1 contain an excision repair cross complementation group 4 (ERCC4) endonuclease domain and helix-hairpin-helix (H) motifs (gray font denotes degenerate/inactive motifs). The MUS81–EME1 heterodimer exhibits activity towards 3′-flaps, RFs, and HJ substrates (red arrowheads). HJs are efficiently resolved if they contain a preexisting nick. GEN1 is a monomeric XPG family nuclease that contains the superfamily-specific N-terminal and internal XPG nuclease motifs (X_N and X_I, respectively). Adjacent to the nuclease domain is a helix-hairpin-helix motif. GEN1 cuts 5′-flaps, RFs, and HJs, as indicated. SLX1 is a small GIY-YIG superfamily nuclease with a PHD-type zinc-finger (ZF) motif at the C-terminus. It forms a heterodimeric structure-specific nuclease with the large SLX4 subunit. SLX4 contains ubiquitin-binding zinc-finger-, MUS312-MEI9 interaction-like (MLR)-, bric-a-brac tramtrack broad complex (BTB)-, SAF-A/B acinus and PIAS (SAP)-, and SLX1-binding (SBD) domains. Human SLX4 interacts with multiple nucleases involved in ICL repair via the Fanconi anemia pathway, as indicated below. The interaction with SNM1B (Salewsky et al. 2012) has not been mapped. SLX1–SLX4 cleaves splayed arm, 5′-flap, RF, and HJ substrates. HJs are cleaved in symmetric fashion by the human protein, but nicked at multiple nonsymmetric positions by yeast Slx1–Slx4 (orange arrowheads)
Yen1/GEN1 belongs to the Rad2/XPG family of nucleases, whose members are characterized by a bipartite N-terminal/internal XPG nuclease domain and exhibit diverse structure-specific nuclease activities (Tomlinson et al. 2010). Human GEN1 is monomeric in solution but dimerizes on HJ DNA in order to introduce symmetric incisions, thus resolving HJs in a manner reminiscent of RuvC (Rass et al. 2010). Other in vitro target structures include 5′-flaps and RFs (Ip et al. 2008; Rass et al. 2010).
Slx1–Slx4/SLX1–SLX4 owes its structure-specific nuclease activity to the Slx1/SLX1 subunit (Fricke and Brill 2003), which is a GIY-YIG superfamily nuclease (Dunin-Horkawicz et al. 2006). Yeast and human Slx1–Slx4/SLX1–SLX4 cleave splayed arm DNA substrates (a duplex with unpaired 3′- and 5′-overhangs on one side), 5′-flaps, RFs, and HJs (Coulon et al. 2004; Fekairi et al. 2009; Fricke and Brill 2003; Muñoz et al. 2009; Svendsen et al. 2009). The human enzyme has been shown to be capable of RuvC-like HJ resolution (Fekairi et al. 2009; Svendsen et al. 2009), while yeast Slx1–Slx4 cuts this substrate at multiple, nonsymmetric sites and with relatively weak activity compared to that exhibited on 5′-flaps and RFs (Fricke and Brill 2003). Interestingly, human SLX4 interacts with MUS81–EME1 (Fekairi et al. 2009; Muñoz et al. 2009; Svendsen et al. 2009), while this interaction has not been found in yeast (Schwartz et al. 2012).
Eukaryotic HJ-resolving enzymes in the processing of late recombination intermediates
The ability of Mus81–Mms4, Yen1, and Slx1–Slx4 to resolve HR-dependent HJ intermediates in vivo has been elegantly demonstrated in budding yeast by physically tracking recombination intermediates during meiosis in various mutant strains (De Muyt et al. 2012; Zakharyevich et al. 2012). Given the difference in substrate specificity, they likely target a mixture of HJ precursors and fully matured (covalently closed) HJs (the exact nature of the targeted intermediates has been discussed recently by Schwartz and Heyer (2011)). Most meiotic recombination intermediates were found to be processed in an Sgs1-dependent manner, either via SDSA or double HJ dissolution, or along a crossover pathway that in addition involves Exo1 and Mlh1-Mlh3, the latter potentially providing the required nuclease activity (De Muyt et al. 2012; Zakharyevich et al. 2012). The contribution of Mus81–Mms4, Yen1, and Slx1–Slx4 is therefore small, and yet important to facilitate meiotic chromosome segregation. Thus, the absence of Mus81 compromises spore viability, and this effect is exacerbated by the additional absence of Slx1 or Slx4. No viable spores are produced when Mus81–Mms4 and Yen1 are absent (Matos et al. 2011).
Mus81–Mms4/Eme1, Yen1, and Slx1–Slx4 also contribute to the resolution of recombination intermediates during the repair of induced DSBs in mitotic cells. This is evidenced by a loss of crossover recombination in their absence in different experimental systems using chromosomal or plasmid-borne substrates (Agmon et al. 2011; Ho et al. 2010; Muñoz-Galván et al. 2012; Sun et al. 2008; Tay and Wu 2010). However, DSB repair in mitotic cells is thought to proceed mainly via SDSA (Fig. 2, step 9), largely avoiding the formation of HJs (Bzymek et al. 2010). This is consistent with the mild sensitivity of mus81 and mms4, yen1, and/or slx1 and slx4 mutant yeast cells to ionizing radiation-induced DSBs (Blanco et al. 2010; Ho et al. 2010; Tay and Wu 2010). In fact, these mutants are much more affected by treatment that compromises replication progression, suggesting that the critical function of Mus81–Mms4, Yen1, and Slx1–Slx4 in mitotic cells is to assist in the repair of perturbed RFs.
Eukaryotic HJ-resolving enzymes and the repair of replication forks
Loss of Mus81–Mms4/MUS81–EME1, Yen1/GEN1, and Slx1–Slx4/SLX1–SLX4 function sensitizes cells to replication stress, i.e., to conditions that lead to frequent RF stalling or arrest. This indicates a defect in the processing of failing RFs and/or the resolution of intermediates of HR-dependent RF recovery pathways. Distinguishing between these possibilities is not easy because a defect in either process can lead to similar phenotypes relating to chromosome segregation failure resulting from unreplicated DNA or persistent recombination intermediates physically linking the sister chromatids (Fig. 1). The interpretation of genetic data is further complicated by the overlapping biochemical activities and functional redundancies that exist between Mus81–Mms4/MUS81–EME1, Yen1/GEN1, and Slx1–Slx4/SLX1–SLX4. Nonetheless, important in vivo targets for these nucleases in replicating cells are being identified.
Slx1–Slx4/SLX1–SLX4 (FANCP)
SLX1 and SLX4 were first identified in yeast as genes with an essential function in the absence Sgs1 (Mullen et al. 2000). Eliminating HR by deletion of RAD52 does not restore viability to sgs1 slx1 or sgs1 slx4 double mutant cells (Bastin-Shanower et al. 2003), indicating that the synthetic lethal relationship is not due to a functional overlap of Slx1–Slx4 and Sgs1 in the resolution/dissolution of recombination intermediates. Instead, the phenotype has been linked to a role of Sgs1 in promoting the replication of the repetitive ribosomal DNA (rDNA) array, which is characterized by a high frequency of replication stalling (Kaliraman and Brill 2002; Versini et al. 2003). Budding yeast harboring a SLX4 gene deletion and a temperature-sensitive sgs1 allele were shown to be unable to complete rDNA replication at the restrictive temperature (Kaliraman and Brill 2002). In fission yeast, Slx1 associates with the rDNA and induces HR repair foci specifically in the nucleolus, which is consistent with a function of Slx1–Slx4 in rDNA maintenance that is upstream—rather than downstream—of HR (Coulon et al. 2004, 2006). A model has been proposed suggesting that Slx1–Slx4 may cleave a pair of converging RFs that have stalled (see Fig. 5, point 2), thereby facilitating replication termination. Because the rDNA is repetitive, break-proximal regions at the ensuing DSB would be homologous. Thus, 5′-resection at the DSB ends would allow repair by annealing of complementary 3′-overhangs as they become exposed (a pathway known as single-strand annealing), albeit at the expense of rDNA contraction (Fricke and Brill 2003).
Fig. 5.
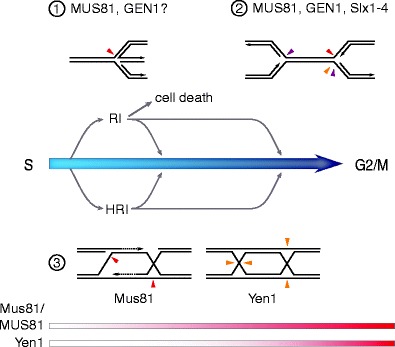
Ways in which the HJ-resolving endonucleases Mus81–Mms4/MUS81–EME1, Yen1/GEN1, and Slx1–Slx4/SLX1–SLX4 may support DNA replication. When cells progress through S phase, RF stalling and arrest can lead to the accumulation of replication intermediates (RIs) not actively engaged in DNA synthesis. 1, MUS81-dependent DSB formation in response to drug-induced (APH, CPT, HU, MMC) or oncogene-induced (overexpressed cyclin D1, E) replication stress indicates active RI cleavage. Subsequent replication restart and increased cell survival has been reported, suggesting that active RF breakage can promote bulk DNA synthesis, while unrestrained RI cleavage may cause cell death. CDK1-dependent stimulation of MUS81 activity may promote DSB-independent RF recovery in early S phase and delay RF breakage until replication is approaching its completion. Reversed RFs with strand interruptions have been observed in unperturbed human cells and may represent an in vivo target for MUS81–EME1 in accordance with the enzyme's in vitro preference for nicked HJs. Given the functional overlap between Mus81 and Yen1 in yeast, it is conceivable that GEN1 also targets RIs. 2, RFs stalled within difficult-to-replicate areas, such as common fragile sites and the rDNA array in yeast, have been shown to require the attention of HJ-resolving enzymes. Fragile site cleavage by MUS81 (red arrowhead) allows the disengagement of the parental DNA strands within unreplicated segments and sister chromatid disjunction. A higher incidence of anaphase bridges in cells depleted for GEN1 indicates the enzyme may fulfill related functions (cutting RFs with the opposite polarity, orange arrowhead). Slx1–Slx4 has been proposed to resolve blocked RFs in the rDNA in yeast (purple arrowheads). A similar reaction may be catalyzed by SLX4 in conjunction with multiple nucleases at RFs arrested at intrastrand crosslinks (FA pathway). 3, HR intermediates (HRIs) arise during RF recovery and DSB repair (see Figs. 2 and 3, steps 7–10). In yeast, Mus81–Mms4 and Yen1 have been shown to resolve HRIs upon replication stress and under unperturbed conditions. Perhaps the most likely scenario is that Mus81–Mms4 targets nicked HJ precursors and D-loop structures by virtue of its 3′-flap cleavage activity. After maturation into fully ligated HJs, Yen1 is the more suitable processing factor (shown here on a double HJ intermediate). This is in agreement with a successive cell cycle-dependent surge in Mus81–Mms4 and Yen1 activity as cells approach G2/M (below, maximal activity bright red)
Outside this context, there is little evidence of any replication-associated role of Slx1/SLX1, while Slx4/SLX4 clearly has further, Slx1/SLX1 nuclease-independent functions. Deletion of SLX1 or SLX4 in yeast does not lead to a general problem in coping with replication stress, and only the loss of Slx4 causes sensitivity to DNA alkylation by MMS associated with unreplicated chromosomal areas (Bastin-Shanower et al. 2003; Flott et al. 2007; Flott and Rouse 2005; Roberts et al. 2005). It has been shown that Slx4 is recruited to stalled RFs in response to MMS treatment, where it appears to act as a scaffold for DNA repair proteins other than Slx1 and to exert a regulatory function on the S phase checkpoint (Ohouo et al. 2010, 2012). In Caenorhabditis elegans, loss of SLX1 function was shown to sensitize germline cells to UV light-induced bulky DNA lesions and TOP1 inhibition by CPT, indicating that in this organism SLX1 may be more widely used in DNA replication-associated repair (Saito et al. 2012).
In keeping with the multifaceted role of Slx4 observed in yeast, depletion of SLX4 in HeLa cells causes a more pronounced sensitivity to drugs that induce DNA interstrand cross-links (ICLs) than depletion of SLX1 (Andersen et al. 2009; Fekairi et al. 2009; Svendsen et al. 2009). The collision of RFs with ICLs triggers a multiprotein repair pathway that is defective in Fanconi anemia (FA), a genome stability disorder associated with bone marrow failure, cancer predisposition, and hypersensitivity to ICL-inducing agents (Constantinou 2012). SLX4 has recently been identified as downstream FA factor FANCP (Kim et al. 2011; Stoepker et al. 2011) and has been shown to interact with multiple nucleases involved in ICL excision (Andersen et al. 2009; Fekairi et al. 2009; Kim et al. 2013; Muñoz et al. 2009; Svendsen et al. 2009) (Fig. 4). A truncated version of SLX4, which is unable to interact with SLX1, was found to complement the MMC sensitivity of SLX4-deficient murine btbd12 −/− (Crossan et al. 2011) and human FA complementation group P cells (Kim et al. 2013). It therefore appears that SLX4 functions independently of SLX1 in ICL repair. The interaction between SLX1–SLX4 and MUS81–EME1 raises the possibility that these nucleases cooperate to resolve HJ-containing DNA intermediates. This appears not to be the case in yeast (Schwartz et al. 2012), while experiments with the human proteins show that the SLX1–SLX4–MUS81–EME1 complex promotes coordinated HJ resolution more efficiently than its component heterodimeric nucleases alone (Stephen C. West, personal communication).
Mus81–Mms4/MUS81–EME1 and Yen1/GEN1
MUS81 and MMS4, like SLX1 and SLX4, have been identified as a gene pair required for viability in sgs1 mutant yeast cells (Boddy et al. 2000; Mullen et al. 2000). In contrast to sgs1 slx1 or sgs1 slx4 cells, the synthetic lethality in mus81 sgs1 cells is suppressed when HR is eliminated by deletion of key RAD52 epistasis group genes such as RAD51 or RAD52 (Fabre et al. 2002). When the Sgs1–Top3–Rmi1 double HJ dissolvasome is compromised, Mus81–Mms4 limits the accumulation of Rad51-dependent HJ-containing DNA at damaged RFs, but when MUS81 or MMS4 are disrupted, these accumulate to levels that preclude chromosome segregation (Ashton et al. 2011). These observations indicate a functional overlap between Mus81–Mms4 and Sgs1–Top3–Rmi1 in the processing of replication-associated recombination intermediates, which in their absence accumulate to toxic levels.
GEN1 was isolated from HeLa cell extracts fractionated extensively on the basis of detectable HJ resolution activity (Ip et al. 2008). In the same study, Yen1 was identified (alongside Slx1–Slx4 and Mus81–Mms4) in a yeast screen for HJ resolvase activities. Subsequently, Yen1 was shown to functionally overlap with Mus81–Mms4, indicating the importance of analyzing these enzymes together.
Loss of Mus81–Mms4 activity sensitizes yeast cells to a large panel of agents whose effects inhibit RF progression, including MMS, UV light, CPT, HU, and DNA cross-linking agents. In all cases, concomitant loss of Yen1 leads to hypersensitivity (not further exacerbated by deletion of SLX1), and mus81 yen1 mutant cells are characterized by growth defects, chronic low-level Rad53 phosphorylation indicative of checkpoint activation, and fail to segregate their chromosomes in the presence of exogenous DNA damage (Agmon et al. 2011; Blanco et al. 2010; Ho et al. 2010; Tay and Wu 2010). Similar phenotypes have been observed for mus81 single mutant fission yeast, an organism lacking a Yen1 ortholog (Boddy et al. 2000). In budding yeast, mus81 yen1 double mutants exhibit a much higher than normal rate of spontaneous chromosome loss, indicating the persistence of DNA structures that link the sister chromatids and cause nondisjunction in these cells (Ho et al. 2010). This defect was greatly reduced by deletion of RAD51, suggesting that unresolved HR intermediates cause the observed mis-segregation in mus81 yen1 cells. Therefore, Mus81 and Yen1 target replication-associated recombination intermediates that arise in unperturbed S phase. This function of Mus81 and Yen1 is subject to intricate cell cycle-dependent control mechanisms to prevent genome instability as a result of spurious cleavage of branched DNA intermediates: Cdk1- and Cdc5-dependent phosphorylation of Mms4 leads to a rise in Mus81–Mms4 nuclease activity during late S phase with a peak in G2/M (Gallo-Fernández et al. 2012; Matos et al. 2011, 2013; Szakal and Branzei 2013). In fission yeast, Mus81–Eme1 can be further stimulated by hyperphosphorylation mediated by the DNA damage checkpoint kinases, if Eme1 is primed by the cell cycle kinases (Dehé et al. 2013). Yen1, on the other hand, is activated with a slightly later timing than Mus81–Mms4 through dephosphorylation (Matos et al. 2011). This gives Sgs1-dependent processing of HR intermediates precedence over nucleolytic cleavage for much of S phase, limiting the risk of crossover formation which can be accompanied by loss of heterozygosity (Matos et al. 2013; Szakal and Branzei 2013). Distinct windows of activation may also explain why endogenous Yen1 does not fully protect mus81 or mms4 mutant cells from exogenous replication stress. Indeed, overexpression of Yen1 (Blanco et al. 2010), or mutations that render Yen1 resistant to inhibitory phosphorylation (Matos et al. 2013), suppresses the damage sensitivity phenotype of mus81 cells to a large extent, indicating that Mus81–Mms4 and Yen1 can resolve equivalent (although not necessarily the same) DNA structures in vivo. In addition to dynamic modulation of nuclease activity, cellular localization may play a part in regulating Mus81–Mms4 and Yen1: phosphorylation-dependent nucleo-cytoplasmic shuttling has been demonstrated for Yen1 (Kosugi et al. 2009), while Mus81–Mms4/MUS81–EME1 in yeast and human cells has consistently been found to localize to the nucleus (Blais et al. 2004; Fu and Xiao 2003; Gao et al. 2003; Naim et al. 2013; Svendsen et al. 2009; Ying et al. 2013; Zhang et al. 2005).
While the interplay of Mus81 and Yen1 in processing HR intermediates is becoming increasing well characterized, it is important to note that chromosome mis-segregation in mus81 and mus81 yen1 mutant yeast was found not to be entirely dependent on Rad51 and, therefore, HR, providing strong evidence that Mus81–Mms4 (and possibly Yen1) has a second physiologically important role associated with the processing of stalled RFs (Ho et al. 2010). This notion is corroborated by a number of very recent reports, which will be discussed in the following sections.
Evidence for the conversion of stalled RFs into DSBs by Mus81–Mms4/MUS81–EME1
Mus81–Mms4/MUS81–EME1 has previously been considered as an important factor in the conversion of stalled or regressed forks into DSBs to enable subsequent HR-mediated repair and replication restart (Haber and Heyer 2001; Osman and Whitby 2007) (Fig. 3). However, as mentioned above, findings regarding the importance of Mus81–Mms4 for viability under conditions of replication stress, induced either by RF blocking agents or disruption of a number of non-essential replication genes in yeast (Collins et al. 2007; Costanzo et al. 2010; Pan et al. 2006), are not easily interpreted with regard to cleavage of stalled or regressed RFs and/or the resolution of HR repair intermediates (Fig. 1). Recent observations of Mus81/MUS81-dependent chromosome breaks, however, provide strong physical evidence for a role in the cleavage of perturbed RFs and provide new insights into pathways of deliberate RF breakage in eukaryotes.
RF cleavage in response to exogenous replication stress
The most overt DNA damage sensitivity exhibited by mammalian cells lacking MUS81–EME1 is against DNA cross-linking agents (Abraham et al. 2003; Dendouga et al. 2005; Hanada et al. 2006; Hiyama et al. 2006; McPherson et al. 2004; Svendsen et al. 2009). As mentioned above, SLX4 appears to coordinate the multiple nucleases required for ICL repair, including MUS81–EME1 and the nucleotide excision repair endonuclease XPF–ERCC1. It has been proposed that MUS81–EME1 might convert RFs blocked at an ICL site into DSBs. Subsequent incision by XPF–ERCC1 could dislodge the cross-linked nucleobase to allow lesion bypass by translesion synthesis, followed by HR-dependent RF reassembly (Niedernhofer et al. 2004). More recently, it was suggested that XPF–ERCC1, together with exonuclease SNM1A, is involved in an initial response to ICL-stalled RFs (Wang et al. 2011). A nick 5′ of the ICL introduced by XPF–ERCC1 could provide an entry point of SNM1A, which is capable of degrading DNA past an ICL lesion to clear the path for translesion synthesis without a need for MUS81–EME1-dependent DSB formation. Consistent with this, cells depleted for XPF–ERCC1 or SNM1A accumulate MUS81-dependent DSBs after ICL induction. These observations led to the proposal that the DSB-dependent MUS81–EME1 response to ICL damage may only come into play if lesions persist or escape the SLX4–XPF–ERCC1–SNM1A pathway (Wang et al. 2011). This fits with an earlier report showing that chronic treatment with DNA cross-linking agents MMC or cisplatin leads to replication-associated DSB formation in wild-type, but not in mus81 −/− mouse embryonic stem cells (Hanada et al. 2006). By analyzing genomic DNA using pulsed field gel electrophoresis, this study provided direct evidence for MUS81–EME1-dependent chromosomal breaks. While the exact interplay of the different nucleases involved in fork repair at ICLs remains to be determined (Cybulski and Howlett 2011), this demonstrates that MUS81–EME1 can indeed be used to cleave (HR-independent) branched DNA replication intermediates. In light of this, the sensitivity of mammalian cells lacking the activities of MUS81–EME1 to ICL-inducing agents may suggest that deliberate RF breakage by MUS81–EME1 contributes to cell survival. This notion became more tangible with a subsequent report linking MUS81-dependent DSBs to RF restart (Hanada et al. 2007). Prolonged replication stress (of 18 h and more) in the presence of HU or DNA polymerase inhibitor aphidicolin (APH) was shown to provoke DSB formation in wild-type but not mus81 −/− mouse embryonic stem cells. Assessment of replication restart on a single molecule level by in vivo labeling of newly synthesized DNA (DNA fiber technique) revealed that the absence of MUS81 did not affect RF restart after short HU treatment (6 h), but reduced restarting events after longer HU exposure (24 h), i.e., at a time when DSBs appeared in the presence of MUS81. One interpretation of these findings is that deliberate RF cleavage promotes the recovery of preexisting RFs after prolonged stalling (as depicted in Fig. 3). The late manifestation of this pathway implies that it might be secondary to other, DSB-independent, means of RF repair.
The positive effect of MUS81 on RF restart seen in mouse cells could not be discerned in U2OS cells treated with siRNA against MUS81 after prolonged HU treatment (Petermann et al. 2010), and extensive treatment periods (36 h) have been shown to lead to massive MUS81-induced DNA breakage and cell death (Fugger et al. 2013). However, under conditions of milder replication stress, the actions of MUS81–EME1 on stalled replication intermediates appear to benefit human cells. Thus, a drastic increase in stalled RFs has been reported after short (2 h) HU treatment of U2OS cells depleted of MUS81 (Ying et al. 2013). Similarly, a decrease of RF restart (as assessed by DNA fiber technique) and phosphorylation of histone H2AX (γH2AX), an event that marks sites of broken DNA (Fernandez-Capetillo et al. 2004), has been observed in a mus81 −/− HCT116 colon carcinoma cell line treated with APH when compared with a MUS81-complemented control (Shimura et al. 2008). In a separate study, acute treatment of control HCT116 cells with the TOP1 poison CPT was found to induce dose-dependent γH2AX focus formation (Regairaz et al. 2011). In mus81 −/− HCT116 cells, γH2AX focus formation was significantly abrogated, specifically in the subset of actively replicating cells. Consistent with this, a decrease in DNA breakage in the absence of MUS81 was detected by neutral comet assay. Importantly, efficient RF recovery after CPT treatment (assessed by DNA fiber technique) in HCT116 cells was dependent upon MUS81 (Regairaz et al. 2011). During replication, DNA supercoiling builds up ahead of RFs, and excessive torsional stress may prohibit RF movement when relaxation by TOP1 is compromised globally in the presence of CPT (Pommier 2006). RF cleavage by MUS81–EME1 might help to dissipate excessive superhelical tension and promote subsequent replication restart (Regairaz et al. 2011).
RF cleavage in response to endogenous replication stress
Cells regularly experience replication stress at difficult-to-replicate common fragile sites, which are prone to breakage if their replication is unfinished at mitotic onset (Durkin and Glover 2007). Two recent studies have linked deliberate cleavage of late replication intermediates at fragile sites by MUS81–EME1 to genome stability in human cells (Naim et al. 2013; Ying et al. 2013). While MUS81–EME1 depletion reduced common fragile site expression (breakage observed on metaphase chromosomes), it caused increased anaphase bridge formation and chromosome mis-segregation. These observations have led to the proposal that active RF cleavage by MUS81–EME1 safeguards sister chromatid disjunction as it allows to resolve the impediment resulting from the intertwined parental DNA strands at underreplicated areas (see Fig. 5, point 2). The DNA breaks and single-stranded gaps formed in the process are likely to be less of a burden to the cell than potential chromosome segregation failure (Naim et al. 2013; Ying et al. 2013).
More pleiotropically acting endogenous replication stress, associated with oncogene activation, has been identified as a candidate driver in the development of cancer (Bartkova et al. 2005, 2006; Di Micco et al. 2006; Gorgoulis et al. 2005). According to the oncogene-induced DNA damage model of tumorigenesis, the activation of oncogenes causes hyperproliferation, RF failure, and replication-associated DNA damage (Halazonetis et al. 2008). Initially, cells react with activation of the S phase checkpoint, leading to p53-induced senescence or apoptosis. This barrier to cancer development is prone to breakdown due to inactivation/mutation of p53, allowing cell proliferation in the presence of DNA damage. The consequence is genome instability and transition of a precancerous lesion to cancer. With regard to the mechanistic basis of replication stress and DNA damage following oncogene activation, it has been shown that forced cell cycle entry involves DNA synthesis in the presence of insufficient dNTP pools (Bester et al. 2011). This may result from failing to adjust the cellular metabolism to the demands of S phase. Alternatively, the presence of supernumerary RFs as a consequence of excessive origin firing has been suggested to lead to the depletion of dNTP pools and cause RF stalling/collapse (Jones et al. 2012). The DSBs that result from experimental oncogene overexpression are in part mediated by MUS81. Thus, depletion of MUS81 was shown to reduce γH2AX focus formation and DNA breaks detected by comet assay in cells overexpressing cyclin E (Murfuni et al. 2013). Similar observations have been made in cells overexpressing cyclin D1 or CDC25A phosphatase. Long-term exposure of human cancer cell lines to small daily doses of ionizing radiation, mimicking fractionated radiotherapy used in cancer treatment, can produce radioresistant cells that overexpress cyclin D1 (Shimura et al. 2010). High levels of cyclin D1 during S phase cause RF slow down and induce MUS81-dependent DSBs (Shimura et al. 2013). MUS81-dependent DSBs have also been detected by pulsed field gel electrophoresis after overexpression of CDC25A (Neelsen et al. 2013). In this study, CDC25A-induced reversed RFs were observed by electron microscopy, and their levels rose dramatically after depletion of MUS81, indicating that reversed fork structures are converted into DSBs by MUS81–EME1. Of note, reversed forks either resembled fully double-stranded four-way HJs or contained nicks or gaps at the branch point, the latter representing a structure that is cleaved with great efficiency by Mus81/MUS81 in vitro (see Fig. 5, point 1).
While chromosome breakage was dramatic after CDC25A overexpression leading to cell cycle arrest, the breaks induced by MUS81 in response to cyclin E or D1 overexpression were found to correlate with increased cell survival. This indicates that deliberate RF cleavage can help cells to overcome endogenous replication stress. A corollary of this is that cells in precancerous lesions may have a heightened dependency on structure-specific nucleases such as MUS81–EME1, raising the interesting possibility that they may provide a target for therapeutic intervention.
RF cleavage versus DSB-independent RF restart
Deliberate RF cleavage in response to replication stress harbors the risk of illegitimate recombination and chromosomal rearrangements. RF regression into an HJ-like intermediate on the other hand may provide space and time to repair/overcome a RF block with the opportunity to restart DNA synthesis in a DSB-independent manner. As mentioned above, a recombination reaction at the DSB end spooled out by fork regression may reestablish a RF through strand invasion at the upstream template (Fig. 2, steps 10–13). Alternatively, simple resetting of the RF by migrating the HJ to reverse regression may be possible if the RF remains replication-competent (Fig. 3, steps 1–4). Regressed replication intermediates that form in response to exogenous DNA damage/induced replication stress in mammalian (Higgins et al. 1976; Neelsen et al. 2013; Ray Chaudhuri et al. 2012) and replication checkpoint deficient yeast cells (Sogo et al. 2002) have been directly visualized by electron microscopy. In E. coli, RF regression is an active process that can be catalyzed by the helicase RecG, which translocates along the leading and lagging strand templates of a RF to displace the nascent DNA (McGlynn and Lloyd 2001; McGlynn et al. 2001). There are no obvious RecG orthologs in eukaryotes, but a number of helicases/translocases have been implicated in the interconversion of three- and four-way DNA junctions and may therefore fulfill an analogous function (Atkinson and McGlynn 2009). RF regression and HJ branch migration activities have been demonstrated for the Bloom's syndrome BLM (Constantinou et al. 2000; Karow et al. 2000; Ralf et al. 2006) and Werner's syndrome WRN (Machwe et al. 2006, 2011) RecQ family helicases, FA protein FANCM (Gari et al. 2008a, b) and its yeast orthologs Fml1 (Sun et al. 2008) and Mph1 (Zheng et al. 2011), the Snf2 family ATP-dependent dsDNA translocases Rad54/RAD54 (Bugreev et al. 2006, 2011) and Rad5/HLTF (Achar et al. 2011; Blastyák et al. 2007, 2010), and the Snf2 family ATP-dependent annealing helicases SMARCAL1 (also known as HARP) (Bétous et al. 2012), mutated in Schimke immuno-osseous dysplasia, and ZRANB3 (Ciccia et al. 2012). Interestingly, two of these fork-remodeling proteins, WRN and SMARCAL1, exhibit functional interactions with MUS81. Assuming that fork regression/reestablishment and fork cleavage represent alternative means to deal with stalling RFs (Fig. 3, steps 1–6), inhibition of the former pathway may be expected to promote fork cleavage. WRN has been shown to promote the stability of arrested RFs and their efficient restart (Franchitto et al. 2008). In Werner's syndrome cells (with a mutation in WRN) or in HeLa cells depleted for WRN, spontaneous and HU-induced DSBs, which were detected by pulsed field gel electrophoresis of genomic DNA, neutral comet assay, and γH2AX focus formation, have been shown to accumulate in a MUS81-dependent manner (Franchitto et al. 2008; Murfuni et al. 2013). Depletion of RAD51 in Werner's syndrome cells had no effect on DSB formation, but led to massive cell death after HU treatment (Murfuni et al. 2012). Therefore, MUS81 appears to operate upstream of HR in the processing of arrested RFs, on a pathway parallel to fork remodeling and restart by WRN. Although concomitant loss of WRN and MUS81 was shown to limit DSB formation in response to HU, it induced cell death, indicating that deliberate RF cleavage functions as a survival mechanism in this setting. This may come at a price, however, as excessive use of the DSB-dependent MUS81 pathway (in the absence of functional WRN) correlates with a hyperrecombination phenotype, which may help explain the genome instability seen in Werner's syndrome (Franchitto et al. 2008).
Like WRN, SMARCAL1 facilitates the restart of RFs under conditions of replication stress. DNA fiber analysis after a temporary replication block has revealed that perturbed forks fail to resume DNA synthesis in SMARCAL1-depleted U2OS cells, while new origin firing was unaffected (Ciccia et al. 2009). Recruitment to sites of stalled replication and DNA damage is mediated by SMARCAL1's interaction with single-stranded DNA binding protein RPA, and the absence of SMARCAL1 causes accumulation of spontaneous DNA damage as indicated by activation of the ATM damage response kinase and γH2AX focus formation (Bansbach et al. 2009; Ciccia et al. 2009; Postow et al. 2009; Yuan et al. 2009; Yusufzai et al. 2009). Strikingly, γH2AX focus formation induced in the absence of SMARCAL1 could be almost entirely suppressed by concomitant depletion of MUS81 (Bétous et al. 2012). SMARCAL1 possess a novel annealing activity rather than conventional strand separation helicase activity, which allows the enzyme to mediate duplex formation from RPA-bound DNA single strands (Yusufzai and Kadonaga 2008). This activity may be required to limit the amount of ssDNA generated at perturbed RFs, thereby protecting the fork from inadvertent nucleolytic processing. Alternatively, the ability of SMARCAL1 to remodel RFs through regression and branch migration could serve to protect and restart stalled RFs, and failure to do so may activate fork cleavage by MUS81 as an alternative route of fork recovery (Bétous et al. 2012).
RF protection by the S phase checkpoint
DNA replication under stress conditions is facilitated by the replication/S phase checkpoint, which initiates a cascade of protein phosphorylation events upon sensing RF stalling (Segurado and Tercero 2009). Importantly, events downstream of the apical kinase Mec1/ATR protect the structural integrity of RFs and help maintain their replication competence to facilitate the timely resumption of DNA synthesis (Cobb et al. 2003; De Piccoli et al. 2012; Lopes et al. 2001; Lucca et al. 2004). Therefore, the replication checkpoint can be expected to antagonize deliberate RF breakage, notwithstanding that cleavage may provide an important alternative to RF preservation when arrest is irreversible (Ciccia and Elledge 2010).
Upon RF stalling, the S phase checkpoint kinase Mec1 and the mammalian ortholog ATR exert many of their functions through the effector kinases Rad53 and CHK1, respectively. Mus81 in fission yeast was originally identified by its interaction with the Rad53 ortholog Cds1 (Boddy et al. 2000). In cds1 mutant cells that proceed through S phase in the presence of HU, excessive Mus81–Eme1-dependent chromosome fragmentation ensues (Froget et al. 2008). This was independent of HR, indicating that Mus81–Eme1 targets stalled RFs rather than recombination intermediates under these conditions. This phenotype is independent of the ability of Cds1 to release Mus81–Eme1 from chromatin in response to replication stress, an event previously suggested to prevent unscheduled cleavage of replication intermediates (Kai et al. 2005), and may relate more directly to fork protection by the S phase checkpoint (Froget et al. 2008). Considering the recent elucidation of CDK activity-dependent control mechanisms to ensure that the Mus81 and Yen1 nucleases are stimulated late in the cell cycle to mediate “late resolution” of HR intermediates (see above) (Dehé et al. 2013; Gallo-Fernández et al. 2012; Matos et al. 2011; 2013; Szakal and Branzei 2013), it appears that checkpoint-mediated RF protection works also indirectly. As checkpoint activation restrains CDK activity, premature stimulation of Mus81 under replication stress conditions is avoided, which may protect replication intermediates from untimely cleavage. This is consistent with the observation in human cells that overexpression of CDC25A, a positive effector of CDK activity that is targeted for degradation by the S phase checkpoint, induces excessive processing of replication intermediates by MUS81–EME1 (Neelsen et al. 2013). Similarly, depletion of the WEE1 kinase, an inhibitor of cell cycle progression to mitosis, causes S phase-associated DNA damage, decreased RF progression, and accumulation of cells in S phase in a MUS81–EME1-dependent manner (Beck et al. 2010; Domínguez-Kelly et al. 2011). Treatment of U2OS cells with a small molecule WEE1 inhibitor (MK-1775) has been shown to result in loss of phosphorylation-dependent CDK1 inhibition and increased rates of DNA synthesis due to excessive replication initiation. As a result, cellular dNTP pools are being depleted, leading to replication stress. Four hours after addition of MK-1775, DNA breakage and concomitant accumulation of MUS81 in actively replicating chromatin was observed. Depletion of MUS81, and interestingly also of SLX4, was shown to curtail DNA breakage, suggesting that MUS81–EME1 in association with SLX4 targets stalled RFs under these conditions (Beck et al. 2012). Consistent with these studies, inhibition or knockdown of CHK1, a positive regulator of WEE1 and negative regulator of CDC25 (Perry and Kornbluth 2007), induces DSBs in human cells and inhibits progression through S phase (Forment et al. 2011; Syljuåsen et al. 2005). Both of these defects are ameliorated by depletion of MUS81 (Forment et al. 2011). CHK1 does not affect the chromatin association of MUS81, and although CHK1 can phosphorylate MUS81–EME1 in vitro, assessment of DNA cleavage (using a 3′-flap substrate) did not reveal any impact on the enzyme's nuclease activity (Forment et al. 2011). It is possible that the absence of CHK1 has a direct impact on the disposition of RFs that renders them more susceptible to nucleolytic attack and/or that the RF protection works indirectly through cell cycle kinase-mediated control of MUS81 activity. These observations highlight an important interplay between the replication/damage S phase checkpoint and cell cycle kinases that promotes normal replication dynamics and limits the actions of structure-specific nucleases on RFs, perhaps to low basal levels or situations of irreversible arrest.
Recovery of broken RFs
In E. coli, replication can be re-initiated from a RF-associated DSB end in a recombination-dependent manner (Fig. 3, steps 7–10). In eukaryotes, a similar process, termed break-induced replication (BIR), has been described (Lydeard et al. 2007; Llorente et al. 2008). Although this pathway has not been studied extensively in the context of broken RFs, analyses of recombination events in mammalian cells induced by the collision of RFs with DNA single-stranded breaks are compatible with BIR (Arnaudeau et al. 2001; Saleh-Gohari et al. 2005). Similarly, recovery of broken RFs in Xenopus laevis egg extracts has been shown to depend on a BIR-like process (Hashimoto et al. 2011). It is reasonable to assume that BIR is also coupled to events of structure-specific nuclease-mediated RF breakage. This could explain the apparent MUS81-dependent restart of preexisting RFs in mammalian cells after treatment with HU or CPT (Hanada et al. 2007; Regairaz et al. 2011; Ying et al. 2013), and the observation that deliberate RF cleavage under replication stress conditions can correlate with better survival. However, it has also been shown that DNA synthesis after prolonged RF stalling by HU is mediated to a large extent by new origin firing rather than reactivated RFs (Petermann et al. 2010). This raises the possibility that broken RFs are stabilized rather than channeled into BIR. Arrival of an oncoming RF would then generate a two-ended DSB, which may be repaired subsequent to bulk DNA synthesis. Such a strategy would be suitable for eukaryotic systems where a large excess of dormant origins is present in the genome, which can be activated to compensate for any shortfall in DNA synthesis caused by RF arrest (McIntosh and Blow 2012). This would also limit the use of BIR forks, which have recently been shown not to deliver the same replication accuracy as regular S phase RFs (Deem et al. 2011).
Conclusion and outlook
In recent years, it has become clear that eukaryotes possess multiple structure-specific nuclease including Mus81–Mms4/MUS81–EME1, Yen1/GEN1, and Slx1–Slx4/SLX1–SLX4, whose combined efforts, rather than their individual contributions, mirror the various actions of RuvC on branched recombination and replication intermediates (Fig. 5). All three resolve HJ-containing recombination intermediates in meiotic and mitotic DSB repair in yeast, while only Mus81–Mms4 and Yen1 have a well-documented role in processing replication-associated recombination intermediates. Slx1–Slx4 has been shown to promote rDNA replication, probably by targeting stalled RFs to facilitate termination, but while Slx4/SLX4 has other S phase roles, these appear largely independent of Slx1. Mus81–Mms4/MUS81–EME1 takes a prominent position as it is known to protect genome stability in unchallenged yeast and mammalian cells (Abraham et al. 2003; Dendouga et al. 2005; Hiyama et al. 2006; McPherson et al. 2004; Zhang et al. 2006). Symington and colleagues found that elevated chromosome mis-segregation in mus81 single mutant yeast is not entirely dependent upon HR, suggesting the phenotype may in part be caused by blocked RFs/unreplicated areas tethering together sister chromatids (Ho et al. 2010). A growing body of physical evidence supports the notion that Mus81–Mms4/MUS81–EME1 cuts branched replication structures such as idling or regressed RFs. However, most of this evidence has been gathered under conditions of massive exogenous DNA damage/replication stress or from cells in which fork protection and restart mechanisms were compromised. It will be interesting to see whether deliberate fork breakage represents an important pathway in the absence of exogenous replication stress in wild-type cells. Frequency of use might not be the yardstick to measure significance considering the detrimental effect that even one underreplicated chromosomal area might have at cell division, and it seems likely that situations will arise in which replication cannot be completed without deliberate RF breakage. Indeed, recent findings that MUS81 limits the number of reversed RFs detectable by electron microscopy under unperturbed conditions in human cells (Neelsen et al. 2013) and promotes DNA breakage at late-replicating fragile sites (Naim et al. 2013; Ying et al. 2013) suggest that RF cleavage is a normal part of every cell cycle.
Defects associated with Yen1 tend to transpire only in the absence of Mus81–Mms4 and it is therefore difficult to pinpoint Yen1-specific functions associated with S phase. However, the functional overlap with Mus81–Mms4 means that both enzymes are implicated in the same set of reactions. Since there are similarities and differences in their in vitro substrate spectra, it is likely that Yen1/GEN1 and Mus81–Mms4/MUS81–EME1 have evolved to perform overlapping as well as distinct tasks. Potential roles of GEN1 in higher eukaryotes have started to emerge, and recent observations appear to confirm a contribution of GEN1 to the resolution of recombination and/or aberrant replication structures. Thus, depletion of GEN1 in human cells causes mild sensitivity to CPT and MMC (Svendsen et al. 2009), the accumulation of spontaneous DNA damage (Gao et al. 2012), and the occurrence of DNA bridges between segregating chromosomes (Rodrigue et al. 2012). Unexpectedly, GEN1 depletion appears to cause centrosome overduplication/fragmentation and multipolar spindle formation in M phase (Gao et al. 2012; Rodrigue et al. 2012), suggesting novel functions that go beyond the resolution of branched DNA intermediates.
Finally, deliberate RF breakage by MUS81–EME1 can promote survival under conditions of endogenous replication stress, an emerging hallmark of early cancerous lesions (Halazonetis et al. 2008). Under these conditions, cells may depend on DSB-associated replication restart mechanisms involving structure-specific nucleases, suggesting that these may offer a point of intervention in the development of cancer.
Acknowledgments
I thank Steve West for communicating unpublished results and my colleagues Nicolas Thomä and Jeffrey Chao for suggestions on the manuscript. This work was supported by the Novartis Research Foundation.
References
- Abraham J, Lemmers B, Hande MP, et al. Eme1 is involved in DNA damage processing and maintenance of genomic stability in mammalian cells. EMBO J. 2003;22:6137–6147. doi: 10.1093/emboj/cdg580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achar YJ, Balogh D, Haracska L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc Natl Acad Sci U S A. 2011;108:14073–14078. doi: 10.1073/pnas.1101951108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agmon N, Yovel M, Harari Y, et al. The role of Holliday junction resolvases in the repair of spontaneous and induced DNA damage. Nucleic Acids Res. 2011;39:7009–7019. doi: 10.1093/nar/gkr277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- Alabert C, Bianco JN, Pasero P. Differential regulation of homologous recombination at DNA breaks and replication forks by the Mrc1 branch of the S-phase checkpoint. EMBO J. 2009;28:1131–1141. doi: 10.1038/emboj.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvino GM, Collingwood D, Murphy JM, et al. Replication in hydroxyurea: it's a matter of time. Mol Cell Biol. 2007;27:6396–6406. doi: 10.1128/MCB.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SL, Bergstralh DT, Kohl KP, et al. Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol Cell. 2009;35:128–135. doi: 10.1016/j.molcel.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307:1235–1245. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- Asai T, Bates DB, Kogoma T. DNA replication triggered by double-stranded breaks in E. coli: dependence on homologous recombination functions. Cell. 1994;78:1051–1061. doi: 10.1016/0092-8674(94)90279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton TM, Mankouri HW, Heidenblut A, et al. Pathways for Holliday junction processing during homologous recombination in Saccharomyces cerevisiae. Mol Cell Biol. 2011;31:1921–1933. doi: 10.1128/MCB.01130-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson J, McGlynn P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009;37:3475–3492. doi: 10.1093/nar/gkp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansbach CE, Bétous R, Lovejoy CA, et al. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Horejsí Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bastin-Shanower SA, Fricke WM, Mullen JR, Brill SJ. The mechanism of Mus81–Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1–Rad10. Mol Cell Biol. 2003;23:3487–3496. doi: 10.1128/MCB.23.10.3487-3496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Nähse V, Larsen MSY, et al. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J Cell Biol. 2010;188:629–638. doi: 10.1083/jcb.200905059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Nähse-Kumpf V, Larsen MSY, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–4236. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson FE, West SC. Substrate specificity of the Escherichia coli RuvC protein. Resolution of three- and four-stranded recombination intermediates. J Biol Chem. 1994;269:5195–5201. [PubMed] [Google Scholar]
- Benson F, Collier S, Lloyd RG. Evidence of abortive recombination in ruv mutants of Escherichia coli K12. Mol Gen Genet. 1991;225:266–272. doi: 10.1007/BF00269858. [DOI] [PubMed] [Google Scholar]
- Bester AC, Roniger M, Oren YS, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétous R, Mason AC, Rambo RP, et al. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais V, Gao H, Elwell CA, et al. RNA interference inhibition of Mus81 reduces mitotic recombination in human cells. Mol Biol Cell. 2004;15:552–562. doi: 10.1091/mbc.E03-08-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco MG, Matos J, Rass U, et al. Functional overlap between the structure-specific nucleases Yen1 and Mus81–Mms4 for DNA-damage repair in S. cerevisiae. DNA Repair (Amst) 2010;9:394–402. doi: 10.1016/j.dnarep.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Blastyák A, Pintér L, Unk I, et al. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol Cell. 2007;28:167–175. doi: 10.1016/j.molcel.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blastyák A, Hajdu I, Unk I, Haracska L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol. 2010;30:684–693. doi: 10.1128/MCB.00863-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy MN, Lopez-Girona A, Shanahan P, et al. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol Cell Biol. 2000;20:8758–8766. doi: 10.1128/mcb.20.23.8758-8766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy MN, Gaillard PH, McDonald WH, et al. Mus81–Eme1 are essential components of a Holliday junction resolvase. Cell. 2001;107:537–548. doi: 10.1016/s0092-8674(01)00536-0. [DOI] [PubMed] [Google Scholar]
- Bugreev DV, Mazina OM, Mazin AV. Rad54 protein promotes branch migration of Holliday junctions. Nature. 2006;442:590–593. doi: 10.1038/nature04889. [DOI] [PubMed] [Google Scholar]
- Bugreev DV, Rossi MJ, Mazin AV. Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 2011;39:2153–2164. doi: 10.1093/nar/gkq1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzymek M, Thayer NH, Oh SD, et al. Double Holliday junctions are intermediates of DNA break repair. Nature. 2010;464:937–941. doi: 10.1038/nature08868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Plank JL, Bachrati CZ, et al. Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1–Top3. Nat Struct Mol Biol. 2010;17:1377–1382. doi: 10.1038/nsmb.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci U S A. 2002;99:16934–16939. doi: 10.1073/pnas.262669299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XB, Melchionna R, Denis CM, et al. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol Cell. 2001;8:1117–1127. doi: 10.1016/s1097-2765(01)00375-6. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Constantinou A, West SC. Identification and characterization of the human Mus81–Eme1 endonuclease. J Biol Chem. 2003;278:25172–25178. doi: 10.1074/jbc.M302882200. [DOI] [PubMed] [Google Scholar]
- Ciccia A, McDonald N, West SC. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem. 2008;77:259–287. doi: 10.1146/annurev.biochem.77.070306.102408. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Bredemeyer AL, Sowa ME, et al. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Nimonkar AV, Hu Y, et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JA, Bjergbaek L, Shimada K, et al. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 2003;22:4325–4336. doi: 10.1093/emboj/cdg391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Miller KM, Maas NL, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806–810. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- Constantinou A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma. 2012;121:21–36. doi: 10.1007/s00412-011-0349-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinou A, Tarsounas M, Karow JK, et al. Werner's syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinou A, Chen X-B, McGowan CH, West SC. Holliday junction resolution in human cells: two junction endonucleases with distinct substrate specificities. EMBO J. 2002;21:5577–5585. doi: 10.1093/emboj/cdf554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon S, Gaillard P-HL, Chahwan C, et al. Slx1–Slx4 are subunits of a structure-specific endonuclease that maintains ribosomal DNA in fission yeast. Mol Biol Cell. 2004;15:71–80. doi: 10.1091/mbc.E03-08-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon S, Noguchi E, Noguchi C, et al. Rad22Rad52-dependent repair of ribosomal DNA repeats cleaved by Slx1–Slx4 endonuclease. Mol Biol Cell. 2006;17:2081–2090. doi: 10.1091/mbc.E05-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, et al. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- Crossan GP, van der Weyden L, Rosado IV, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet. 2011;43:147–152. doi: 10.1038/ng.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski KE, Howlett NG. FANCP/SLX4: a Swiss army knife of DNA interstrand crosslink repair. Cell Cycle. 2011;10:1757–1763. doi: 10.4161/cc.10.11.15818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Muyt A, Jessop L, Kolar E, et al. BLM helicase ortholog Sgs1 is a central regulator of meiotic recombination intermediate metabolism. Mol Cell. 2012;46:43–53. doi: 10.1016/j.molcel.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Piccoli G, Katou Y, Itoh T, et al. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol Cell. 2012;45:696–704. doi: 10.1016/j.molcel.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, et al. Break-induced replication is highly inaccurate. PLoS Biol. 2011;9:e1000594. doi: 10.1371/journal.pbio.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehé P-M, Coulon S, Scaglione S, et al. Regulation of Mus81–Eme1 Holliday junction resolvase in response to DNA damage. Nat Struct Mol Biol. 2013;20:598–603. doi: 10.1038/nsmb.2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dendouga N, Gao H, Moechars D, et al. Disruption of murine Mus81 increases genomic instability and DNA damage sensitivity but does not promote tumorigenesis. Mol Cell Biol. 2005;25:7569–7579. doi: 10.1128/MCB.25.17.7569-7579.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Diffley JFX. Quality control in the initiation of eukaryotic DNA replication. Philos Trans R Soc Lond B Biol Sci. 2011;366:3545–3553. doi: 10.1098/rstb.2011.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe CL, Ahn JS, Dixon J, Whitby MC (2002) Mus81–Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J Biol Chem 277:32753–32759. doi:10.1074/jbc.M202120200 [DOI] [PubMed]
- Domínguez-Kelly R, Martín Y, Koundrioukoff S, et al. Wee1 controls genomic stability during replication by regulating the Mus81–Eme1 endonuclease. J Cell Biol. 2011;194:567–579. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunin-Horkawicz S, Feder M, Bujnicki JM. Phylogenomic analysis of the GIY-YIG nuclease superfamily. BMC Genomics. 2006;7:98. doi: 10.1186/1471-2164-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- Ehmsen KT, Heyer WD. Saccharomyces cerevisiae Mus81–Mms4 is a catalytic, DNA structure-selective endonuclease. Nucleic Acids Res. 2008;36:2182–2195. doi: 10.1093/nar/gkm1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmsen KT, Heyer WD. A junction branch point adjacent to a DNA backbone nick directs substrate cleavage by Saccharomyces cerevisiae Mus81–Mms4. Nucleic Acids Res. 2009;37:2026–2036. doi: 10.1093/nar/gkp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico A, Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Crit Rev Biochem Mol Biol. 2012;47:222–235. doi: 10.3109/10409238.2012.655374. [DOI] [PubMed] [Google Scholar]
- Essers J, Hendriks RW, Swagemakers SM, et al. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 1997;89:195–204. doi: 10.1016/s0092-8674(00)80199-3. [DOI] [PubMed] [Google Scholar]
- Fabre F, Chan A, Heyer W-D, Gangloff S. Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci U S A. 2002;99:16887–16892. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekairi S, Scaglione S, Chahwan C, et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009;138:78–89. doi: 10.1016/j.cell.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Fingerhut R, Kiefer J, Otto F. Cell cycle parameters in radiation sensitive strains of Saccharomyces cerevisiae. Mol Gen Genet. 1984;193:192–194. doi: 10.1007/BF00327437. [DOI] [PubMed] [Google Scholar]
- Flott S, Rouse J. Slx4 becomes phosphorylated after DNA damage in a Mec1/Tel1-dependent manner and is required for repair of DNA alkylation damage. Biochem J. 2005;391:325. doi: 10.1042/BJ20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flott S, Alabert C, Toh GW, et al. Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol Cell Biol. 2007;27:6433–6445. doi: 10.1128/MCB.00135-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forment JV, Blasius M, Guerini I, Jackson SP. Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLoS ONE. 2011;6:e23517. doi: 10.1371/journal.pone.0023517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchitto A, Pirzio LM, Prosperi E, et al. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J Cell Biol. 2008;183:241–252. doi: 10.1083/jcb.200803173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke WM, Brill SJ. Slx1–Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1–Top3. Genes Dev. 2003;17:1768–1778. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke WM, Bastin-Shanower SA, Brill SJ. Substrate specificity of the Saccharomyces cerevisiae Mus81–Mms4 endonuclease. DNA Repair (Amst) 2005;4:243–251. doi: 10.1016/j.dnarep.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Froget B, Blaisonneau J, Lambert S, Baldacci G. Cleavage of stalled forks by fission yeast Mus81/Eme1 in absence of DNA replication checkpoint. Mol Biol Cell. 2008;19:445–456. doi: 10.1091/mbc.E07-07-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Xiao W. Functional domains required for the Saccharomyces cerevisiae Mus81–Mms4 endonuclease complex formation and nuclear localization. DNA Repair (Amst) 2003;2:1435–1447. doi: 10.1016/j.dnarep.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Fugger K, Kit Chu W, Haahr P et al (2013) FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nat Commun 4:1423. doi:10.1038/ncomms2395 [DOI] [PubMed]
- Gallo-Fernández M, Saugar I, Ortiz-Bazán MÁ, et al. Cell cycle-dependent regulation of the nuclease activity of Mus81–Eme1/Mms4. Nucleic Acids Res. 2012;40:8325–8335. doi: 10.1093/nar/gks599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Chen X-B, McGowan CH. Mus81 endonuclease localizes to nucleoli and to regions of DNA damage in human S-phase cells. Mol Biol Cell. 2003;14:4826–4834. doi: 10.1091/mbc.E03-05-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Rendtlew Danielsen J, Wei L-Z, et al. A novel role of human Holliday junction resolvase GEN1 in the maintenance of centrosome integrity. PLoS ONE. 2012;7:e49687. doi: 10.1371/journal.pone.0049687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Décaillet C, Delannoy M, et al. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci U S A. 2008;105:16107–16112. doi: 10.1073/pnas.0804777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Décaillet C, Stasiak AZ, et al. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell. 2008;29:141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- Ghosal G, Chen J. DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res. 2013;2:107–129. doi: 10.3978/j.issn.2218-676X.2013.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou L-VF, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Haber JE, Heyer WD. The fuss about Mus81. Cell. 2001;107:551–554. doi: 10.1016/s0092-8674(01)00593-1. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Hanada K, Budzowska M, Modesti M, et al. The structure-specific endonuclease Mus81–Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 2006;25:4921–4932. doi: 10.1038/sj.emboj.7601344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Budzowska M, Davies SL, et al. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol. 2007;14:1096–1104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Puddu F, Costanzo V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat Struct Mol Biol. 2011;19:17–24. doi: 10.1038/nsmb.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol. 2006;7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- Higgins NP, Kato K, Strauss B. A model for replication repair in mammalian cells. J Mol Biol. 1976;101:417–425. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- Hiyama T, Katsura M, Yoshihara T, et al. Haploinsufficiency of the Mus81–Eme1 endonuclease activates the intra-S-phase and G2/M checkpoints and promotes rereplication in human cells. Nucleic Acids Res. 2006;34:880–892. doi: 10.1093/nar/gkj495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CK, Mazón G, Lam AF, Symington LS. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol Cell. 2010;40:988–1000. doi: 10.1016/j.molcel.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- Holliday R. A mechanism for gene conversion in fungi. Genet Res Camb. 1964;5:282–304. doi: 10.1017/S0016672308009476. [DOI] [PubMed] [Google Scholar]
- Ip SCY, Rass U, Blanco MG, et al. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008;456:357–361. doi: 10.1038/nature07470. [DOI] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, et al. Srs2 and Sgs1–Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RM, Mortusewicz O, Afzal I, et al. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene. 2012 doi: 10.1038/onc.2012.387. [DOI] [PubMed] [Google Scholar]
- Kai M, Boddy MN, Russell P, et al. Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes Dev. 2005;19:919–932. doi: 10.1101/gad.1304305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliraman V, Brill SJ. Role of SGS1 and SLX4 in maintaining rDNA structure in Saccharomyces cerevisiae. Curr Genet. 2002;41:389–400. doi: 10.1007/s00294-002-0319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliraman V, Mullen JR, Fricke WM, et al. Functional overlap between Sgs1–Top3 and the Mms4–Mus81 endonuclease. Genes Dev. 2001;15:2730–2740. doi: 10.1101/gad.932201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karow JK, Constantinou A, Li JL, et al. The Bloom's syndrome gene product promotes branch migration of Holliday junctions. Proc Natl Acad Sci U S A. 2000;97:6504–6508. doi: 10.1073/pnas.100448097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SR, Kuzminov A. Replication forks stalled at ultraviolet lesions are rescued via RecA and RuvABC protein-catalyzed disintegration in Escherichia coli. J Biol Chem. 2012;287:6250–6265. doi: 10.1074/jbc.M111.322990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lach FP, Desetty R, et al. Mutations of the SLX4 gene in Fanconi anemia. Nat Genet. 2011;43:142–146. doi: 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Spitz GS, Veturi U, et al. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood. 2013;121:54–63. doi: 10.1182/blood-2012-07-441212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev. 1997;61:212–238. doi: 10.1128/mmbr.61.2.212-238.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Froget B, Carr AM. Arrested replication fork processing: interplay between checkpoints and recombination. DNA Repair (Amst) 2007;6:1042–1061. doi: 10.1016/j.dnarep.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Liu Y, West SC. Happy Hollidays: 40th anniversary of the Holliday junction. Nat Rev Mol Cell Biol. 2004;5:937–944. doi: 10.1038/nrm1502. [DOI] [PubMed] [Google Scholar]
- Llorente B, Smith CE, Symington LS. Break-induced replication: what is it and what is it for? Cell Cycle. 2008;7:859–864. doi: 10.4161/cc.7.7.5613. [DOI] [PubMed] [Google Scholar]
- Lloyd RG. Conjugational recombination in resolvase-deficient ruvC mutants of Escherichia coli K-12 depends on recG. J Bacteriol. 1991;173:5414–5418. doi: 10.1128/jb.173.17.5414-5418.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG, Benson FE, Shurvinton CE. Effect of ruv mutations on recombination and DNA repair in Escherichia coli K12. Mol Gen Genet. 1984;194:303–309. doi: 10.1007/BF00383532. [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, et al. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- Lucca C, Vanoli F, Cotta-Ramusino C, et al. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene. 2004;23:1206–1213. doi: 10.1038/sj.onc.1207199. [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE (2007) Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448:820–823. doi:10.1038/nature06047 [DOI] [PubMed]
- Machwe A, Xiao L, Groden J, Orren DK. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006;45:13939–13946. doi: 10.1021/bi0615487. [DOI] [PubMed] [Google Scholar]
- Machwe A, Karale R, Xu X, et al. The Werner and Bloom syndrome proteins help resolve replication blockage by converting (regressed) Holliday junctions to functional replication forks. Biochemistry. 2011;50:6774–6788. doi: 10.1021/bi2001054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos J, Blanco MG, Maslen S, et al. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011;147:158–172. doi: 10.1016/j.cell.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos J, Blanco MG, West SC. Cell-cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Reports. 2013;4:1–11. doi: 10.1016/j.celrep.2013.05.039. [DOI] [PubMed] [Google Scholar]
- Mazón G, Mimitou EP, Symington LS (2010) SnapShot: homologous recombination in DNA double-strand break repair. Cell 142:646, 646.e1. doi:10.1016/j.cell.2010.08.006 [DOI] [PubMed]
- McGlynn P, Lloyd RG. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc Natl Acad Sci U S A. 2001;98:8227–8234. doi: 10.1073/pnas.111008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P, Lloyd RG. Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol. 2002;3:859–870. doi: 10.1038/nrm951. [DOI] [PubMed] [Google Scholar]
- McGlynn P, Lloyd RG, Marians KJ. Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc Natl Acad Sci U S A. 2001;98:8235–8240. doi: 10.1073/pnas.121007798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh D, Blow JJ. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb Perspect Biol. 2012;4:a012955. doi: 10.1101/cshperspect.a012955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson JP, Lemmers B, Chahwan R, et al. Involvement of mammalian Mus81 in genome integrity and tumor suppression. Science. 2004;304:1822–1826. doi: 10.1126/science.1094557. [DOI] [PubMed] [Google Scholar]
- Michel B, Boubakri H, Baharoglu Z, et al. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst) 2007;6:967–980. doi: 10.1016/j.dnarep.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics. 2000;157:103–118. doi: 10.1093/genetics/157.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz IM, Hain K, Déclais A-C, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell. 2009;35:116–127. doi: 10.1016/j.molcel.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Muñoz-Galván S, Tous C, Blanco MG, et al. Distinct roles of Mus81, Yen, 1, Slx1–Slx4 and Rad1 nucleases in the repair of replication-born double strand breaks by sister chromatid exchange. Mol Cell Biol. 2012;32:1592–1603. doi: 10.1128/MCB.00111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murfuni I, De Santis A, Federico M, et al. Perturbed replication induced genome wide or at common fragile sites is differently managed in the absence of WRN. Carcinogenesis. 2012;33:1655–1663. doi: 10.1093/carcin/bgs206. [DOI] [PubMed] [Google Scholar]
- Murfuni I, Nicolai S, Baldari S, et al. The WRN and MUS81 proteins limit cell death and genome instability following oncogene activation. Oncogene. 2013;32:610–620. doi: 10.1038/onc.2012.80. [DOI] [PubMed] [Google Scholar]
- Naim V, Wilhelm T, Debatisse M, Rosselli F (2013) ERCC1 and MUS81–EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol 15:1008–1015. doi:10.1038/ncb2793 [DOI] [PubMed]
- Neelsen KJ, Zanini IMY, Herrador R, Lopes M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol. 2013;200:699–708. doi: 10.1083/jcb.201212058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, de Oliveira FMB, Almeida BS, Smolka MB. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell. 2010;39:300–306. doi: 10.1016/j.molcel.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Liu Y, et al. DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature. 2012;493:120–124. doi: 10.1038/nature11658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman F, Whitby MC. Exploring the roles of Mus81–Eme1/Mms4 at perturbed replication forks. DNA Repair (Amst) 2007;6:1004–1017. doi: 10.1016/j.dnarep.2007.02.019. [DOI] [PubMed] [Google Scholar]
- Osman F, Dixon J, Doe CL, Whitby MC. Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81–Eme1 in meiosis. Mol Cell. 2003;12:761–774. doi: 10.1016/s1097-2765(03)00343-5. [DOI] [PubMed] [Google Scholar]
- Pan X, Ye P, Yuan DS, et al. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006;124:1069–1081. doi: 10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons AB, Brost RL, Ding H, et al. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 2004;22:62–69. doi: 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- Perry JA, Kornbluth S. Cdc25 and Wee1: analogous opposites? Cell Div. 2007;2:12. doi: 10.1186/1747-1028-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol. 2010;11:683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, et al. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem. 2009;284:35951–35961. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralf C, Hickson ID, Wu L. The Bloom's syndrome helicase can promote the regression of a model replication fork. J Biol Chem. 2006;281:22839–22846. doi: 10.1074/jbc.M604268200. [DOI] [PubMed] [Google Scholar]
- Rass U, Compton SA, Matos J, et al. Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev. 2010;24:1559–1569. doi: 10.1101/gad.585310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, et al. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- Regairaz M, Zhang Y-W, Fu H, et al. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J Cell Biol. 2011;195:739–749. doi: 10.1083/jcb.201104003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TM, Kobor MS, Bastin-Shanower et al (2005) Slx4 regulates DNA damage checkpoint-dependent phosphorylation of the BRCT domain protein Rtt107/Esc4. Mol Biol Cell 17:539–548. doi:10.1091/mbc.E05-08-0785 [DOI] [PMC free article] [PubMed]
- Rodrigue A, Coulombe Y, Jacquet K, et al. The RAD51 paralogs ensure cellular protection against mitotic defects and aneuploidy. J Cell Sci. 2012 doi: 10.1242/jcs.114595. [DOI] [PubMed] [Google Scholar]
- Saintigny Y, Delacôte F, Varès G, et al. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. doi: 10.1093/emboj/20.14.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito TT, Mohideen F, Meyer K, et al. SLX-1 is required for maintaining genomic integrity and promoting meiotic noncrossovers in the Caenorhabditis elegans germline. PLoS Genet. 2012;8:e1002888. doi: 10.1371/journal.pgen.1002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh-Gohari N, Bryant HE, Schultz N, et al. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol. 2005;25:7158–7169. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salewsky B, Schmiester M, Schindler D, et al. The nuclease hSNM1B/Apollo is linked to the Fanconi anemia pathway via its interaction with FANCP/SLX4. Hum Mol Genet. 2012;21:4948–4956. doi: 10.1093/hmg/dds338. [DOI] [PubMed] [Google Scholar]
- Schwartz EK, Heyer W-D. Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma. 2011;120:109–127. doi: 10.1007/s00412-010-0304-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz EK, Wright WD, Ehmsen KT, et al. Mus81–Mms4 functions as a single heterodimer to cleave nicked intermediates in recombinational DNA repair. Mol Cell Biol. 2012;32:3065–3080. doi: 10.1128/MCB.00547-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segurado M, Tercero JA. The S-phase checkpoint: targeting the replication fork. Biol Cell. 2009;101:617–627. doi: 10.1042/BC20090053. [DOI] [PubMed] [Google Scholar]
- Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- Seigneur M, Ehrlich SD, Michel B. RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol Microbiol. 2000;38:565–574. doi: 10.1046/j.1365-2958.2000.02152.x. [DOI] [PubMed] [Google Scholar]
- Sharples GJ, Benson FE, Illing GT, Lloyd RG. Molecular and functional analysis of the ruv region of Escherichia coli K-12 reveals three genes involved in DNA repair and recombination. Mol Gen Genet. 1990;221:219–226. doi: 10.1007/BF00261724. [DOI] [PubMed] [Google Scholar]
- Shimura T, Torres MJ, Martin MM, et al. Bloom's syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J Mol Biol. 2008;375:1152–1164. doi: 10.1016/j.jmb.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura T, Kakuda S, Ochiai Y, et al. Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3beta-mediated cyclin D1 overexpression. Oncogene. 2010;29:4826–4837. doi: 10.1038/onc.2010.238. [DOI] [PubMed] [Google Scholar]
- Shimura T, Ochiai Y, Noma N, et al. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle. 2013;12:773–782. doi: 10.4161/cc.23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Buerstedde JM, et al. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoepker C, Hain K, Schuster B, et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet. 2011;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- Su X, Bernal JA, Venkitaraman AR. Cell-cycle coordination between DNA replication and recombination revealed by a vertebrate N-end rule degron-Rad51. Nat Struct Mol Biol. 2008;15:1049–1058. doi: 10.1038/nsmb.1490. [DOI] [PubMed] [Google Scholar]
- Sun W, Nandi S, Osman F, et al. The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol Cell. 2008;32:118–128. doi: 10.1016/j.molcel.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen JM, Smogorzewska A, Sowa ME, et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138:63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syljuåsen RG, Sørensen CS, Hansen LT, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–3562. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakal B, Branzei D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J. 2013;32:1155–1167. doi: 10.1038/emboj.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- Takahagi M, Iwasaki H, Shinagawa H. Structural requirements of substrate DNA for binding to and cleavage by RuvC, a Holliday junction resolvase. J Biol Chem. 1994;269:15132–15139. [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Tachiiri S, et al. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol. 2001;21:2858–2866. doi: 10.1128/MCB.21.8.2858-2866.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay YD, Wu L. Overlapping roles for Yen1 and Mus81 in cellular Holliday junction processing. J Biol Chem. 2010;285:11427–11432. doi: 10.1074/jbc.M110.108399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson CG, Atack JM, Chapados B, et al. Substrate recognition and catalysis by flap endonucleases and related enzymes. Biochem Soc Trans. 2010;38:433. doi: 10.1042/BST0380433. [DOI] [PubMed] [Google Scholar]
- Versini G, Comet I, Wu M, et al. The yeast Sgs1 helicase is differentially required for genomic and ribosomal DNA replication. EMBO J. 2003;22:1939–1949. doi: 10.1093/emboj/cdg180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AT, Sengerová B, Cattell E, et al. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011;25:1859–1870. doi: 10.1101/gad.15699211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SC. Processing of recombination intermediates by the RuvABC proteins. Annu Rev Genet. 1997;31:213–244. doi: 10.1146/annurev.genet.31.1.213. [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- Ying S, Minocherhomji S, Chan KL et al (2013) MUS81 promotes common fragile site expression. Nat Cell Biol 15:1001–1007. doi:10.1038/ncb2773 [DOI] [PubMed]
- Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–2399. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, Kadonaga JT. HARP is an ATP-driven annealing helicase. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, Kong X, Yokomori K, Kadonaga JT. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009;23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharyevich K, Tang S, Ma Y, Hunter N. Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell. 2012;149:334–347. doi: 10.1016/j.cell.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Sengupta S, Yang Q, et al. BLM helicase facilitates Mus81 endonuclease activity in human cells. Cancer Res. 2005;65:2526–2531. doi: 10.1158/0008-5472.CAN-04-2421. [DOI] [PubMed] [Google Scholar]
- Zhang C, Roberts TM, Yang J, et al. Suppression of genomic instability by SLX5 and SLX8 in Saccharomyces cerevisiae. DNA Repair (Amst) 2006;5:336–346. doi: 10.1016/j.dnarep.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Zheng X-F, Prakash R, Saro D, et al. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein. DNA Repair (Amst) 2011;10:1034–1043. doi: 10.1016/j.dnarep.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]