

Abstract
Ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist, has well-described rapid antidepressant effects in clinical studies of individuals with treatment-resistant major depressive disorder (MDD). Preclinical studies investigating the effects of ketamine on brain-derived neurotrophic factor (BDNF) and on sleep slow wave activity (SWA) support its use as a prototype for investigating the neuroplastic mechanisms presumably involved in the mechanism of rapidly acting antidepressants. This review discusses human EEG slow wave sleep parameters and plasma BDNF as central and peripheral surrogate markers of plasticity, and their use in assessing ketamine’s effects. Acutely, ketamine elevates BDNF levels, as well as early night SWA and high-amplitude slow waves; each of these measures correlates with change in mood in depressed patients who respond to ketamine. The slow wave effects are limited to the first night post-infusion, suggesting that their increase is part of an early cascade of events triggering improved mood. Increased total sleep and decreased waking occur during the first and second night post infusion, suggesting that these measures are associated with the enduring treatment response observed with ketamine.
Keywords: Ketamine, NMDA antagonist, Sleep, Major depressive disorder, MDD, Synaptic plasticity, Slow wave sleep, Slow wave activity, SWA, AMPA, Mood, Rapid antidepressant response, Neurotrophic factors, Brain-derived neurotrophic factor, BDNF, Treatment-resistant depression, Bipolar depression, Sleep disorders, Psychiatry
Introduction
Delayed onset of antidepressant action is a major limitation of existing antidepressant therapies. Several large effectiveness studies conducted in patients with major depressive disorder (MDD) indicate that less than 40 % of patients treated with standard antidepressants—either mono- or combined therapy—achieved remission within 10–14 weeks [1–3], thus highlighting the need for novel treatment approaches. Novel treatment strategies as well as greater understanding of their molecular underpinnings might be identified by examining the molecular mechanisms that mediate the antidepressant effects of existing rapid-acting interventions such as sleep deprivation (SD), ketamine, scopolamine and electroconvulsive therapy (ECT) [4–8].
Given that SD and ketamine enhance slow wave effects, an initial approach for uncovering the molecular underpinnings of these rapid antidepressant treatments might include examining the effects of ketamine and other rapidly acting drugs on electroencephalographic (EEG) sleep measures and brain-derived neurotrophic factor (BDNF), which are central and peripheral putative surrogate biomarkers of synaptic plasticity. Key to the themes explored in this review, sleep interventions (e.g., SD) have been linked to synaptic plasticity, as suggested by increased sleep slow wave activity (SWA) and slow wave amplitude during recovery sleep. Further, both SD and ketamine increase the EEG SWA range (1–4 Hz) during non-REM (NREM) sleep and increase levels of BDNF [9]. Thus, these interventions may involve mechanisms key to understanding novel rapid antidepressant mechanisms.
Since the publication of the initial study identifying ketamine’s rapid antidepressant effects [4], numerous preclinical and clinical studies have examined its cellular and molecular effects as well as which clinical populations respond to ketamine, which drugs and doses are most effective, and what interventions might be used to delay relapse. Notably, clinical trials found that a single dose of sub-anesthetic ketamine induces a rapid (within 2 h) and sustained (1 to 2 weeks) antidepressant effect in patients with treatment-resistant MDD [10, 11] as well as bipolar depression [10, 12]. Additional glutamatergic plasticity-enhancing agents have been studied for the treatment of mood disorders, including subunit selective NMDA antagonists, glutamate release-reducing agents, and (2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid) (AMPA) potentiators. Evidence suggests that all of these agents have potential as fast-acting antidepressants. Further, preclinical studies using different behavioral paradigms have similarly shown that the noncompetitive NMDA receptor antagonist ketamine has robust antidepressant properties [13, 14]. Combining these behavioral findings with those from cellular and molecular studies has significantly advanced our understanding of ketamine’s antidepressant properties.
The purpose of this article is to briefly review the cellular and molecular mechanisms of ketamine’s rapid antidepressant effects while highlighting the association between biomarkers of synaptic plasticity, sleep slow waves, and BDNF. The article will also discuss recent findings of current clinical investigations of ketamine’s rapid antidepressant effects on BDNF and sleep slow waves in MDD and bipolar depression.
Ketamine: Cellular and Molecular Mechanisms
Ketamine exerts its initial rapid antidepressant properties via the NMDA receptor blockade and a prolonged change in glutamatergic signaling downstream of the NMDA blockade, ultimately resulting in increased synaptic strength and plasticity. Current research efforts have focused on uncovering the sequence of events that lead from blockade to rapid improvement in mood, cognition and behavior. Experiments investigating the role of glutamatergic transmission indicate that blockade of NMDA receptor-induced firing of gamma aminobutyric acid (GABA)ergic interneurons disinhibits glutamatergic pyramidal cells [15], thus increasing glutamate release and activation of AMPA receptors, and culminating in an activity-dependent release of BDNF [13, 16••]. Ultimately changes in glutamatergic transmission activate the mammalian target of rapamycin (mTOR) signaling pathway and affect downstream structural changes in dendritic spines and local synaptic protein synthesis, including BDNF [17•]. BDNF secretion, activation of the tropomyosin-receptor-kinase B (TrkB) receptor, and downstream trafficking lead to further dendritic structural complexity, spine and BDNF synthesis, and synaptic plasticity. The functional effects of BDNF trafficking are compromised by polymorphisms such as BDNF Val66Met [18, 19]. Ultimately changes in critical local neuronal circuits converge via enhanced synaptic plasticity and neuronal synchronization, especially in areas involved in mood and behavior, to produce rapid antidepressant effects [11, 13].
Notable in these observations is that manipulation of many steps in this signaling cascade either replicate or reverse the effects of ketamine in various rodent models of depression. For instance, the multi-day antidepressant effects of ketamine, as well as the synaptic spine production associated with its use, are blocked by rapamycin, which prevents activation of the mTOR pathway [13, 16••]. In addition, the effects of chronic unpredictable stress (decreased spine density, depressive-like behavior) are known to be reversed with ketamine [20]; rapamycin similarly blocks ketamine’s effects. An alternative interpretation is that the blockade of spontaneous (rather than evoked) glutamatergic activation of NMDA receptor blockade is critical to ketamine’s mechanism of action [21••], resulting in the inability to activate eukaryotic elongation factor 2 (eEF2) kinase, blunting of eFF2 phosphorylation, and BDNF derepression [22]. Further research is needed to discriminate between these mechanisms. Finally, inhibition of rapid (30 min) BDNF synthesis by anisomycin after ketamine treatment prevented the long-term behavioral effects of ketamine measured with the forced swim test [21••].
Sleep and Plasticity
Demonstrating increased synaptic plasticity in humans following ketamine administration is a challenging undertaking, especially in a clinical population of patients with severe treatment-resistant depression. However, work from the Tononi laboratory at the University of Wisconsin suggests that some electrophysiological measures—such as sleep EEG and evoked potentials—are reliable indicators of increased synaptic plasticity in humans. In fact, high-density EEG studies have shown that manipulations leading to synaptic potentiation in local cortical circuits [rotation learning, high-frequency transcranial magnetic stimulation (TMS)] lead to a local increases in SWA during subsequent sleep [23]; in contrast, manipulations leading to synaptic depression (arm immobilization) lead to a local reduction in SWA [24]. Large-scale computer simulations, validated by experimental studies in both rats and animals, have demonstrated that sleep SWA directly reflects synaptic strength due to changes in neural synchronization and recruitment [25, 26]. Moreover, these studies have shown that the slopes of sleep slow waves represent a highly sensitive marker of synaptic strength in underlying circuits [27].
Several studies that directly examined the effect of BDNF on EEG sleep slow waves have also noted a close relationship between SWA and BDNF [9, 28]. Interestingly, these studies found that SWA is increased by intrahemispheric infusion of BDNF, diminished by BDNF antagonism [9], and increased by behavioral interventions that increase central levels of BDNF [28] as well as the plasticity-related genes Arc, Homer, and NGFI-A [28]. Acoustic suppression of SWA activity and its capacity to diminish perceptual learning [29] further supports the possibility that decreased levels of slow wave sleep per se may contribute to cognitive and memory deficits in some depressed patients. Recent clinical studies have established another link between BDNF, sleep slow wave activity, and mood by showing that human carriers of the BDNF Met allele of the Val66Met polymorphism have reduced production of sleep slow waves [30]. Another study found that individuals with this polymorphism were less likely to respond to ketamine than the Val/Val allele [31].
Sleep Deprivation (And Other Sleep Interventions) as Rapidly Acting Interventions and Extenders of Remission
The robust and rapid antidepressant efficacy of SD therapy underlies its continued clinical and research use for over 5 decades [32, 33]. SD leads to increased slow wave sleep during recovery sleep, suggesting that the deficient production of sleep slow waves in many patients with depression may be part of a pathology that can be briefly reversed by the homeostatic processes activated by SD [34]. More recently, the synaptic homeostasis hypothesis [35], which extends the two-process model of sleep regulation described above [36], has provided a conceptual and cellular framework to understand the possible molecular underpinnings of SD therapy. Specifically, SD is associated with increased neurotrophic factors such as BDNF and VEGF [37, 38] as well as with synaptic plasticity and rapid remission of depression; notably, many of the same effects are observed with ketamine treatment. However, it is currently not known whether response to SD therapy predicts response to ketamine, which would provide support for a common mechanism underlying rapid antidepressant effects.
Interestingly, while SD therapy and ketamine treatment both exert rapid antidepressant effects, these interventions differ with regard to course of remission and relapse. One distinction concerns the depressogenic role of sleep in relapse, a core problem with SD therapy. After the rapid response to SD therapy, sleep per se contributes to relapse, an effect often seen following the first night of recovery sleep, or possibly sooner following a daytime nap. In contrast, the rapid antidepressant effects of ketamine are not rapidly or robustly reversed by sleep. However, careful evaluation of this point is needed since a differential response to recovery sleep would suggest that SD and ketamine differ with respect to synaptic downscaling, the process associated with restorative function of sleep, sleep homeostasis [35], and the course of remission.
Relatedly, time to relapse can be rapid (<1 day) after SD therapy, but often extends past 4 to 7 days in those patients who respond to a single infusion of ketamine. Several adjunct interventions [lithium, selective serotonin reuptake inhibitors (SSRIs), phototherapy, chronotherapy] have been shown to prolong response to SD therapy [39–41]. While repeated ketamine infusions extend the antidepressant response [42–44], concurrent treatment with riluzole, a presynaptic inhibitor of glutamate release and enhancer of AMPA trafficking and glial glutamate reuptake, does not alter the course of remission [45, 46]. Another study noted that treatment with the mood stabilizers lithium or valproate did not alter the relapse rate in individuals with bipolar depression treated with ketamine [10], but it remains unknown whether they may extend the interval to relapse.
The contribution of sleep in affecting the response to ketamine has not been directly evaluated. Microsleep episodes [47] as well as nighttime sleep restriction [48] have been shown to blunt and extend response to SD therapy [47] respectively, but similar research with ketamine has not been conducted. Taken together with the fact that altered glutamate transmission contributes to the rapid antidepressant mechanism of both SD therapy [40, 49, 50] and ketamine [13, 16••, 21••], suggests that sleep restriction and careful management of sleep after ketamine may prolong remission by affecting glutamatergic transmission.
Effects of Ketamine on Sleep and Slow Waves in Treatment-Resistant MDD
Earlier studies indicated that the NMDA antagonists ketamine [51] and MK-801 [52] increased SWA. Given the rapid anti-depressant effects associated with SD, as well as its ability to increase neurotrophins and SWA, we hypothesized that similar changes might also be associated with rapid mood change after ketamine infusion. To test the hypothesis that ketamine increased synaptic strength in conjunction with its rapid effects on mood, sleep slow waves (a putative central marker of plasticity) and BDNF (a peripheral marker of plasticity) were measured [53•]. Montgomery-Åsberg Depression Rating Scale (MADRS) scores rapidly decrease and remain low for several days following ketamine treatment in patients with treatment-resistant MDD. In our recent clinical investigation of ketamine and sleep, plasma BDNF levels, SWA, and high amplitude waves increased after ketamine [53•]. SWA and amplitude effects were increased during early sleep (first nREM period). These slow wave effects were limited to the first night after ketamine infusion. Importantly, the first-night EEG slow wave changes were accompanied by an increase in slow wave slope, consistent with increased synaptic strength [27]. Further, in those patients who responded to ketamine (defined as a greater than 50 % reduction in MADRS score 230 min after infusion), changes in BDNF levels were proportional to changes in EEG slow wave parameters, suggesting that enhanced synaptic plasticity is part of the physiological mechanism underlying the rapid antidepressant effects of NMDA antagonists.
Ketamine’s effects on sleep EEG were specific to low frequencies corresponding to the SWA range. Further, ketamine did not increase SWA in waking epochs prior to sleep onset, indicating that the change was sleep-specific. Negligible effects of the ketamine infusion on sleep EEG were measured in bands corresponding to sleep spindles, and alpha or theta frequencies [53•].
In addition to significant ketamine-induced increases in levels of BDNF, SWA, high amplitude waves, and slope, the observed correlations between BDNF levels and slow wave parameters indicate a strong relationship between these measures. Specifically, the positive correlations between changes in logBDNF and changes in relative SWA, as well as between changes in logBDNF and the incidence of high amplitude sleep slow waves, are consistent with the association between these markers of synaptic plasticity [9, 28]. Interestingly, most patients classified as ketamine responders had low levels of SWA and BDNF at baseline, followed by large increases in both of these measures after ketamine infusion [53•]. Indeed, the low baseline production of SWA during the first nREM period predicted later response to ketamine in responders [54], suggesting that lower brain plasticity is a marker of ketamine response, which is reversed by ketamine in responders [53•, 55].
Significant day-to-day changes in sleep variables appear to provide markers of an underlying sequence of signaling cascades associated with immediate (day one) and continued (day 2) mood response [53•] involving synaptic plasticity and BDNF. Of special interest is the observation that ketamine-induced increases in sleep slow waves were confined to the first night after treatment but were then decreased toward baseline levels on night two (Fig. 1). The timing of a day 1 increase in slow waves, followed by a decline toward baseline levels on day 2, is consistent with preclinical findings of Autry et al. [21••] of an acute day 1 (30 min) increase in BDNF levels, followed by a day 2 decrease, and may be particularly important given the previously described association between BDNF and slow wave production [9, 28]. Thus, the decline of sleep slow waves on day 2 would be consistent with a decline in BDNF levels. However, because BDNF was not measured in the clinical study, this point requires further investigation.
Fig. 1.
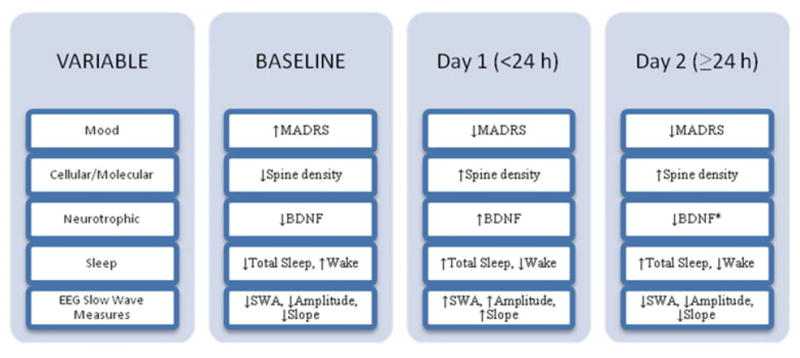
A schematic model of acute and subchronic (day 1 and day 2) ketamine-induced changes in mood, molecular, sleep, and slow-wave variables in MDD with treatment-resistant depression. Note the parallel change of BDNF and EEG slow wave measures on day 1 and 2, as well as the parallel change of mood and sleep measures on day 1 and 2. Baseline refers to effects prior to ketamine intervention. Day 1 refers to the interval within 24 h after ketamine treatment. ‘Day 1’ clinical sleep changes are measured over 8 h on the first night after ketamine. ‘Day 2’ sleep changes are measured on the second night after ketamine. ‘Day 1’ and ‘Day 2’ EEG sleep slow wave effects are measured during the first non-REM sleep period of night 1 and 2. The mood, sleep, and EEG slow wave measures are based on clinical findings [53•]. The cellular/molecular measures are based on preclinical findings [16••, 17•]. BDNF BL and Day 1 measures are based on both clinical [53•] and preclinical [21••] findings; the BDNF Day 2 measure is based on preclinical [21••] findings
It is also important to note that there was an increase in total sleep and a decrease in waking during both the first and second nights following ketamine infusion, indicating that persisting changes related to sleep consolidation may contribute to continued antidepressant mood effects. Overall, the increase in sleep slow waves appears to be a marker of the acute increase in BDNF and rapid antidepressant effects, whereas improved sleep quality is associated with extended mood response. Studies are ongoing to identify additional mediators of the extended mood response.
Effect of Ketamine on Sleep and Slow Waves in Patients with Bipolar Disorder Treated with Mood Stabilizers
Sleep slow waves were examined in a group of patients with bipolar depression (n=15, mean baseline MADRS=34.5) during a baseline night and on the first night after ketamine infusion; these patients were also receiving maintenance doses of the mood stabilizers lithium (n=11) or valproate (n=4). Baseline (BL) sleep measures were similar between BP and MDD [Total Sleep Time=374.5±14.2; 368.1±10.4; Wake%=11.65±3.0; 11.14±2.1; REM%=18.1±1.7; 25.3±1.3; Slow Wave Sleep%=5.2±1.4; 3.5±0.6; (BP, MDD, respectively, mean±SEM)].
Although MADRS scores rapidly decreased after ketamine infusion in BP, relative to BL sleep measures, ketamine had little effect on standard measures of Total Sleep (BL=374.5±14.2; Day 1=406.2±14.7; p=0.13; mean±SEM), Wake % (BL=11.6±3.1; Day 1=9.8±2.2; p=0.62), REM % (BL=18.1±1.7; Day 1=21.3±2.1; p=0.26) and Slow Wave Sleep% (BL=5.2±1.4; Day 1=5.2±1.6; p=0.74).
Importantly, ketamine effects on slow wave activity differed from patients with MDD. In contrast to the ketamine-induced increases in sleep slow waves described above in individuals with MDD, ketamine did not increase EEG sleep slow waves relative to baseline in individuals with bipolar depression [56]. Spectral and slow wave analysis showed that ketamine had no effect on slow wave power or the amplitude distribution of slow waves, and no correlation was observed between high (>80 μV) amplitude wave count and MADRS score. Interestingly in BP, ketamine decreased early (nREM1) relative SWA (BL nR1=1.15±0.1; Day1 nR1=0.98±0.1; BL nR2=1.14±0.08; Day 1=1.21±0.09; BL nR3=0.9±0.07; Day 1=0.8±0.07; mean±SEM). Several factors might contribute to this discrepancy between BP and MDD. First, individuals with bipolar depression and MDD may differ in sleep slow wave responses to ketamine because of biological differences in neuroplastic and sleep homeostatic mechanisms (i.e., our BP patients tended to have greater SWS than the MDD). Secondly, suppression of SWA is also associated with an antidepressant response [57]. Thus, in BP, the ketamine-induced decrease in early SWA may be related to antidepressant effects in BP, but further research is required to evaluate this possibility, as well as the association among BDNF, SWA, and mood. Alternatively, maintenance doses of lithium or valproate may have altered the response to ketamine. Both lithium and valproate are known to increase slow wave sleep and enhance plasticity [58–63], as well as inhibit glycogen synthase kinase-3 (GSK3) [64], which is necessary for ketamine’s antidepressant effects [65]. Their effects on second messenger systems and associated synaptic strength during maintenance therapy are also likely to increase baseline levels of SWA, thus creating a ceiling effect, so that ketamine-induced slow wave increases could not occur. A critical finding here is that ketamine’s ability to enhance sleep slow waves is not necessary for mood response (e.g., blocking production has no effect on mood). Rather, the enhanced production of slow waves present in MDD is a marker of plasticity and increased BDNF, but might be masked by drug effects. Alternately, the marker may vary across different patient groups. This study highlights the need for further studies to confirm the link between mood and central (sleep slow waves) or peripheral (BDNF) markers of synaptic plasticity. Critical to these studies is the need to examine ketamine challenges in both drug-free depressed and healthy populations. It is important to note that while biomarkers such as sleep slow waves are promising tools while developing novel drugs and identifying target populations, ongoing drug use might compromise their utility.
Conclusion
Remarkable progress has been made in the last 5 years to identify molecular and cellular mechanisms critical to ketamine’s rapid antidepressant effects. In particular, combining two markers of synaptic plasticity, BDNF levels and EEG sleep slow waves, has proven to be an effective approach for identifying ketamine’s capacity to increase synaptic strength. Nevertheless, numerous fundamental questions remain to be answered regarding the association between sleep, slow waves, and the time course of mood response to ketamine because these measures appear to have different response times. Identifying markers of ketamine’s prolonged mood effects as well as the utility of baseline slow wave sleep—a possible marker of brain plasticity—to predict mood response to ketamine will be important for optimizing treatment outcome. It should also be noted that, despite the promising nature of the research investigating ketamine’s effects on sleep slow waves, the subject groups have largely comprised individuals with treatment-resistant depression; thus, replication of these findings is critical. Future studies will need to address the effects of ketamine on sleep in other drug-free patient and healthy control populations.
As noted above, sleep slow wave measures (SWA, amplitude, and slope) appear to be related to an acute day one rise in BDNF levels as well as rapid antidepressant response; these measures (and preclinical BDNF levels) decline on day 2 during remission. In contrast, increased total sleep and improved sleep continuity on day 2 are associated with the extended remission. Further studies are needed to identify additional markers of plasticity and extended mood response. Finally, if interventions that produce rapid antidepressant effects share a common mechanism that increase synaptic strength, one might anticipate that such treatments also have similar effects on EEG sleep slow waves measures. Such studies are essential to uncovering key components of the rapid antidepressant response mechanism as well as to identifying additional novel treatment interventions.
Footnotes
Conflict of Interest Wallace C. Duncan, Jr., declares that he has no conflict of interest.
Carlos A. Zarate, Jr., is listed as a coinventor on a patent application for the use of ketamine and its metabolites in major depression. Dr. Zarate has assigned his rights in the patent to the US government but will share a percentage of any royalties that may be received by the government.
Human Rights and Informed Consent The MDD and BP patients that are discussed in this review were part of studies that were approved by the Combined Neuroscience Institutional Review Board of the National Institutes of Health. All subjects provided written informed consent before entry into the studies.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Rush AJ, Trivedi MH, Stewart JW, Nierenberg AA, Fava M, Kurian BT, et al. Combining medications to enhance depression outcomes (CO-MED): acute and long-term outcomes of a single-blind randomized study. Am J Psychiatry. 2011;168:689–701. doi: 10.1176/appi.ajp.2011.10111645. [DOI] [PubMed] [Google Scholar]
- 2.Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–17. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- 3.Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 4.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 5.Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63:1121–9. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hemmeter UM, Hemmeter-Spernal J, Krieg JC. Sleep deprivation in depression. Expert Rev Neurother. 2010;10:1101–15. doi: 10.1586/ern.10.83. [DOI] [PubMed] [Google Scholar]
- 7.Husain MM, Rush AJ, Fink M, Knapp R, Petrides G, Rummans T, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65:485–91. doi: 10.4088/jcp.v65n0406. [DOI] [PubMed] [Google Scholar]
- 8.Pagnin D, de Queiroz V, Pini S, Cassano GB. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20:13–20. doi: 10.1097/00124509-200403000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Faraguna U, Vyazovskiy VV, Nelson AB, Tononi G, Cirelli C. A causal role for brain-derived neurotrophic factor in the homeostatic regulation of sleep. J Neurosci. 2008;28:4088–95. doi: 10.1523/JNEUROSCI.5510-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67:793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 12.Machado-Vieira R, Ibrahim L, Henter ID, Zarate CA., Jr Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol, Biochem Behav. 2012;100:678–87. doi: 10.1016/j.pbb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeng S, Zarate CA., Jr The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9:467–74. doi: 10.1007/s11920-007-0063-1. [DOI] [PubMed] [Google Scholar]
- 14.Yilmaz A, Schulz D, Aksoy A, Canbeyli R. Prolonged effect of an anesthetic dose of ketamine on behavioral despair. Pharmacol, Biochem Behav. 2002;71:341–4. doi: 10.1016/s0091-3057(01)00693-1. [DOI] [PubMed] [Google Scholar]
- 15.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–64. doi: 10.1126/science.1190287. This study shows that in rats, ketamine rapidly activates the mTOR pathway, thereby increasing synaptic signaling proteins, spine density, and function. Blocking the mTOR pathway negated these effects as well as ketamine’s antidepressant-like effects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17•.Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338:68–72. doi: 10.1126/science.1222939. This review summarizes preclinical work showing that ketamine rapidly induces synaptogenesis and reverses synaptic deficits caused by chronic stress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, et al. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci. 2004;24:4401–11. doi: 10.1523/JNEUROSCI.0348-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–69. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 20.Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–61. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21••.Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–5. doi: 10.1038/nature10130. This study shows that NMDA antagonists cause fast-acting antidepressant-like effects in mouse models and that such effects depend on rapid synthesis of BDNF. Spontaneous neurotransmission effects on protein synthesis are viable targets of fast-acting antidepressants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kavalali ET, Monteggia LM. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry. 2012;169:1150–6. doi: 10.1176/appi.ajp.2012.12040531. [DOI] [PubMed] [Google Scholar]
- 23.Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- 24.Huber R, Ghilardi MF, Massimini M, Ferrarelli F, Riedner BA, Peterson MJ, et al. Arm immobilization causes cortical plastic changes and locally decreases sleep slow wave activity. Nat Neurosci. 2006;9:1169–76. doi: 10.1038/nn1758. [DOI] [PubMed] [Google Scholar]
- 25.Esser SK, Hill SL, Tononi G. Sleep homeostasis and cortical synchronization: I. Modeling the effects of synaptic strength on sleep slow waves. Sleep. 2007;30:1617–30. doi: 10.1093/sleep/30.12.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vyazovskiy VV, Riedner BA, Cirelli C, Tononi G. Sleep homeostasis and cortical synchronization: II. A local field potential study of sleep slow waves in the rat. Sleep. 2007;30:1631–42. doi: 10.1093/sleep/30.12.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–8. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- 28.Huber R, Tononi G, Cirelli C. Exploratory behavior, cortical BDNF expression, and sleep homeostasis. Sleep. 2007;30:129–39. doi: 10.1093/sleep/30.2.129. [DOI] [PubMed] [Google Scholar]
- 29.Aeschbach D, Cutler AJ, Ronda JM. A role for non-rapid-eye-movement sleep homeostasis in perceptual learning. J Neurosci. 2008;28:2766–72. doi: 10.1523/JNEUROSCI.5548-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bachmann V, Klein C, Bodenmann S, Schafer N, Berger W, Brugger P, et al. The BDNF Val66Met polymorphism modulates sleep intensity: EEG frequency- and state-specificity. Sleep. 2012;35:335–44. doi: 10.5665/sleep.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laje G, Lally N, Mathews D, Brutsche N, Chemerinski A, Akula N, et al. Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol Psychiatry. 2012;72:e27–8. doi: 10.1016/j.biopsych.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostenfeld I. Abstinence from night sleep as a treatment for endogenous depressions. The earliest observations in a Danish mental hospital (1954) and the analysis of the causal mechanism. Dan Med Bull. 1986;33:45–9. [PubMed] [Google Scholar]
- 33.Schulte W. Sequelae of sleep deprivation. Medizinische Klinik (Munich) 1959;54:969–73. [PubMed] [Google Scholar]
- 34.Borbely AA, Wirz-Justice A. Sleep, sleep deprivation and depression. A hypothesis derived from a model of sleep regulation. Hum Neurobiol. 1982;1:205–10. [PubMed] [Google Scholar]
- 35.Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Borbely AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- 37.Gorgulu Y, Caliyurt O. Rapid antidepressant effects of sleep deprivation therapy correlates with serum BDNF changes in major depression. Brain Res Bull. 2009;80:158–62. doi: 10.1016/j.brainresbull.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 38.Ibrahim L, Duncan W, Luckenbaugh DA, Yuan P, Machado-Vieira R, Zarate CA., Jr Rapid antidepressant changes with sleep deprivation in major depressive disorder are associated with changes in vascular endothelial growth factor (VEGF): a pilot study. Brain Res Bull. 2011;86:129–33. doi: 10.1016/j.brainresbull.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baxter LR., Jr Can lithium carbonate prolong the antidepressant effect of sleep deprivation? Arch Gen Psychiatry. 1985;42:635. doi: 10.1001/archpsyc.1985.01790290117017. [DOI] [PubMed] [Google Scholar]
- 40.Bunney BG, Bunney WE. Rapid-acting antidepressant strategies: mechanisms of action. Int J Neuropsychopharmacol. 2011:1–19. doi: 10.1017/S1461145711000927. [DOI] [PubMed] [Google Scholar]
- 41.Wu JC, Kelsoe JR, Schachat C, Bunney BG, DeModena A, Golshan S, et al. Rapid and sustained antidepressant response with sleep deprivation and chronotherapy in bipolar disorder. Biol Psychiatry. 2009;66:298–301. doi: 10.1016/j.biopsych.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 42.aan het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67:139–45. doi: 10.1016/j.biopsych.2009.08.038. [DOI] [PubMed] [Google Scholar]
- 43.Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, Aan Het Rot M, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry. 2013;74:250–256. doi: 10.1016/j.biopsych.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rasmussen KG, Lineberry TW, Galardy CW, Kung S, Lapid MI, Palmer BA, et al. Serial infusions of low-dose ketamine for major depression. J Psychopharmacol. 2013;27:444–50. doi: 10.1177/0269881113478283. [DOI] [PubMed] [Google Scholar]
- 45.Ibrahim L, Diazgranados N, Franco-Chaves J, Brutsche N, Henter ID, Kronstein P, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37:1526–33. doi: 10.1038/npp.2011.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathew SJ, Murrough JW, aan het Rot M, Collins KA, Reich DL, Charney DS. Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: a pilot randomized, placebo-controlled continuation trial. Int J Neuropsychopharmacol. 2010;13:71–82. doi: 10.1017/S1461145709000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemmeter U, Bischof R, Hatzinger M, Seifritz E, Holsboer-Trachsler E. Microsleep during partial sleep deprivation in depression. Biol Psychiatry. 1998;43:829–39. doi: 10.1016/s0006-3223(97)00297-7. [DOI] [PubMed] [Google Scholar]
- 48.Van Bemmel A, van den Hoofdakker R. Maintenance of therapeutic effects of total sleep deprivation by limitation of subsequent sleep. A pilot study. Acta Psychiatr Scand. 1981;63:453–62. doi: 10.1111/j.1600-0447.1981.tb00695.x. [DOI] [PubMed] [Google Scholar]
- 49.Hefti K, Holst SC, Sovago J, Bachmann V, Buck A, Ametamey SM, et al. Increased metabotropic glutamate receptor subtype 5 availability in human brain after one night without sleep. Biol Psychiatry. 2013;73:161–8. doi: 10.1016/j.biopsych.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 50.John J, Ramanathan L, Siegel JM. Rapid changes in glutamate levels in the posterior hypothalamus across sleep-wake states in freely behaving rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R2041–9. doi: 10.1152/ajpregu.90541.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feinberg I, Campbell IG. Ketamine administration during waking increases delta EEG intensity in rat sleep. Neuropsychopharmacology. 1993;9:41–8. doi: 10.1038/npp.1993.41. [DOI] [PubMed] [Google Scholar]
- 52.Campbell IG, Feinberg I. NREM delta stimulation following MK-801 is a response of sleep systems. J Neurophysiol. 1996;76:3714–20. doi: 10.1152/jn.1996.76.6.3714. [DOI] [PubMed] [Google Scholar]
- 53•.Duncan WC, Sarasso S, Ferrarelli F, Selter J, Riedner BA, Hejazi NS, et al. Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int J Neuropsychopharmacol. 2013;16:301–11. doi: 10.1017/S1461145712000545. This clinical study of ketamine’s antidepressant effects in treatment-resistant depression shows that ketamine acutely increases BDNF, slow wave activity, the occurrence of high amplitude waves, and slow wave slope, consistent with increased synaptic strength. Changes in BDNF levels are proportional to changes in EEG parameters in patients who responded to ketamine treatment, suggesting that enhanced synaptic plasticity is part of the rapid antidepressant. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duncan WC, Jr, Selter J, Brutsche N, Sarasso S, Zarate CA., Jr Baseline delta sleep ratio predicts acute ketamine mood response in major depressive disorder. J Affect Disord. 2013;145:115–9. doi: 10.1016/j.jad.2012.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cornwell BR, Salvadore G, Furey M, Marquardt CA, Brutsche NE, Grillon C, et al. Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry. 2012;72:555–61. doi: 10.1016/j.biopsych.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selter J, Duncan WC, Luckenbaugh D, Chen G, Zarate C. Abstracts of the Society for Neuroscience. Washington DC: Nov 12–16, 2011. Differential slow wave sleep response to ketamine in MDD versus BP Disorder. 2011. [Google Scholar]
- 57.Landsness EC, Goldstein MR, Peterson MJ, Tononi G, Benca RM. Antidepressant effects of selective slow wave sleep deprivation in major depression: a high-density EEG investigation. J Psychiatr Res. 2011;45:1019–26. doi: 10.1016/j.jpsychires.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friston KJ, Sharpley AL, Solomon RA, Cowen PJ. Lithium increases slow wave sleep: possible mediation by brain 5-HT2 receptors? Psychopharmacology (Berl) 1989;98:139–40. doi: 10.1007/BF00442020. [DOI] [PubMed] [Google Scholar]
- 59.Kupfer DJ, Reynolds CF, 3rd, Weiss BL, Foster FG. Lithium carbonate and sleep in affective disorders. Further considerations. Arch Gen Psychiatry. 1974;30:79–84. doi: 10.1001/archpsyc.1974.01760070061009. [DOI] [PubMed] [Google Scholar]
- 60.Lanoir J, Lardennois D. The action of lithium carbonate on the sleep-waking cycle in the cat. Electroencephalogr Clin Neurophysiol. 1977;42:676–90. doi: 10.1016/0013-4694(77)90284-x. [DOI] [PubMed] [Google Scholar]
- 61.Son H, Yu IT, Hwang SJ, Kim JS, Lee SH, Lee YS, et al. Lithium enhances long-term potentiation independently of hippocampal neurogenesis in the rat dentate gyrus. J Neurochem. 2003;85:872–81. doi: 10.1046/j.1471-4159.2003.01725.x. [DOI] [PubMed] [Google Scholar]
- 62.Gray NA, Zhou R, Du J, Moore GJ, Manji HK. The use of mood stabilizers as plasticity enhancers in the treatment of neuropsychiatric disorders. J Clin Psychiatry. 2003;64 (Suppl 5):3–17. [PubMed] [Google Scholar]
- 63.Harding GF, Alford CA, Powell TE. The effect of sodium valproate on sleep, reaction times, and visual evoked potential in normal subjects. Epilepsia. 1985;26:597–601. doi: 10.1111/j.1528-1157.1985.tb05698.x. [DOI] [PubMed] [Google Scholar]
- 64.Beaulieu JM. Not only lithium: regulation of glycogen synthase kinase-3 by antipsychotics and serotonergic drugs. Int J Neuropsychopharmacol. 2007;10:3–6. doi: 10.1017/S1461145706006857. [DOI] [PubMed] [Google Scholar]
- 65.Beurel E, Song L, Jope RS. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry. 2011;16:1068–70. doi: 10.1038/mp.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]