

Abstract
LP-BM5, a retroviral isolate, induces a disease featuring an acquired immunodeficiency syndrome termed murine AIDS (MAIDS). Many of the features of the LP-BM5-initiated disease are shared with HIV/AIDS. Our lab has shown that the interaction of B and CD4 T cells that is central to MAIDS pathogenesis requires ligation of CD40 on B cells by CD154 on CD4 T cells. Despite this strict requirement for CD154 expression, whether CD4 T cell receptor (TCR) occupany is essential for the induction of MAIDS is unknown. To block TCR engagement, Tg mouse strains with monoclonal TCR of irrelevant peptide/MHC specificities, all on MAIDS-susceptible genetic backgrounds, were tested: the study of a panel of TCR Tg CD4 T cells controlled for the possibility of serendipitous crossreactive recognition of virus-associated or induced-self peptide, or superantigen, MHC complexes by a given TCR. The results argue that TCR engagement is not necessary for the induction of MAIDS.
Keywords: Murine AIDS (MAIDS), LP-BM5 retrovirus, pathogenesis, immunodeficiency, CD4 T cells, CD154, CD40, T cell receptor (TCR), Signal 1, transgenic (Tg)
Introduction
CD40 ligand (CD40L or CD154) is a 33-kDa transmembrane glycoprotein that is transiently expressed on the surface of activated T cells, predominantly CD4 T cells. Its receptor, CD40, is found on B cells, dendritic cells, monocytes, and some other cell types. The CD154/CD40 system is crucially involved in immunity because cognate interactions between CD154 and CD40 generate intra- (through CD40) and inter-cellular signals that result in up-regulation of a variety of cell surface and soluble molecules that ultimately impact humoral and cellular immunity, as well as inflammation and other disease processes (e. g. Laman et al., 1996; Danese et al., 2006). For example, CD154 and CD40 are overexpressed in both forms of inflammatory bowel disease (IBD) (Liu et al., 1999; Danese et al., 2006), Crohn's disease (CD) (Battaglia et al., 1999), and ulcerative colitis (Polese et al., 2002). These studies and others strongly indicate that the CD154/CD40 pathway plays a key pathogenic role in intestinal inflammation. A number of studies have focused on the impact of CD154/CD40 interactions on the immunopathogenesis of other autoimmune diseases, such as systemic lupus erythematosus (SLE) (Desai-Mehta et al., 1996; Koshy et al., 1996), rheumatoid arthritis (RA) (MacDonald et al., 1997), multiple sclerosis (Gerritse et al., 1996), and Graves' disease (Faure et al., 1997); as well as on allograft rejection (Reul et al., 1997).
The interaction of CD154 and CD40 is not restricted to the regulation of immune responses and dysfunction resulting in autoimmunity, but also often plays an important role in the development of effector cells that function at local sites of infection and inflammation (Reichmann et al., 2000). In vivo studies have indicated the important role of CD40 ligation by CD154 in thymus-dependent humoral immune responses and germinal center formation (Allen et al., 1993; Xu et al., 1994; Liu et al., 1999). CD154 also is a critical molecular player in antigen-specific T-cell responses. Thus, adoptively transferred antigen-specific CD4 T cells lacking CD154 failed to expand upon antigen challenge of the recipients, showing that expression of CD154 is required for in vivo priming of CD4 T cells and therefore for the initiation of specific T-cell immune responses (Grewal et al., 1995). Similarly, CD154-/- mice display a deficiency in T cell-dependent macrophage-mediated immune responses (Stout et al., 1996). Thus, CD154 ligation of CD40 is important in the protective cell-mediated immune responses conferred by T cell-directed activated macrophage effector function against intracellular parasite infections such as Pneumocystis, Listeria monocytogenes, Leishmania (Grewal et al., 1997; Soong et al., 1996; Campbell et al., 1996), and a T. gondii-driven experimental model of acute ileitis (Li et al., 2002). CD154/CD40 interactions are also required for the priming and expansion of antigen-specific CD4 T cells and in the induction of co-stimulatory activity on antigen-presenting cells (APCs) (Grewal et al., 1997). CD154/CD40 binding and signaling are also well documented to have a key role in APC maturation and survival in the induction of adaptive immune responses, partially the activation of naïve T cells by dendritic cells (Grewal et al., 1997; Caux et al., 1994; Peguet-Navarro et al., 1995; Cella et al., 1996).
The LP-BM5 retroviral isolate consists of a pathogenic defective murine retrovirus (BM5def) that requires replication-competent ecotropic helper viruses (e.g. BM5eco) for its entry into cells and spread in vivo. The ensuing murine acquired immunodeficiency syndrome, termed murine AIDS (MAIDS) (Latarjet and Duplan, 1962; Mosier et al., 1985), exhibits many features shared with HIV-induced disease in man (Aziz et al., 1989; Jolicoeur, 1991; Morse et al., 1992; Tayar et al., 1999; Casabianca et al., 2003). Similarities include early onset hypergammaglobulinemia (hyper-Ig), splenomegaly, and lymphadenopathy; dependence on CD4 T cells for initiation of disease; loss of CD4 T cell function; severely depressed T and B cell responses; increased susceptibility to infection and death when exposed to normally nonpathogenic microorganisms; and the development of terminal B-cell lymphomas. Although there are also differences between MAIDS and human AIDS, the study of MAIDS, as the most commonly used murine system of retrovirus-induced immunodeficiency, offers many advantages. Thus, the availability of informative inbred, Tg, knockout, and other mouse strains, and ability to do in vivo pathogenesis experiments allows direct investigation of the cellular and molecular mechanisms required for the development of retroviral pathogenesis, including the associated immunodeficiency.
Both CD4 T cells and B cells are required for disease induction and progression. Severe combined immunodeficient (SCID) mice (lacking mature T and B cells) do not develop MAIDS (Simard et al., 1997). B6 mice that were depleted of B cells from birth by chronic administration of rabbit anti-mouse IgM antibodies (μ-suppressed) are resistant to LP-BM5 MAIDS (Cerny et al., 1990). It has been reported that the main targets of initial LP-BM5 retrovirus infection are B cells (Kim et al., 1994; K. Green, unpublished data), and to some extent macrophages (Cheung et al., 1991) and T cells (Kim et al., 1994; K. Green, unpublished data). However, B-cell infection by itself is not progressive and does not lead to disease in the absence of mature CD4 T cells (Klinman and Morse, 1989; Cerny et al., 1990). Indeed, an early hyperactivity of CD4 T cells may contribute to the pathogenesis of MAIDS (Mosier et al., 1987; Yetter et al., 1988). Thus, in vivo depletion of CD4 T cells by treatment with the anti-CD4-specific mAb GK1.5, before LP-BM5 infection rendered susceptible B6 mice resistant to the development of MAIDS (Yetter et al., 1988). Similarly, congenitally-athymic nude B6 mice become infected but do not develop significant LP-BM5 induced MAIDS (Mosier at al., 1987). However, if reconstituted with CD4 T cells from B6 donor mice, B6.nude recipients develop MAIDS upon LP-BM5 infection (Yetter et al., 1988; Giese et al., 1994). In contrast, by comparing the adoptive transfer of T cell subsets into B6.nude mice, it was demonstrated that CD8 T cells are not required for LP-BM5 induced pathogenesis (Green et al., 2001).
These and other results suggested that an interaction between B and CD4 T cells is critical for the pathogenesis of MAIDS. Thus, in additional adoptive transfer experiments, our lab showed that if B6.nude recipients were reconstituted with CD154+/+, but not CD154-/-, CD4 T cells, they converted to disease susceptibility (Green et al., 2001). Reciprocally, with regard to the cell type interacting with CD154+ CD4 T cells, B6 CD40-/- mice, which are resistant to LP-BM5-induced MAIDS (Green et al., 2001; Yu et al., 1999), became susceptible to LP-BM5-induced disease after reconstitution with highly purified wild-type (w.t.) (CD40+) B cells, but not after receiving purified wild-type dendritic cells (DC) or a combined CD40+ population composed of DC and macrophages obtained from B6.SCID mouse donors (Green et al., 2001). Indeed, by F2 genetic crossing and screening to generate a B6.CD40-/- SCID recipient, combined transfer of only w.t. (CD154+) CD4 T cells and w.t. (CD40+) B cells allowed for LP-BM5 infection to cause MAIDS (K. Green, unpublished data). In a follow-up study, we demonstrated that B6 mice deficient for both the CD80/CD86 (B7.1/B7.2) co-stimulatory ligands are susceptible to LP-BM5 induction of MAIDS (Green et al., 2002). These results on the APC (B cell) expressed B7 ligands, coupled with data from another laboratory on the CD80/CD86 receptor-CD4T cells expressed CD28 (Morawetz et al., 1998), suggested that CD28/CD80-CD86 interactions are not absolutely required for the initiation of MAIDS or the commitment to disease progression. Although the classic upregulation of CD80/CD86 after CD40 ligation is thus not necessary, we have used a panel of chimeric CD40 Tg mice on the CD40-/- background to confirm that the CD40 signaling is needed for MAIDS induction and is differentially mediated by TRAF binding to the TRAF 6 site (vs the TRAF 2/3/5 site) on the CD40 cytoplasmic tail (Green et al., 2004). Collectively, these findings provide clear evidence that activated CD4 T cell CD154 ligation of B cell CD40 and downstream signaling events are required for MAIDS (Green et al., 2001; Green et al., 2004).
In view of the absolute requirement for CD154 expression, it is not clear, however, whether there is a role for CD4 T cell antigen receptor (TCR) engagement of peptide or superantigen/MHC complexes in the induction of MAIDS. It is well accepted that, in general, the induction of CD154 depends on TCR occupancy and Signal 1 in the T cell activation paradigm. But it is unknown if ligation of the TCR expressed by the required CD4 T cells is necessary for MAIDS pathogenesis. If TCR is indeed engaged, then it would follow that a viral peptide, induced self peptide, or superantigen (SAg) complexed with MHC class II would be the TCR ligand driving Signal 1 propagation. However, as no LP-BM5 virus-encoded or -associated peptide, or SAg, has yet been defined that could provide the ligand for the CD4 TCR in the context of MAIDS (Hugin et al, 1991; Muralidhar et al., 1992; Gilmore at al., 1993; McCarty et al., 1996; Doyon et al., 1996; Hayashi and Kanagawa, 1999), the question of a requirement for CD4 TCR engagement in MAIDS can not be approached using antibody or other reagents to block the ligation of the TCR by these putative peptide or SAg -MHC complexes. Also, it is not possible to study the need for the TCR by use of TCR-/- mice because the required CD4 T cells will not be present. Thus, this key question of the role, if any, of the CD4 TCR is addressed here by utilizing a panel of CD4 T cells that express class II-restricted transgenic (Tg) TCR that are specific for different peptides totally unrelated to LP-BM5 virus, and are of various Vβ chain compositions. To determine the ability of these CD4 T cells to support LP-BM5 induction of MAIDS in the absence of classic TCR ligation, it is crucial that only TCR Tg CD4 T cells are present. To this end, one or both of two approaches were utilized. First, highly purified TCR Tg CD4 T cells were separated and adoptively transferred into either B6.nude or B6.TCRα-/- recipients, that have no pre-existing CD4 T cell compartment. Second and alternatively, to avoid the presence of non-Tg CD4 T cells, these TCR Tg strains were backcrossed onto the B6.TCRα-/- or B6.RAG-1-/- background and infected directly.
Results and Discussion
Adoptive transfer of polyclonal CD4 T cells converts MAIDS resistant B6.nude recipients to a MAIDS susceptible phenotype
To set up the adoptive transfer system, in initial experiments CD4 T cells were obtained by either positive or negative selection methods of purifying mature splenic CD4 T cells from normal donor C57BL/6 mice. After adoptive transfer of such purified CD4 T cells into B6.nude recipients, a portion of the recipients was infected i.p. with LP-BM5 virus 48 hours after reconstitution, while the others served as non-infected control groups. Progression to MAIDS was evaluated 8 to 12.5 weeks later by the following standard readouts of MAIDS-associated symptoms, which we and others have previously established (Mosier at al., 1987; Klinman and Morse, 1989; Morse et al., 1992; Green et al., 1996; Green, at al., 1998; Green et al., 2001; Green et al., 2002; Green et al., 2004): (i) spleen size, with enlargement indicating MAIDS-associated B- and T-cell lymphoproliferation; (ii) serum IgG2a and IgM levels, with increases due to MAIDS polyclonal B-cell activation; and (iii) splenic B-cell responses to lipopolysaccharide (LPS) stimulation, with decreases indicative of B-cell-associated MAIDS-induced immunodeficiency. For all the experiments, the early activational MAIDS disease parameter, hypergammaglobulinemia (serum IgM and IgG2a levels), was also assessed at 7 to 8 weeks post infection (p.i.) by analysis of serum taken without sacrifice of the mouse. We have previously shown that the adoptive transfer of CD4 T cells converted B6.nude mice to MAIDS susceptibility (Green et al., 2001; Li and Green, 2006). Comparing positive selection and negative selection approaches used to purify the CD4 T cells, there were no substantial differences in the pattern of the results obtained, which were also roughly comparable to those observed in intact B6 mice (Li and Green, 2006).
The MAIDS susceptibility of B6.nude vs B6.TCRα-/- recipients reconstituted with polyclonal CD4 T cells appears to be equivalent
Like B6.nude mice, B6.TCRα-/- mice are also resistant to LP-BM5 induced disease due to lack of the required CD4 T cells. Here, we compared head to head B6.nude recipients and B6.TCRα-/- recipients reconstituted with total polyclonal CD4 T cells purified from B6 donors. Upon LP-BM5 virus infection, these two recipient groups developed essentially the same amount of MAIDS, with all the disease parameters in agreement; here for the sake of brevity, we show only one activational parameter: spleen weight (Fig. 1A), and an immunodeficiency parameter: splenic mitogen response to LPS (Fig. 1B). Thus, B6.nude and B6.TCRα-/- mice can be used interchangeably as recipients of CD4 T cell adoptive transfer.
FIG. 1. Both B6.nude and B6.TCRα-/- recipients equally supported MAIDS.

CD4 T cells purified from B6 donors were adoptively transferred into either B6.nude or B6.TCRα-/- recipients. After LP-BM5 inoculation, disease was assessed in infected or control mice at 10 weeks p.i. by the standard MAIDS readout panel. Shown here are one representative activational parameter: spleen weight (A) and one immunodeficiency parameter: splenic mitogenic response to LPS (B), compared to uninfected control mice, with all other parameters also demonstrating that B6.nude and B6.TCRα-/- recipients reconstituted with B6 CD4 T cells are equivalently MAIDS susceptible. The results were significant (*, P < 0.05) or not significant (NS, P ≥ 0.05). The data shown are representative of a total of three experiments, with the same pattern of results always obtained.
Adoptive transfer of OT-II TCR Tg+ CD4 T cells converts B6.nude recipients to susceptibility to MAIDS pathogenesis
To determine whether CD4 T cells of monoclonal specificity could mediate LP-BM5 induced MAIDS, we first studied OT-II TCR Tg mice, whose CD4 T cells are specific for the irrelevant antigen ovalbumin, as presented by MHC class II (I-Ab) (Table 1). By flow cytometric analysis of OT-II TCR Tg mice, we found that only ∼73% of splenic CD4 T cells were actually TCR Tg+ (Fig. 2A, upper right). It was important to avoid the remaining ∼27% CD4 T cells that express non-Tg TCR chains, because it may well be that these cells would satisfy the CD4 T-cell requirement for MAIDS induction. Thus, our first approach was to purify OT-II TCRVβ5.1, 5.2 Tg+ CD4 T cells by use of the corresponding anti-Vβ mAb and Miltenyi bead selection (see Materials and Methods) prior to their adoptive transfer. In this way highly purified (≥ 98%) TCR Tg positive CD4 T cells (Fig. 2A, lower right), vs purified polyclonal B6 CD4 T cells (Fig 2A, lower left) from B6 donors (Fig 2A, upper left) as a control, were transferred into B6.nude recipients. Following LP-BM5 infection 48 hours after adoptive transfer, induction of MAIDS was subsequently determined by assessing the standard panel of disease-related parameters. However, for the sake of brevity, here we only show one activational parameter: spleen weight (Fig. 2B), and an immunodeficiency readout: mitogen response to LPS (Fig. 2C). All the other disease parameters agreed (data not shown) in that: the B6.nude recipients reconstituted with OT-II TCR Tg CD4 T cells mediated significant MAIDS, compared to their non-infected control group (Figs. 2B, C) or infected B6.nude mice not receiving CD4 T-cell transfer (data not shown). However, significantly less disease was observed with OT-II TCR Tg CD4 transfer, compared to the infected recipients previously reconstituted with total polyclonal CD4 T cells purified from normal B6 donors (Figs. 2B, C).
Table 1.
A panel of TCR-transgenic strains to address the role of the TCR.
| Tg TCR mouse strain | Background strain | MHC restriction | Foreign peptide specificity | TCRVα/Vβ subunits |
|---|---|---|---|---|
| OT-II | B6 | I-Ab | Ovalbumin: OVA323-339 | Vα2/Vβ5.1, 5.2 |
| TEa | B6 | I-Ab | Eα52-68, class II I-Eαd-chain | Vα2/Vβ6 |
| ANDb/b | B6 | I-Ab | Pigeon cytochrome C: PPC81-104 | Vα11/Vβ3 |
| ANDk/k | B10.BR | I-Ek | Pigeon cytochrome C: PPC81-104 | Vα11/Vβ3 |
FIG. 2. Purified OT-II TCR Tg+ CD4 T cells mediate MAIDS.
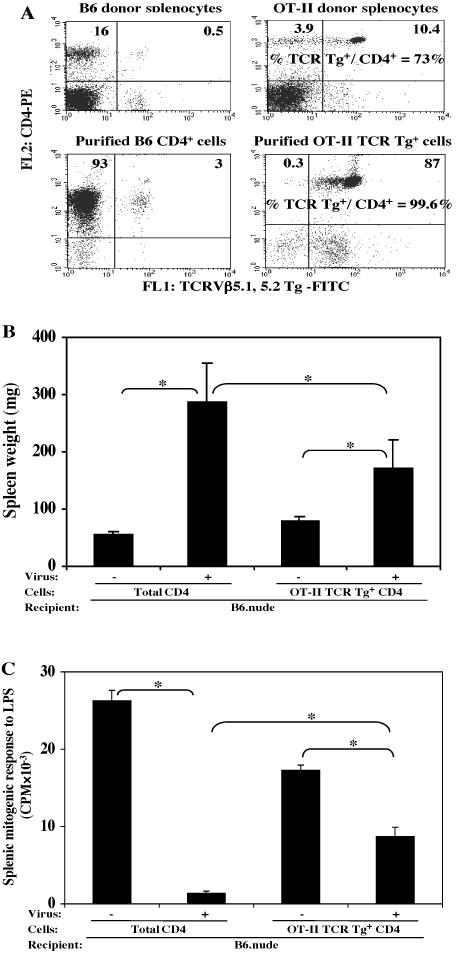
CD4 T cells (A, lower left) purified from B6 donors (A, upper left), or OT-II TCR Tg+ CD4 T cells (A, lower right) purified from OT-II TCR Tg donors (A, upper right) were adoptively transferred into B6.nude recipients. Mice were assayed for disease at 9 weeks p.i. with LP-BM5. Here, one activational parameter: spleen weight (B), and one immunodeficiency parameter: splenic mitogenic response to LPS (C). are depicted; with all other parameters consistent in demonstrating that B6.nude recipients reconstituted with OT-II TCR Tg+ CD4 T cells mediated MAIDS. The results were significant (*, P < 0.05) or not significant (NS, P ≥ 0.05). This experiment has been done a total of three times, with the same pattern of results always obtained.
OT-II TCR Tg TCRα-/- mice: Susceptibility to LP-BM5-induced MAIDS and the effect of OVA immunization
Alternatively to eliminate non-TCR-Tg CD4 T cells, we crossed these OT-II TCR Tg mice onto a B6.TCRα-/- background, and screened the offspring by flow cytometry to determine Tg positivity (Fig. 3A) and by PCR to determine the endogenous TCRα-/- status (Fig. 3B). We thus obtained OT-II TCR Tg+ TCRα-/- mice, whose CD4 T-cell compartment consisted of essentially 100% TCR Tg cells, and directly infected these mice, vs w.t. B6 controls, with LP-BM5 virus. Similar to the results obtained by the adoptive transfer approach (Fig. 2B, C), again, there was clear evidence that OT-II TCR Tg CD4 T cells mediated substantial LP-BM5-induced MAIDS -- here as depicted by one activational parameter: hyper IgG2a levels (Fig. 3C), and one immunodeficiency parameter: the splenic mitogenic response to LPS (Fig. 3D).
FIG. 3. The OT-II TCR Tg strain on the B6.TCRα-/- background is MAIDS susceptible.
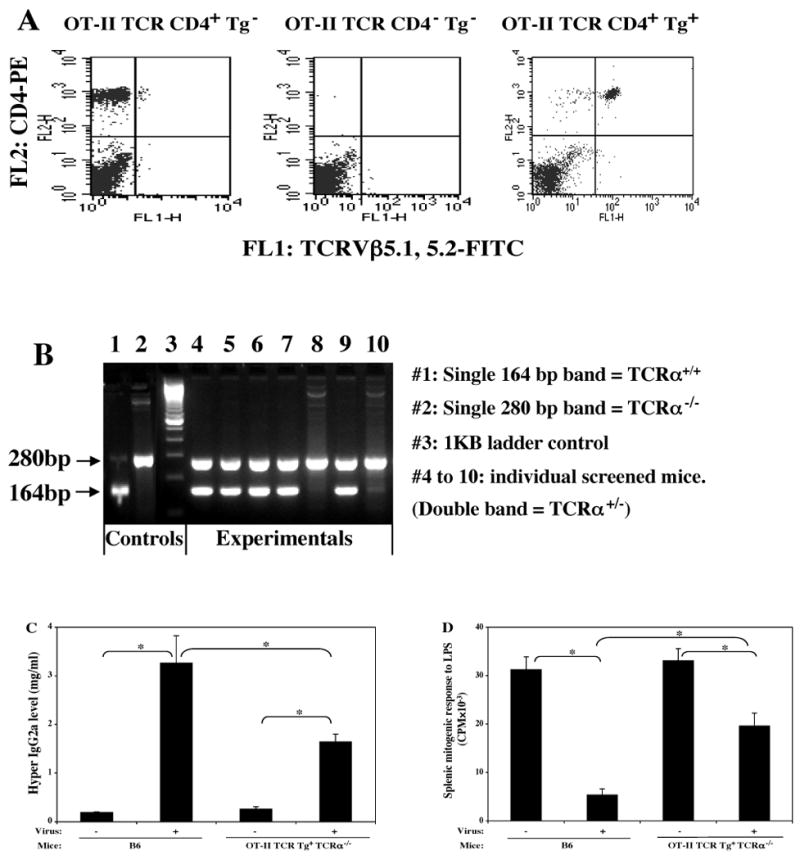
The OT-II TCR Tg strain was crossed onto the B6.TCRα-/- background (see Materials and Methods). The offspring were screened for Tg positivity (A) by use of FITC anti-TCRVβ5.1, 5.2, and PE anti-CD4 mAbs; and also screened for TCRα-/- status (B) by PCR. Experimental mice that were both Tg positive and also on a TCRα-/- background, versus B6 control mice, were infected with LP-BM5. Disease was assessed at 10 weeks p.i. by the standard MAIDS readout panel. Shown here are one representative activational parameter: hyper IgG2a level (C) and one immunodeficiency parameter: splenic mitogenic response to LPS (D), with all other parameters demonstrating that OT-II TCR Tg TCRα-/- mice are MAIDS susceptible. The results were significant (*, P < 0.05) or not significant (NS, P ≥ 0.05). This experiment was done a total of three times, with the same pattern of results always obtained.
Thus, OT-II TCR Tg CD4 T cells, when studied in isolation from non-Tg CD4 T cells, do mediate LP-BM5 induced MAIDS, as determined by both the adoptive transfer and TCRα-/- backcross approaches. Therefore, given the absence of the OVA protein antigen source of the corresponding peptide epitope, TCR occupancy by peptide/MHC seems to not be absolutely required for induction of MAIDS by LP-BM5 infection. However, in view of the partial disease observed, it seemed possible that if we provided the cognate OVA peptide for TCR ligation and Signal 1 by injecting OVA in vivo, this strategy might lead to more profound disease. Thus, OT-II TCR Tg TCRα-/- mice received multiple doses of OVA plus adjuvant immunization prior to, and subsequent to, LP-BM infection (see Materials and Methods).
To make sure that the OVA delivered was sufficient to have activated the OT-II TCR Tg cells in vivo and provided TCR Signal 1, peripheral blood lymphocyte samples were obtained at several time points p.i. These CD4 TCR Tg cells were specifically analyzed by flow cytometry via staining for the activational markers CD62L and CD69; and functional assessment of their ability to proliferate in vitro in response to OVA re-stimulation. The flow cytometric results indicated that immunizing with OVA in vivo provided clear evidence for activation specifically of OT-II TCR Tg CD4 T cells at days 2, 5, and 14 p.i.: e.g. at day 14 p.i., there was a two-fold decrease of CD62L expression and a three-fold increase in CD69 expression among the TCR Tg CD4 T cells from the OT-II TCR Tg+ TCRα-/- mice that were immunized with OVA and infected by LP-BM5 virus versus mice that were only infected. In addition, a strong OVA-dependent in vitro recall proliferative response was observed at several time points at days 2, 5 and 14, p.i. (data not shown). In this context of documented TCR-mediated activation and presumed Signal 1 generation, the development of LP-BM5-induced MAIDS was subsequently assessed as shown here by one activational parameter: spleen weight (Fig. 4A), and an immunodeficiency parameter: the mitogen response to LPS (Fig. 4B). In agreement with the other disease parameters not shown, and the results of Figs. 3A and 3B above, OT-II TCR Tg CD4 T cells mediated clear MAIDS. However, there was no evidence supporting an increased degree of MAIDS by OVA immunization in vivo.
FIG. 4. In vivo activation of TCR Tg CD4 T cells with cognate antigen did not enhance the degree of LP-BM5 retrovirus-induced MAIDS.

OT-II TCR Tg+ TCRα-/- mice received multiple doses of OVA protein antigen in vivo (see Materials and Methods). Disease was assessed at 8.5 weeks p.i., with one activational parameter (spleen weight) (A), and one immunodeficiency parameter (splenic mitogenic response to LPS) (B) depicted as representative of all other disease read-outs, which showed the same pattern of results. The results were significant (*, P < 0.05) or not significant (NS, P ≥ 0.05). This experiment has been done twice; the same pattern of result was always obtained.
Other I-Ab restricted TCR Tg strains and the ability of their CD4 T cells to mediate MAIDS
Despite our repeatable results obtained from the OT-II TCR Tg mice, it was important to study additional TCR Tg strains. First, although the specificity of the Tg OT-II TCR for OVA, as an irrelevant antigen, would appear to eliminate TCR ligation, signaling through the TCR could have occurred due to a random cross-reactive recognition of a virus-encoded, or –induced self, peptide. If more than one irrelevant TCR Tg strain mediated LP-BM5-induced MAIDS, the possibility that this was dependent on another crossreactive stimulation of the TCR seemed unlikely. Second, considering differing Vβ and Vα TCR subunit compositions, multiple TCR Tg strains needed to be assessed to test the possibility that the TCR might be ligated by a virus-encoded, or –induced-self superantigen (SAg). Third, the observation that OT-II TCR Tg CD4 T cells mediated MAIDS, but only to a partial extent compared to polyclonal CD4 T cells, begged the question as to whether all TCR Tg CD4 T cells were capable, but limited, in their ability to support MAIDS pathogenesis.
To address these questions, the ANDb/b TCR Tg (specific for PCC81-104) and TEa TCR Tg (specific for Eα52-68) strains (Table 1) were studied in parallel. Both the ANDb/b and TEa TCR Tg strains were crossed onto the B6.RAG-1-/- background (see Materials and Methods) to eliminate/limit the presence of non-Tg CD4 T cells (Figs. 5A, B). As RAG-1-/- background mice are also deficient in the B-cell compartment that is necessary for MAIDS development (Cerny et al., 1990), polyclonal CD19 B cells purified from w.t. B6 donors (see Materials and Methods) were adoptively transferred into the ANDb/b TCR Tg+ RAG-1-/- (Fig. 5A, left panel) and TEa TCR Tg+ RAG-1-/- recipient mice (Fig. 5B, left panel) as the experimental groups. As positive controls for this transfer, aliquots of the same purified CD19 B cells (plus polyclonal CD4 T cells) from B6 donors were adoptively transferred in parallel into either ANDb/b TCR Tg- RAG-1-/- (Fig. 5A, right panel) or TEa TCR Tg- RAG-1-/- (Fig. 5B, right panel) littermate TCR Tg negative recipients. Again, showing only one of the standard panel of activational parameters: spleen weight (Fig. 5C) and an immunodeficiency parameter: mitogen response to LPS (Fig. 5D) for brevity, the results were clear and contrasting. ANDb/b TCR Tg+ RAG-1-/- mice reconstituted with CD19 B cells clearly developed LP-BM5-induced MAIDS, with a roughly similar degree of penetrance relative to the non-Tg recipient controls that received polyclonal B6 CD4 T cells and B cells. Despite thus proving that the quality and quantity of B6 CD19 B cells transferred was sufficient for disease induction, in sharp contrast the TEa TCR Tg+ RAG-1-/- B-cell reconstituted mice did not develop significant MAIDS, compared to their non-infected control group, for either the activational or immunosuppressive disease parameters (Fig. 5C, D, and data not shown). These results were highly reproducible: ANDb/b TCR Tg mice were MAIDS susceptible in 4/4 experiments of this type, and the same conclusion was reached both by infection of the intact original Tg strain, and via adoptive transfer of ANDb/b TCR Tg CD4 T cells into B6.TCRα-/- recipients (see below). On the other hand, TEa TCR Tg mice failed to develop LP-BM5-induced MAIDS (Fig. 5C, D) in a total of four experiments utilizing the adoptive transfer protocol.
FIG. 5. The ANDb/b TCR Tg strain, but not the TEa TCR Tg strain, crossed onto a B6.RAG-1-/- background is MAIDS susceptible.
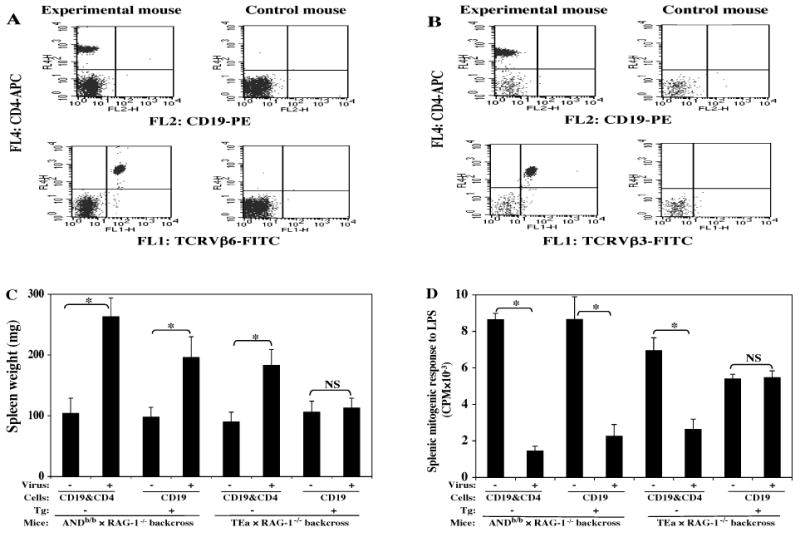
The offspring from ANDb/b TCR Tg and TEa TCR Tg strains crossed onto a B6.RAG-1-/- background were screened for Tg positivity (A and B) by use of FITC anti-TCRVβ6 (A) or Vβ3 (B), respectively; PE anti-CD19; and APC anti-CD4. The experimental Tg+ mice on the RAG-1-/- background (A and B, left panel) were recipients of adoptive transfer of CD19 B cells, while the control Tg- mice on the RAG-1-/- background (A and B, right panel) received both CD4 T and CD19 B cells (see Materials and Methods). Disease parameters were assayed at 12.5 weeks p.i. by the standard MAIDS readout panel. Shown here are one representative activational parameter: spleen weight (C), and one immunodeficiency parameter: splenic mitogenic response to LPS (D), with all other parameters clearly demonstrating that ANDb/b TCR Tg RAG-1-/- mice are MAIDS susceptible but TEa TCR Tg RAG-1-/- mice are MAIDS resistant. The results were significant (*, P < 0.05) or not significant (NS, P ≥ 0.05). This experiment has been performed a total of four times, with the same pattern of results always obtained.
Evidence against superantigen involvement in MAIDS pathogenesis
The observations that the OT-II and ANDb/b TCR Tg strains were MAIDS susceptible, but not the TEa TCR Tg strain, could be interpreted to suggest that a SAg, binding to Vβ5.1, 5.2 and Vβ3, but not Vβ6 (Table 1), selectively ligated these Tg TCR and thus was critically involved in MAIDS pathogenesis. To approach the possibility of SAg involvement as the basis for the differential ability of TCR Tg CD4 T cells to mediate MAIDS, an additional highly related TCR Tg strain, was studied in parallel to the ANDb/b TCR Tg strain just shown to be MAIDS susceptible. Thus, the AND TCR Tg has also been bred onto, and thus selected on, the H-2k/B10.BR background. Importantly, CD4 T cells of the ANDb/b and ANDk/k TCR Tg strains, are specific for the same peptide of the MAIDS-irrelevant antigen pigeon cytochrome C: PCC81-104, and express the same TCR subunits. Thus, as long as the putative SAg is presented by both MHC class II I-Ab and I-Ak/I-Ek molecules, the ANDk/k TCR Tg strain should be MAIDS susceptible as is the ANDb/b TCR Tg strain (Figs. 5C, D).
After LP-BM5 infection, these two TCR Tg strains, and their respective B6 and B10.BR background control strains, were assessed for MAIDS development by the standard disease parameter panel. Depicting data representative of all disease parameters, shown here are one activational parameter: spleen weight (Fig. 6A), and one immunodeficiency read-out: mitogen response to LPS (Fig. 6B). ANDb/b TCR Tg mice were MAIDS susceptible as expected; in fact, their extent of MAIDS splenomegaly (and hyper-Ig/data not shown) was, if anything, more pronounced compared to their B6 background control mice. However, clearly, there was no evidence that ANDk/k TCR Tg mice developed any MAIDS symptoms, considering all disease parameters at 10 weeks p.i. Given that the control for the ANDk/k background, the B10.BR strain, displayed significant MAIDS symptomatology upon LP-BM5 infection, importantly it could be inferred that any SAg necessarily involved could be presented by H-2k class II MHC. Thus, the lack of disease in infected ANDk/k TCR Tg mice was not simply due to the consequences of the selection of this Tg TCR on the H-2k (vs H-2b) haplotype, or other genetic background effects, and these results thus agued against TCR ligation by a SAg as a requisite for MAIDS.
FIG. 6. The ANDb/b TCR Tg strain is MAIDS susceptible, while the ANDk/k TCR Tg strain is not.

ANDb/b TCR Tg and ANDk/k TCR Tg mice, plus their respective B6 and B10.BR control groups, were infected with LP-BM5 and studied in parallel. Disease parameters were assayed 10 weeks p.i., and shown here are one activational parameter: spleen weight (A), and one immunodeficiency parameter: mitogen response to LPS (B), with all other parameters clearly demonstrating that B6, ANDb/b TCR Tg, and B10.BR mice are MAIDS susceptible but ANDk/k TCR Tg mice are MAIDS resistant. The results were significant (*, P < 0.05) or not significant (NS, P ≥ 0.05). This experiment has been performed a total of three times, with the same pattern of results consistently obtained.
For two reasons it was important to follow up this direct infection type of experiment with the adoptive transfer approach. First, the B10.BR (H-2k) strain, which controlled for the differential genetic background of the ANDk/k TCR Tg strain, appeared to be somewhat less susceptible to MAIDS induction than the B6 (H-2b) background strains, at least based upon splenomegaly (Fig. 6A), although the immunodeficiency parameter (Fig. 6B) suggested essentially full disease. This is in keeping with previous reports that, with regard to MHC-control of MAIDS susceptibility, H-2b is more permissive than H-2k (Makino et al., 1990). Thus, if a MAIDS-associated SAg is critical to disease pathogenesis, it may be less well presented by, or recognized in association with, H-2k class II MHC than H-2b class II. Second, the ANDb/b vs ANDk/k TCR Tg strains have different levels of “contaminating” non-TCR Tg CD4 T cells present, and this could account for the differential ability of the ANDb/b, but not the ANDk/k, strain to develop LP-BM5-induced MAIDS. Thus, by flow cytometric analysis there was on average only about 80 to 83% of total CD4 T cells in ANDb/b TCR Tg mice that were actually Tg+ (Fig. 7A, left panel), whereas there was an average of 90 to 93% of CD4 T cells that were ANDk/k TCR Tg+ (Fig. 7A, right panel). To make sure that the disease induced in the ANDb/b, but not the ANDk/k, TCR Tg strain was not due to the higher percentage of contaminating non-Tg CD4 T cells which could mediate MAIDS, and to provide for a common MHC class II presentation of the putative SAg, TCR Tg+ CD4 T cells were purified from either ANDb/b, or ANDk/k, TCR Tg donors (purity ≥ 98%, data not shown). These TCR Tg CD4 T cells were adoptively transferred into B6.TCRα-/- recipients, followed by LP-BM5 infection.
FIG. 7. The difference in MAIDS susceptibility mediated by TCR Tg+ CD4 T cells of the ANDb/b and ANDk/k TCR Tg strains as determined by parallel adoptive transfer experiments.
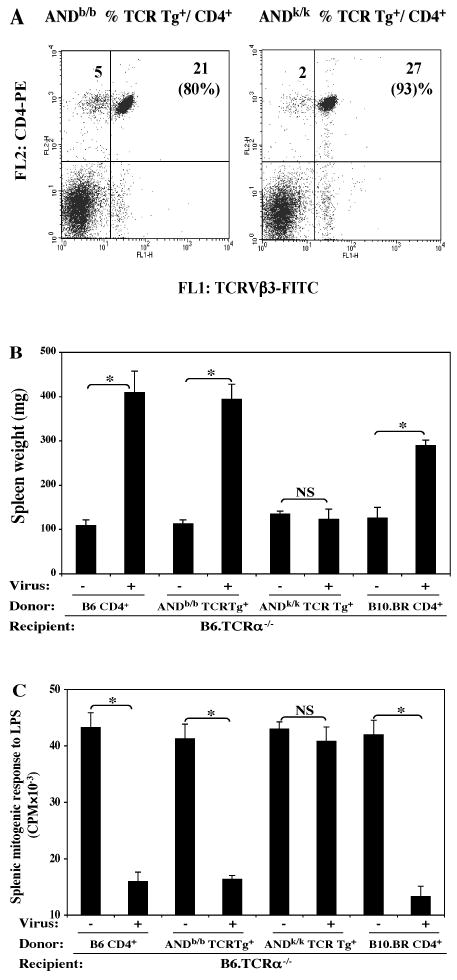
The ANDb/b and ANDk/k TCR Tg mice were phenotyped for Tg status by using FITC anti-TCRVβ3 and PE-anti-CD4 mAbs (A). Purified (≥ 98%) ANDb/b and ANDk/k TCR Tg+ CD4 T cells (2 × 107) isolated from the respective TCR Tg donors, or control w. t. B6 and B10.BR CD4 T cells, were adoptively transferred into B6.TCRα-/- recipients, followed by LP-BM5 infection (2 × 105 PFU). Disease parameters were assayed 10.5 weeks p.i.; here shown are one representative activational parameter: spleen weight (B), and one immunodeficiency parameter: splenic mitogenic response to LPS (C), with all other MAIDS read-outs in agreement: only when ANDk/k TCR Tg CD4 T cells were used to reconstitute B6.TCRα-/- recipients was MAIDS development not observed. The results were significant (*, P < 0.05) or not significant (NS, P ≥ 0.05). This experiment has been performed twice, with the same panel of result has always obtained.
Here, we show one representative activational parameter: spleen weight (Fig. 7B), and an immunodeficiency parameter: the mitogen response to LPS (Fig. 7C). B6.TCRα-/- recipients reconstituted with purified ANDb/b TCR Tg+ CD4 T cells again mediated significant MAIDS as expected from the results of Fig. 6. In sharp contrast, B6.TCRα-/- recipients reconstituted in parallel with ANDk/k TCR Tg+ CD4 T cells failed to demonstrate any signs of the disease, considering all tested MAIDS parameters (Fig. 7B, C, and data not shown). To control for the differences in genetic backgrounds in these adoptive transfer experiments, we adoptively transferred polyclonal CD4 T cells from either B6 or B10.BR donors into B6.TCRα-/- recipients. The results clearly showed that CD4 T cells of both control groups readily mediated substantial MAIDS (Figs. 7B, C). Thus, the resistance to MAIDS observed for the ANDk/k donor strain in these adoptive transfer experiments (or when disease was assessed in the direct infection experiments of this TCR Tg strain, see Fig. 6), was not due to the B10.BR background.
Lack of viral pathogenesis mediated by TEa and ANDk/k TCR Tg strains does not correlate with reduced viral load
It was important to assess whether a differential ability to become infected and/or to allow the spread of the component viruses was the basis for the resistance to MAIDS by the TEa and ANDk/k TCR Tg strains. To that end, the expression of the pathogenic BM5def and helper BM5eco viruses was determined by validated quantitative RT-PCR assays (Cook et al., 2003; Green et al., 2004) for the experiments depicted in Figs. 5 to 7. First, compared to the undetectable level of expression of the non-infected groups, all infected mouse groups demonstrated substantial expression of BM5def and BM5eco. Second, however, such virus expression data indicated that viral load in the infected TEa (Fig. 8A) and ANDk/k (Fig. 8B) TCR Tg strains was not significantly lower compared to the other infected strains in which MAIDS was observed. Thus, the resistance of these two strains to MAIDS was therefore not simply due to a selective inability of these two TCR Tg strains to become infected by, or to propagate, the LP-BM5 retroviruses. Rather, it appeared that these CD4 TCR Tg T cells were unable to mediate viral pathogenesis.
Fig. 8. The resistance of the TEa and ANDk/k TCR Tg strains to MAIDS is not due to insufficient viral infection, or a reduction in viral load.

Expression of the BM5def and BM5eco viruses was determined by separate real-time RT-PCR assays with normalization to the expression of β-actin as we previously published (Cook et al., 2003; Green et al., 2004). Shown here are the results of the viral load for the mouse groups of Fig. 5 (Fig. 8A); and Figs. 6 and 7 (Fig. 8B), with all the non-infected groups' data pooled and presented together as “control”, which exhibited no detectable (ND) viral load. The results were not significantly (NS) different among all the infected groups of both Fig. 8A as shown, and also Fig. 8B, although here, for simplicity, only the most important statistical comparisons of the infected ANDb/b versus ANDk/k groups are shown.
Concluding Remarks
Our studies herein provide strong evidence against a need for TCR ligation, by complexes of either a specific peptide or a superantigen (SAg) with class II MHC, for CD4 T cells to fulfill their required role in mediating pathogenesis caused by the LP-BM5 retroviral complex. This conclusion was reached by careful analysis of a panel of TCR Tg strains of irrelevant specificities to avoid the possibility of random crossreactive recognition with a putative LP-BM5 encoded or induced peptide or SAg. In addition, the presence of contaminating non-Tg CD4 T cells, which might have mediated disease as they are present in significant numbers in many TCR Tg strains, was countered by either the use of highly purified TCR Tg-positive CD4 T cells in adoptive transfer experiments, and/or by crossing the TCR Tg strains onto a TCRα-/- or RAG-1-/- genetic background. This backcrossing approach also controlled for any possible confounding effects if different TCR Tg CD4 T cell populations exhibited variable survival characteristics after their adoptive transfer into B6.nude or B6.TCRα-/- recipients. In addition, by direct analyses, we saw no evidence for such a differential persistence of TCR Tg CD4 T cells that correlated with their ability to mediate MAIDS.
Regardless of the methods utilized, the results were consistent and clear in showing that the I-Ab restricted OT-II (anti-OVA) and ANDb/b (anti-PCC) CD4 T cells mediated MAIDS as assessed by all disease readouts. These data not only indicate that CD4 T-cell TCR ligation is not a necessity for their role in disease initiation and progression, but also lead to the surprising suggestion that the induction of CD154 expression that is required of the “pathogenic” CD4 T cells must be occurring in the absence of TCR occupancy and classic generation on Signal 1. Taken together, these findings thus imply that there may be novel pathways of induction of CD154 expression by CD4 T cells during the early-mid course of LP-BM5 infection. Thus, by in vivo mAb blocking experiments with anti-CD154, we have previously determined that the requirement for CD154/CD40 interactions persisted from the time of infection through at least 3 to 4 weeks p.i., but was not involved at later time points p.i. (Green et al., 1996; Green at al., 1998). This disconnect between TCR ligation and the ability of the pathogenic CD4 T cells to mediate MAIDS, presumably via CD154 induction (at least in part), was underscored by the results obtained with OT-II TCR Tg CD4 T cells. These OVA/I-Ab specific CD4 T cells clearly mediated LP-BM5 induced MAIDS, but did so only to a partial extent (Figs. 2, 3). However, the in vivo introduction of cognate antigen in the form of OVA injections, which were shown to indeed activate OT-II CD4 T cells, apparently via Signal 1, etc., did not enhance the degree of disease (Fig. 4).
An alternative explanation is that CD4 T cell TCR ligation was accomplished instead by a possible SAg that had specificity for the Vβ TCR chains of the MAIDS-susceptible OT-II and ANDb/b (but not the MAIDS-insusceptible TEa, Fig. 5) TCR Tg strains – i.e. Vβ3, 5.1/5.2. However, comparison of the ANDb/b stain, which is on the B6 background, to the identical Tg TCR selected instead on the B10.BR (H-2k) background, i. e. the ANDk/k stain, provided clear evidence that a SAg does not seem to be involved. Thus, in a set of experiments that appropriately controlled for both genetic background differences that could have affected overall MAIDS susceptibility and a variable percentage of contaminating non Tg CD4 T cells, only the ANDb/b TCR Tg CD4 T cells were able to mediate LP-BM5 retrovirus induced MAIDS (Figs. 5 to 7). The inability of the ANDk/k TCR Tg CD4 T cells, expressing the exact same TCR, to mediate disease was inconsistent with the possibility that a SAg existing in this system drives TCR ligation and generation of Signal 1 to induce CD154 expression. In addition, our assessment of viral load by quantative RT-PCR (qRT-PCR) assays for the BM5def and BM5eco viruses indicated that the MAIDS resistance of the ANDk/k TCR Tg strain was not due to an inability of the virus to initially infect or spread in this strain, or in TCRα-/- recipients reconstituted with ANDk/k TCR Tg CD4 T cells (Fig. 8). Also of interest and related to the nature of the interaction in MAIDS between CD4 CD154+ T cells and CD40+ B cells, our ability to induce disease by adoptive transfer of control polyclonal B10.BR (H-2k) CD4 T cells into B6. TCRα-/- recipient providing B cells of H-2b origin, is consistent with a study by Gilmore et al. This study showed MAIDS development in LP-BM5 infected allochimeric reconstituted SCID mice, where the CD4 T cells and B cells were also MHC-incompatible in the sense that the T cells were restricted by an MHC-haplotype not expressed by the B cells (Gilmore at al., 1993). These studies argue, as do the present data, against conventional TCR recognition of MHC class II/peptide complexes on a B-cell “APC” in MAIDS development. In addition, both this report and several others (Hugin et. al, 1991; Muralidhar et al., 1992; Gilmore at al., 1993; McCarty et al., 1996; Doyon et al., 1996; Hayashi and Kanagawa, 1999) have argued against the presence of a SAg in MAIDS, particularly the one proposed early on as expressed on a terminal B-cell lymphoma line from a B6 mouse with end-stage MAIDS (Hugin at al., 1991).
Our results on these two AND TCR Tg strains are interesting to compare to those of Koch et al (Koch at al., 1994) who utilized AND TCR transgenes crossed onto a (B10.BR×B6) F1, H-2b/k, background in an attempt to maximize the percent of naïve CD4 T cells on a MAIDS-susceptible genetic background. Thus, presumably both I-Ab (ANDb/b) and I-Ek (ANDk/k) restricted TCR Tg CD4 T cells were present in these F1 mice. Thus, while the MAIDS susceptibility of these AND TCR Tg F1 mice allowed conclusions to be drawn suggesting that naive CD4 T cells were as efficient as memory CD4 T cells (obtained via other approaches) in their ability to mediate MAIDS, it was not possible to distinguish whether the class II MHC restricted H-2b vs H-2k AND TCR Tg CD4 T cells were the ones mediating LP-BM5-induced disease. Of note also, the percentage of Tg (Vβ3Vα11) CD4 T cells in uninfected F1 mice in these experiments was reported to be 79-88%, leaving open the possibility that non-Tg CD4 T cells may have participated in disease pathogenesis.
If TCR ligation is not required to activate CD4 T cells to express CD154 and otherwise program them so that they can mediate LP-BM5-induced MAIDS, then how does CD4 T-cell activation and CD154 expression occur? Although there are many possibilities, one intriguing mechanism would center on direct effects of infection by the BM5def virus. Thus, whereas early reports focused on B cells as the predominant cell type that is infected and expresses def-gag (Kim et al., 1994; Klinken et al., 1988), we and others have shown that other types of cells are also infected, including CD4 T cells as assessed by qRT-PCR (Cheung et al., 1991; Kim et al., 1994; K. Green, unpublished data). However, infection of a cell and expression of viral products does not in itself guarantee that infection of this cell is central to viral pathogenesis. Rather, such correlations must be extended to experiments testing the functional significance of infection/virus expression, and further studies will be required in which “infectable” versus “non-infectable” CD4 T cells can be compared in parallel. Another informative approach may be the comparison of those TCR Tg CD4 T cells that support LP-BM5 pathogenesis (OT-II and ANDb/b) versus those that do not (ANDk/k and TEa). Our preliminary flow cytometric analyses have not revealed any consistent differences relating to markers (CD44, CD69, CD45RB, and CD62L) that are together indicative of the naïve versus an activated phenotype, etc. for these two functional sets of TCR Tg strains. Similarly, although we were unable to consistently demonstrate expression of cell surface CD154 by any of the polyclonal or TCR Tg CD4 T cells when examined immediately ex vivo from infected (or uninfected) mice, after anti-CD3 mAb stimulation in vitro (Signal 1), equivalent CD154 expression was detected across the TCR Tg sources of CD4 T cells that did, or did not, mediate MAIDS. Thus, there was no evidence for intrinsic differences in the ability of the different TCR Tg CD4 T cells to be induced to express CD154. More in-depth analyses are required, however, which may add further insight into the mechanisms by which CD4 T cells are stimulated during LP-BM5 infection, induce CD154 expression, and develop into pathogenic effector cells that mediate MAIDS.
Materials and Methods
Mice
B6, B6.nude, B6.TCRα-/- and B6.RAG-1-/- breeder mice were purchased from Jackson labs (Bar Harbor, ME) and were expanded at the Dartmouth Medical School Animal Resource Center. Breeding pairs for the following TCR Tg strains on MAIDS genetic susceptible backgrounds with irrelevant peptide/MHC specificity were obtained (Table 1): OT-II mice (Dr. Alexander Y. Rudensky, University of Washington School of Medicine, Seattle, WA), specific for OVA323-339; TEa mice (Dr. Randolph J. Noelle, Dartmouth Medical School, Hanover, NH), specific for Eα52-68; and AND (H-2b/b) and AND (H-2k/k) mice (Dr. Susan Swain, Trudeau Institute, Saranac Lake, NY), specific for PCC81-104. These TCR Tg strains were used as a source of TCR Tg CD4 T cells, or were crossed onto either a B6.TCRα-/- or a B6.RAG-1-/- background.
LP-BM5 virus inoculations
LP-BM5 virus was prepared as previously described (Klinken et al., 1988). Briefly, G6 cells, cloned from SC-1 fibroblasts infected with the LP-BM5 virus mixture, and originally provided by Janet Hartley and Herbert Morse (NIH/NIAID, Bethesda, MD), were cocultured with uninfected SC-1 cells, and viral supernates were obtained. Mice were infected i.p. with 1 × 105 ecotropic PFU virus stock unless otherwise indicated.
Purification of CD19 B cells or CD4 T cells from B6 or TCR Tg donors
CD4 T cells or CD19 B cells from normal w.t. B6 donors were purified with Miltenyi beads, using either anti-CD4 or -CD19 beads following the manufacturer's protocol, with cell subset purity ≥ 98%, as detected by flow cytometric analysis. Purified CD4 T cells (1 × 107, unless otherwise indicated) were adoptively transferred (i.v.) into either B6.nude or B6.TCRα-/- recipients; or CD4 T cells (2 × 107) and CD19 B cells (1 × 108) were adoptively transferred into either the B6.RAG-1-/- strain, or B6.RAG-1-/- littermate offspring from crosses of B6.RAG-1-/- mice with either TEa or ANDb/b TCR Tg mice. TCR Tg+ CD4 T cells, derived from TCR Tg strains (Table 1), were purified and used in similar reconstitution experiments. A portion of the recipients was infected with LP-BM5 48 hours after adoptive transfer, and the remaining mice receiving adoptive cell transfer served as uninfected controls.
(i) Purification of OT-II TCR Tg CD4 T cells
Splenocytes from OT-II TCR Tg donors were labeled with FITC anti-TCRVβ5.1, 5.2, followed by anti-FITC beads for Miltenyi separation. Purified OT-II TCR Tg CD4 T cells were stained by use of FITC anti-TCRVβ5.1, 5.2 and PE anti-CD4, with analysis by FACSCalibur (BD Bioscience) to check the purity (≥ 98%). Appropriate FITC and PE conjugated Ig isotypes of irrelevant specificity were used as controls.
(ii) Purification of CD4 T cells from ANDb/b and ANDk/k TCR Tg donors
Splenocytes from ANDb/b and ANDk/k TCR Tg donors were labeled with FITC anti-TCRVβ3, followed by anti-FITC beads for Miltenyi separation. Purified (≥ 98%) TCR Tg+ CD4 T cells, or w.t. polyclonal B6 CD4 T cells purified as positive controls, were adoptively transferred into B6.TCRα-/- recipients.
Crossing various TCR Tg strains onto either a B6.TCRα-/- or a B6.RAG-1-/- background
(i) The OT-II TCR Tg strain onto a B6.TCRα-/- background
OT-II TCR Tg male and B6.TCRα-/- female mice were bred, and the male Tg+ F1 offspring were bred back to B6.TCRα-/- females. The backcrossed offspring were screened by flow cytometry for Tg positivity via use of FITC anti-TCRVβ5.1,5.2; PE anti-CD4; and appropriate FITC and PE conjugated Ig isotypes of irrelevant specificity; and for TCRα-/- status by PCR (Mombaerts et al., 1992). To study the effect of ovalbumin (OVA) immunization, these OT-II TCR Tg+ TCRα-/- experimental mice were injected i.p. with OVA during the course of LP-BM5 infection: 50/ug of OVA in CFA per mouse at day -1 p.i., and either 50/ug of OVA in IFA at days 10 and 20 p.i., or at days 12, 24 and 36 p.i. Peripheral blood lymphocyte samples, obtained at days 2, 5, 14, 25 and 39 p.i., were analyzed by flow cytometry for markers of CD4 T cell activation: CD62L and CD69; and functional assessment of their ability to proliferate in response to OVA stimulation.
(ii) The ANDb/b TCR Tg strain onto a B6.RAG-1-/- background
ANDb/b TCR Tg mice were crossed with B6.RAG-1-/- mice, and F1 Tg+ offspring were bred back to B6.RAG-1-/- mice, with the resulting offspring screened by flow cytometry using the following mAbs: FITC anti-TCRVβ3, PE anti-CD19, APC anti-CD4 and appropriate FITC, PE, and APC conjugated Ig isotypes controls. Of the offspring approximately 1/4 were ANDb/b TCR Tg+ RAG-1-/- (Tg+, CD4+, CD19-) mice; these mice were recipients of adoptive transfer of CD19 B cells from B6 donors as the experimental group. Another 1/4 of the offspring were ANDb/b TCR Tg- RAG-1-/- (Tg-, CD4-, CD19-) mice; these mice were recipients of adoptive transfer of CD19 B cells plus polyclonal CD4 T cells from w.t. B6 donors as a positive control group.
(iii) The TEa TCR Tg strain onto a B6.RAG-1-/- background
TEa TCR Tg mice, already crossed onto a B6.RAG-1-/- background, were bred with B6.RAG-1-/- mice and the progeny screened for Tg status and RAG-1-/- background with flow cytometry by use of FITC anti-TCRVβ6, PE anti-CD19, APC anti-CD4 mAbs, and appropriate FITC, PE, and APC conjugated Ig isotype of controls. Of the offspring about 1/2 were TEa TCR Tg+ RAG-1-/- (Tg+, CD4+, CD19-) mice; these mice were recipients of adoptive transfer of CD19 B cells from B6 donors as the experimental group. The other ∼1/2 of the offspring were TEa TCR Tg- RAG-1-/- (Tg-, CD4-, CD19-) mice; these mice were recipients of adoptive transfer of CD19 B cells plus CD4 T cells from B6 donors as a positive control group.
ELISA determinations of serum Ig
For measuring hyper-Ig, goat anti-mouse IgM and IgG2a antibody (Southern Biotechnology, Birmingham, AL), was coated in 96-well enzyme-linked immunosorbent assay (ELISA)-grade plates (Becton Dickinson, Oxford, CA) as previously published (Li and Green, 2006). Sera, from control uninfected mice, or obtained at various times post LP-BM5 infection, including at the final termination time point, were then plated and followed with alkaline phosphatase-labeled goat anti-mouse Ig (Southern Biotechnology). After incubation, addition of p-nitrophenyl phosphate as substrate (Sigma) provided a colorimetric change, which was then quantitated at 405 nm by an ELISA reader (Dynatech Laboratories, South Hampton, UK).
Spleen cell responses to mitogen
As previously detailed (Li and Green, 2006), spleen cells (4 × 105/per well) were plated into 96-well plates with a final concentration of 10 μg/ml LPS. After 72 h, wells were pulsed with 1 μCi of [3H] thymidine (Dupont NEN, Boston, Mass.) and harvested 6 h later onto Unifilter 96 well GF/C plates for assessment of thymidine incorporation by scintillation counting (Packard MicroSant NXT Counter).
RNA isolation and real-time quantitative RT-PCR
Viral load was determined for BM5def and BM5eco as previously described (Cook et al., 2003; Green et al., 2004). Briefly, total RNA was isolated from spleen tissue using Tri-Reagent (Molecular Research Center, Cincinnati, OH) and treated with DNA-free kit (Ambion, Austin, TX). Following reverse transcriptase amplification of cDNA (Bio-Rad iScript cDNA Synthesis kit), quantitative RT-PCR was performed using iQ SYBR Green Supermix and iCycler software (Bio-Rad, Hercules, CA).
Statistical analyses
Multiple group comparisons have been conducted to test possible group differences using one-way ANOVA and SPSS software. To control for type I error due to multiple tests, the BONFERRONI correction has been applied. The results were significant (*, P < 0.05) or not significant (NS, P ≥ 0.05).
Acknowledgments
We wish to thank Kathy Green, who prepared the LP-BM5 viral stocks, and On Ho for helpful discussions. We also thank Dr. Alexander Y. Rudensky for the OT-II TCR Tg mice, Dr. Randolph J. Noelle for the TEa TCR Tg mice, and Dr. Susan Swain for the ANDb/b and ANDk/k TCR Tg mice; and Alice Givan and Gary Ward for help with flow cytometry.
This work was supported by National Institutes of Health Grants CA50157 and AI059580 (to W.R.G.); NIH/NIAID T32 Training Grant AI07363 supported W.L. Flow cytometry was performed at Dartmouth Medical School in the Herbert C. Englert Cell Analysis Laboratory, which was established by equipment grants from the Fannie E. Rippel Foundation, the NIH Shared Instrument Program, and Dartmouth Medical School; and is supported in part by the Core Grant (CA 23108) from the National Cancer Institute to the Norris Cotton Cancer Center.
Footnotes
The authors have no conflicting financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen RC, Armitage RJ, Conley ME, Rosenblatt H, Jenkins NA, Copeland NG, Bedell MA, Edelhoff S, Disteche CM, Simoneaux DK, et al. CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science. 1993;259(5097):990–993. doi: 10.1126/science.7679801. [DOI] [PubMed] [Google Scholar]
- Aziz DC, Hanna Z, Jolicoeur P. Severe immunodeficiency disease induced by a defective murine leukaemia virus. Nature. 1989;338(6215):505–508. doi: 10.1038/338505a0. [DOI] [PubMed] [Google Scholar]
- Battaglia E, Biancone L, Resegotti A, Emanuelli G, Fronda GR, Camussi G. Expression of CD40 and its ligand, CD40L, in intestinal lesions of Crohn's disease. Am J Gastroenterol. 1999;94(11):3279–3284. doi: 10.1111/j.1572-0241.1999.01538.x. [DOI] [PubMed] [Google Scholar]
- Campbell KA, Ovendale PJ, Kennedy MK, Fanslow WC, Reed SG, Maliszewski CR. CD40 ligand is required for protective cell-mediated immunity to Leishmania major. Immunity. 1996;4(3):283–289. doi: 10.1016/s1074-7613(00)80436-7. [DOI] [PubMed] [Google Scholar]
- Casabianca A, Orlandi C, Fraternale A, Magnani M. A new one-step RT-PCR method for virus quantitation in murine AIDS. J Virol Methods. 2003;110(1):81–90. doi: 10.1016/s0166-0934(03)00104-6. [DOI] [PubMed] [Google Scholar]
- Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I, Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J Exp Med. 1994;180(4):1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184(2):747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerny A, Hugin AW, Hardy RR, Hayakawa K, Zinkernagel RM, Makino M, Morse HC., 3rd B cells are required for induction of T cell abnormalities in a murine retrovirus-induced immunodeficiency syndrome. J Exp Med. 1990;171:315–320. doi: 10.1084/jem.171.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung SC, Chattopadhyay SK, Hartley JW, Morse HC, 3rd, Pitha PM. Aberrant expression of cytokine genes in peritoneal macrophages from mice infected with LP-BM5 MuLV, a murine model of AIDS. J Immunol. 1991;146(1):121–127. [PubMed] [Google Scholar]
- Cook WJ, Green KA, Obar JJ, Green WR. Quantitative analysis of LP-BM5 murine leukemia retrovirus RNA using real-time RT-PCR. J Virol Methods. 2003;108(1):49–58. doi: 10.1016/s0166-0934(02)00256-2. [DOI] [PubMed] [Google Scholar]
- Danese S, Sans M, Scaldaferri F, Sgambato A, Rutella S, Cittadini A, Pique JM, Panes J, Katz JA, Gasbarrini A, Fiocchi C. TNF-alpha blockade down-regulates the CD40/CD40L pathway in the mucosal microcirculation: a novel anti-inflammatory mechanism of infliximab in Crohn's disease. J Immunol. 2006;176(4):2617–2624. doi: 10.4049/jimmunol.176.4.2617. [DOI] [PubMed] [Google Scholar]
- Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest. 1996;97(9):2063–2073. doi: 10.1172/JCI118643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon L, Simard C, Sekaly RP, Jolicoeur P. Evidence that the murine AIDS defective virus does not encode a superantigen. J Virol. 1996;70(1):1–9. doi: 10.1128/jvi.70.1.1-9.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure GC, Bensoussan-Lejzerowicz D, Bene MC, Aubert V, Leclere J. Coexpression of CD40 and class II antigen HLA-DR in Graves' disease thyroid epithelial cells. Clin Immunol Immunopathol. 1997;84(2):212–215. doi: 10.1006/clin.1997.4391. [DOI] [PubMed] [Google Scholar]
- Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJ, Claassen E. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A. 1996;93(6):2499–2504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese NA, Giese T, Morse HC., 3rd Murine AIDS is an antigen-driven disease: requirements for major histocompatibility complex class II expression and CD4+ T cells. J Virol. 1994;68(9):5819–5824. doi: 10.1128/jvi.68.9.5819-5824.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore GL, Cowing C, Mosier DE. LP-BM5 murine retrovirus-induced immunodeficiency disease in allogeneic SCID chimeric mice. Inability to recognize a putative viral superantigen does not prevent induction of disease. J Immunol. 1993;150(1):185–189. [PubMed] [Google Scholar]
- Green KA, Ahonen CL, Cook WJ, Green WR. CD40-associated TRAF 6 signaling is required for disease induction in a retrovirus-induced murine immunodeficiency. J Virol. 2004;78(11):6055–6060. doi: 10.1128/JVI.78.11.6055-6060.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KA, Cook WJ, Sharpe AH, Green WR. The CD154/CD40 interaction required for retrovirus-induced murine immunodeficiency syndrome is not mediated by upregulation of the CD80/CD86 costimulatory molecules. J Virol. 2002;76(24):13106–13110. doi: 10.1128/JVI.76.24.13106-13110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KA, Crassi KM, Laman JD, Schoneveld A, Strawbridge RR, Foy TM, Noelle RJ, Green WR. Antibody to the ligand for CD40 (gp39) inhibits murine AIDS-associated splenomegaly, hypergammaglobulinemia, and immunodeficiency in disease-susceptible C57BL/6 mice. J Virol. 1996;70(4):2569–2575. doi: 10.1128/jvi.70.4.2569-2575.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KA, Noelle RJ, Durell BG, Green WR. Characterization of the CD154-positive and CD40-positive cellular subsets required for pathogenesis in retrovirus-induced murine immunodeficiency. J Virol. 2001;75(8):3581–3589. doi: 10.1128/JVI.75.8.3581-3589.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KA, Noelle RJ, Green WR. Evidence for a continued requirement for CD40/CD40 ligand (CD154) interactions in the progression of LP-BM5 retrovirus-induced murine AIDS. Virology. 1998;241(2):260–268. doi: 10.1006/viro.1997.8970. [DOI] [PubMed] [Google Scholar]
- Grewal IS, Borrow P, Pamer EG, Oldstone MB, Flavell RA. The CD40-CD154 system in anti-infective host defense. Curr Opin Immunol. 1997;9(4):491–497. doi: 10.1016/s0952-7915(97)80100-8. [DOI] [PubMed] [Google Scholar]
- Grewal IS, Xu J, Flavell RA. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature. 1995;378(6557):617–620. doi: 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- Hayashi RJ, Kanagawa O. Unique CD4(+) T cells in TCR alpha chain-deficient class I MHC-restricted TCR transgenic mice: role in a superantigen-mediated disease process. Int Immunol. 1999;11(9):1581–1590. doi: 10.1093/intimm/11.9.1581. [DOI] [PubMed] [Google Scholar]
- Hugin AW, Vacchio MS, Morse HC., 3rd A virus-encoded “superantigen” in a retrovirus-induced immunodeficiency syndrome of mice. Science. 1991;252(5004):424–427. doi: 10.1126/science.1850169. [DOI] [PubMed] [Google Scholar]
- Jolicoeur P. Murine acquired immunodeficiency syndrome (MAIDS): an animal model to study the AIDS pathogenesis. Faseb J. 1991;5(10):2398–2405. doi: 10.1096/fasebj.5.10.2065888. [DOI] [PubMed] [Google Scholar]
- Kim WK, Tang Y, Kenny JJ, Longo DL, Morse HC., 3rd In murine AIDS, B cells are early targets of defective virus and are required for efficient infection and expression of defective virus in T cells and macrophages. J Virol. 1994;68(10):6767–6769. doi: 10.1128/jvi.68.10.6767-6769.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinken SP, Fredrickson TN, Hartley JW, Yetter RA, Morse HC., 3rd Evolution of B cell lineage lymphomas in mice with a retrovirus-induced immunodeficiency syndrome, MAIDS. J Immunol. 1988;140(4):1123–1131. [PubMed] [Google Scholar]
- Klinman DM, Morse HC., 3rd Characteristic of B cell proliferation and action in murine AIDS. J Immunol. 1989;142(4):1144–1149. [PubMed] [Google Scholar]
- Koch S, Muralidhar G, Swain SL. Both naive and memory CD4 T cell subsets become anergic during MAIDS and each subset can sustain disease. J Immunol. 1994;152(11):5548–5556. [PubMed] [Google Scholar]
- Koshy M, Berger D, Crow MK. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. J Clin Invest. 1996;98(3):826–837. doi: 10.1172/JCI118855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laman JD, Claassen E, Noelle RJ. Functions of CD40 and its ligand, gp39 (CD40L) Crit Rev Immunol. 1996;16(1):59–108. doi: 10.1615/critrevimmunol.v16.i1.40. [DOI] [PubMed] [Google Scholar]
- Latarjet R, Duplan JF. Experiments and discussion on leukamogenesis by cell-free extracts of radiation-induced leukemia in mice. Int J Radiat Biol. 1962;(5):339–344. doi: 10.1080/09553006214550911. [DOI] [PubMed] [Google Scholar]
- Li W, Buzoni-Gatel D, Debbabi H, Hu MS, Mennechet FJ, Durell BG, Noelle RJ, Kasper LH. CD40/CD154 ligation is required for the development of acute ileitis following oral infection with an intracellular pathogen in mice. Gastroenterology. 2002;122(3):762–773. doi: 10.1053/gast.2002.31888. [DOI] [PubMed] [Google Scholar]
- Li W, Green WR. The role of CD4 T cells in the pathogenesis of murine AIDS. J Virol. 2006;80(12):5777–5789. doi: 10.1128/JVI.02711-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Colpaert S, D'Haens GR, Kasran A, de Boer M, Rutgeerts P, Geboes K, Ceuppens JL. Hyperexpression of CD40 ligand (CD154) in inflammatory bowel disease and its contribution to pathogenic cytokine production. J Immunol. 1999;163(7):4049–4057. [PubMed] [Google Scholar]
- MacDonald KP, Nishioka Y, Lipsky PE, Thomas R. Functional CD40 ligand is expressed by T cells in rheumatoid arthritis. J Clin Invest. 1997;100(9):2404–2414. doi: 10.1172/JCI119781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino M, Morse HC, 3rd, Fredrickson TN, Hartley JW. H-2-associated and background genes influence the development of a murine retrovirus-induced immunodeficiency syndrome. J Immunol. 1990;144(11):4347–4355. [PubMed] [Google Scholar]
- McCarty TC, Chattopadhyay SK, Scherer MT, Fredrickson TN, Hartley JW, Morse HC., 3rd Endogenous Mtv-encoded superantigens are not required for development of murine AIDS. J Virol. 1996;70(11):8148–8150. doi: 10.1128/jvi.70.11.8148-8150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ichikawa Y, Jaenisch R, Hooper ML, et al. Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature. 1992;360(6401):225–231. doi: 10.1038/360225a0. [DOI] [PubMed] [Google Scholar]
- Morawetz RA, Giese NA, Gabriele L, Rothman P, Horak I, Ozato K, Morse HC., 3rd Relationship of cytokines and cytokine signaling to immunodeficiency disorders in the mouse. Braz J Med Biol Res. 1998;31(1):61–67. doi: 10.1590/s0100-879x1998000100008. [DOI] [PubMed] [Google Scholar]
- Morse HC, 3rd, Chattopadhyay SK, Makino M, Fredrickson TN, Hugin AW, Hartley JW. Retrovirus-induced immunodeficiency in the mouse: MAIDS as a model for AIDS. Aids. 1992;6(7):607–621. doi: 10.1097/00002030-199207000-00001. [DOI] [PubMed] [Google Scholar]
- Mosier DE, Yetter RA, Morse HC., 3rd Retroviral induction of acute lymphoproliferative disease and profound immunosuppression in adult C57BL/6 mice. J Exp Med. 1985;161(4):766–784. doi: 10.1084/jem.161.4.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosier DE, Yetter RA, Morse HC., 3rd Functional T lymphocytes are required for a murine retrovirus-induced immunodeficiency disease (MAIDS) J Exp Med. 1987;165(6):1737–1742. doi: 10.1084/jem.165.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidhar G, Koch S, Haas M, Swain SL. CD4 T cells in murine acquired immunodeficiency syndrome: polyclonal progression to anergy. J Exp Med. 1992;175(6):1589–1599. doi: 10.1084/jem.175.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peguet-Navarro J, Dalbiez-Gauthier C, Rattis FM, Van Kooten C, Banchereau J, Schmitt D. Functional expression of CD40 antigen on human epidermal Langerhans cells. J Immunol. 1995;155(9):4241–4247. [PubMed] [Google Scholar]
- Polese L, Angriman I, Cecchetto A, Norberto L, Scarpa M, Ruffolo C, Barollo M, Sommariva A, D'Amico DF. The role of CD40 in ulcerative colitis: histochemical analysis and clinical correlation. Eur J Gastroenterol Hepatol. 2002;14(3):237–241. doi: 10.1097/00042737-200203000-00006. [DOI] [PubMed] [Google Scholar]
- Reichmann G, Walker W, Villegas EN, Craig L, Cai G, Alexander J, Hunter CA. The CD40/CD40 ligand interaction is required for resistance to toxoplasmic encephalitis. Infect Immun. 2000;68(3):1312–1318. doi: 10.1128/iai.68.3.1312-1318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reul RM, Fang JC, Denton MD, Geehan C, Long C, Mitchell RN, Ganz P, Briscoe DM. CD40 and CD40 ligand (CD154) are coexpressed on microvessels in vivo in human cardiac allograft rejection. Transplantation. 1997;64(12):1765–1774. doi: 10.1097/00007890-199712270-00025. [DOI] [PubMed] [Google Scholar]
- Simard C, Klein SJ, Mak T, Jolicoeur P. Studies of the susceptibility of nude, CD4 knockout, and SCID mutant mice to the disease induced by the murine AIDS defective virus. J Virol. 1997;71(4):3013–3022. doi: 10.1128/jvi.71.4.3013-3022.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong L, Xu JC, Grewal IS, Kima P, Sun J, Longley BJ, Jr, Ruddle NH, McMahon-Pratt D, Flavell RA. Disruption of CD40-CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity. 1996;4(3):263–273. doi: 10.1016/s1074-7613(00)80434-3. [DOI] [PubMed] [Google Scholar]
- Stout RD, Suttles J, Xu J, Grewal IS, Flavell RA. Impaired T cell-mediated macrophage activation in CD40 ligand-deficient mice. J Immunol. 1996;156(1):8–11. [PubMed] [Google Scholar]
- Tayar L, Higo K, Kubo Y, Wang Y, Lu LM, Zhang F, Iwatani Y, Wang L, Ono T, Maeda M, Sakai H, Ishimoto A. Induction of B-cell lymphoma in BALB/c nude mice with an ecotropic, B-tropic helper virus present in the murine AIDS virus stock. J Virol. 1999;73(2):1640–1644. doi: 10.1128/jvi.73.2.1640-1644.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, Flavell RA. Mice deficient for the CD40 ligand. Immunity. 1994;1(5):423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Yetter RA, Buller RM, Lee JS, Elkins KL, Mosier DE, Fredrickson TN, Morse HC., 3rd CD4+ T cells are required for development of a murine retrovirus-induced immunodeficiency syndrome (MAIDS) J Exp Med. 1988;168(2):623–635. doi: 10.1084/jem.168.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Morawetz RA, Chattopadhyay S, Makino M, Kishimoto T, Kikutani H. CD40-deficient mice infected with the defective murine leukemia virus LP-BM5def do not develop murine AIDS but produce IgE and IgG1 in vivo. Eur J Immunol. 1999;29(2):615–625. doi: 10.1002/(SICI)1521-4141(199902)29:02<615::AID-IMMU615>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]