

Abstract
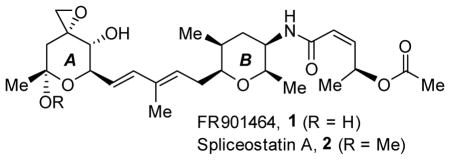
Enantioselective syntheses of FR901464 and Spliceostatin A, potent spliceosome inhibitors are described. The synthesis of FR901464 has been accomplished in a convergent manner in 10 linear steps (20 total steps). The synthesis of the A-tetrahydropyran ring was constructed from (R)-isopropylidene glyceraldehyde. The functionalized tetrahydropyran B-ring was synthesized utilizing a Corey-Bakshi-Shibata reduction, an Achmatowicz reaction, and a stereoselective Michael addition as the key steps. Coupling of A- and B-ring fragments was accomplished via cross-metathesis.
In 1996, Nakajima and co-workers from the Fujisawa Pharmaceutical Co. isolated FR901464 1 (Scheme 1) from the fermentation broth, Pseudomonas sp. No. 2663.1 FR901464 exhibited enhanced transcriptional activity of promoter SV40 at a very low concentration (10 nM). It exhibited remarkable antitumor activity, displaying IC50 values ranging from 0.6 – 3.4 nM against multiple human cancer cell lines. Furthermore, it showed significant effectiveness against human solid tumors implanted in mice at a dose range of 0.056 – 1 mg/kg.1b Subsequently, Yoshida and co-workers reported that a more stable methylated derivative of FR901464, named spliceostatin A 2, retained similar potent antitumor activity as FR90146. 2 More significantly, both FR901464 and spliceostatin A potently inhibited in vitro splicing and promoted pre-mRNA accumulation by binding to SF3b, a ribonuclear protein in the spliceosome.2b Thus, structural analogs of FR901464 may have potential clinical applications with a novel mechanism of action. The biology and chemistry of FR901464 attracted much interest among the synthetic community. The first total synthesis of FR901464 was accomplished by Jacobsen and co-workers.3 Two other syntheses were reported later by Kitahara and co-workers and Koide and co-workers. 4,5 The reported syntheses were carried out in a total of 29 to 41 chemical steps. In our continuing interests in natural products 6 that inhibit splicing activity, we sought to develop a convergent synthesis of FR901464 and spliceostatin A in an effort to facilitate the synthesis of structural variants. Herein, we report a concise, enantioselective synthesis of FR901464 which has been accomplised in 10 linear steps.
Scheme 1.

Retrosynthesis of FR901464.
As shown in Scheme 1, we planned to utilize cross-metathesis 7 to couple the epoxy alcohol segment 3 and the amide segment 4 at a late stage of the synthesis. A related strategy was utilized by Koide and co-workers.5b The highly functionalized tetrahydropyran ring (A) could be constructed by utilizing a versatile chiral pool (R)-isopropylidene glyceraldehyde 5. 8 The amide fragment 4 could be formed via coupling of amine 6 and acid 7. Amine 6 could be obtained by a reductive amination of pyranone derivative 8. The functionalized pyranone ring (B) would be derived from furan derivative 9 via an Achmatowicz rearrangement 9 as the key step. This optically active furan derivative could be obtained by CBS-reduction of a commercially available ketone. Acid 7 would be derived from the known optically active alcohol 10.10
The synthesis of epoxy alcohol segment 3 is shown in Scheme 2. Commercially available bromoketone 11 was protected as its dithiane derivative. Lithiation of the resulting dithiane with t-BuLi at −78 °C for 1h followed by reaction with (R)-isopropylidene glyceraldehyde provided a mixture (1:1) of diastereomers 12 and 13 in 61% yield in two steps.11 This lack of stereoselectivity was somewhat unexpected, especially given the presence of chelating atoms at both α and β positions of (R)-isopropylidene glyceraldehyde. In an attempt to improve anti-diastereoselectivity, we investigated this addition reaction in the presence of a number of Lewis acids such as CeCl312, ZnCl213 and MgBr214, in THF and ether. However, there was no further improvement in the diastereomeric ratio. The isomers were separated by silica gel chromatography. The syn-isomer 12 was converted to desired anti-isomer 13 by a Mitsunobu reaction in the presence of p-nitrobenzoic acid followed by NaOH-mediated hydrolysis of the benzoate ester. 15, 16 The hydroxyl group of 13 was protected as a PMB-ether and subsequent removal of the isopropylidene group was carried out by the addition of p-TsOH in a one-pot operation to provide diol 14. The primary alcohol was selectively mono-tosylated using TsCl and Et3N in the presence of dibutyltin oxide. Reaction of the resulting mono-tosylate with an excess of Corey-Chaykovsky dimethylsulfonium methylide,17 prepared by treatment of trimethylsulfonium iodide with n-BuLi, furnished allylic alcohol 15 in 84% yield. A similar functional group transformation was previously reported by Carreira and co-workers. 18 The dithiane group of 15 was then removed by using an excess of Hg(ClO4)2 in methanol in the presence of dry 2,6-lutidine. This condition resulted in the formation of the corresponding methyl ketal as a mixture of anomers, which upon treatment with a catalytic amount of p-TsOH in methanol at 0°C provided a single diastereomer 16.19 Removal of the PMB group in 16 with DDQ20 followed by alcohol directed epoxidation with m-CPBA afforded the desired epoxy alcohol segment 3 stereoselectively as a white solid in 19% overall yield from 11 (8 steps). The methyl ketal 3 is quite stable and easy to handle for subsequent reactions.
Scheme 2.

Synthesis of epoxy alcohol Segment 3
The preparation of the Z-allylic acetate side chain 7 is shown in Scheme 3. Optically active alcohol 9 was efficiently prepared by utilizing a catalytic asymmetric addition protocol reported by Trost and co-workers to provide 9 in 98% ee.10 Saponification of methyl ester 9 with aqueous LiOH followed by acetylation with acetyl chloride provided acetate 17 in excellent yield. Hydrogenation over Lindlar’s catalyst afforded the desired cis-alkene 7.
Scheme 3.

The synthesis of acid 7
The synthesis of amide segment 4 is shown in Scheme 4. Enantioselective reduction of commercially available acetyl furan 18 with (S)-2-Me-CBS and BH3•Me2S afforded chiral alcohol 9 in 94% yield (93% ee).21 An Achmatowicz rearrangement was then carried out by treatment of alcohol 9 with tBuO2H in the presence of a catalytic amount of VO(acac)2 to furnish a hemiketal,9 which was directly reduced to enone 19 as a single diastereomer by employing the protocol described by Kishi and co-workers.22 Our subsequent synthetic plan required installation of the C20 (S)-methyl-bearing stereocenter. We elected to carry out a 1,4-additon to enone 19. Accordingly, treatment of 19 with MeLi/CuBr•Me2S at −78 °C for 2h provided the desired pyranone 8 in excellent yield (92%) and diastereoselectivity (25:1 dr, by 1H and 13C-NMR analysis). The observed diastereoselectivity can be explained based upon the conformational analysis of enone 19. The stereochemical outcome of Michael addition can be rationalized by assuming stereoelectronically favorable axial attack of the cuprate as shown in the transition state model 20. 23 Further studies regarding the origin of high diastereoselectivity is under investigation.
Scheme 4.

Synthesis of Amide 4
Pyranone 8 and known alkene 21 24 were then subjected to cross-metathesis conditions using Grubbs’ 2nd generation catalyst 25 to provide the corresponding terminal tosylate. Treatment of the resulting tosylate with t-BuOK in DMSO at −75 °C for 12h resulted in diene 22 via base-promoted elimination in 41% yield over two steps.26 For the introduction of the amine group at C-14, we anticipated that a reductive amination could proceed stereoselectively via substrate control. Indeed, reductive amination of 22 with ammonium acetate and NaBH3CN afforded the corresponding primary amine 6 as a major product (6:1 dr by 1H- and 13C-NMR analysis). 27 The crude amine 6 and its epimer were directly treated with acid 7 using standard amidation conditions to give the amide 4 along with minor C-14 epimer, which were separated by column chromatography.
With the stereoselective syntheses of segments epoxy alcohol 3 and amide 4, we then turned our attention to construct the C6-C7 double bond of the target molecules. As shown in Scheme 5, cross-metathesis of the two fragments proceeded smoothly in the presence of Grubbs’ 2nd generation catalyst to afford spliceostatin A, 2 as a white solid in 57% isolated yield based upon one recycle of unreacted 3 and 4 under the same conditions. The removal of the methyl ketal in 2 was achieved by exposure of 2 to PPTS in wet THF28 at 0 °C which provided FR901464, 1 as a white powder in good yield. The 1H and 13C NMR of our synthetic FR901464 {[α]D23-13.0 (c 0.45, CH2Cl2)} is identical to the reported spectra of natural {[α]D23-12.0 (c 0.5, CH2Cl2)}1a and synthetic3–5 FR901464.
Scheme 5.

Synthesis of FR901464
In summary, we have accomplished a concise and enantioselective strategy for the syntheses of FR901464 and spliceostatin A in 20 and 19 total steps with the longest linear sequence of 10 and 9 steps, respectively. This represents a significant improvement over previously reported routes. The key features in our syntheses include the use of readily availalable chiral pool (R)-isopropylidene glyceraldyhyde 5 to form an A-ring fragment, a CBS reduction, an Achmatowicz rearrangement, and a stereoselective Michael addition for the construction of a B-ring fragment, and a cross-metathesis reaction for coupling the two fragments. The synthesis is short, convergent and amenable to the synthesis of structural variants. Further syntheses of structural variants and biological studies are in progress.
Supplementary Material
Acknowledgments
Financial support by the National Institutes of Health is gratefully acknowledged.
Footnotes
Supporting Information Available General experimental procedures, characterization data for all products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M. J Antibiot. 1996;49:1196–1203. doi: 10.7164/antibiotics.49.1196. [DOI] [PubMed] [Google Scholar]; (b) Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K. J Antibiot. 1996;49:1204–1211. doi: 10.7164/antibiotics.49.1204. [DOI] [PubMed] [Google Scholar]; (c) Nakajima H, Takase S, Terano H, Tanaka H. J Antibiot. 1997;50:96–99. doi: 10.7164/antibiotics.50.96. [DOI] [PubMed] [Google Scholar]
- 2.(a) Motoyoshi H, Horigome M, Ishigami K, Yoshida T, Horinouchi S, Yoshida M, Watanabe H, Kitahara T. Biosci Biotechnol Biochem. 2004;68:2178–2182. doi: 10.1271/bbb.68.2178. [DOI] [PubMed] [Google Scholar]; (b) Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, Tani T, Horinouchi S, Yoshida M. Nature Chem Biol. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]; (c) Zhang F, He HY, Tang MC, Tang YM, Zhou Q, Tang GL. J Am Chem Soc. 2011;133:2452–2462. doi: 10.1021/ja105649g. [DOI] [PubMed] [Google Scholar]; (d) Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. ACS Chem Biol. 2011;6:582–589. doi: 10.1021/cb100356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Thompson CF, Jamison TF, Jacobsen EN. J Am Chem Soc. 2000;122:10482–10483. [Google Scholar]; (b) Thompson CF, Jamison TF, Jacobsen EN. J Am Chem Soc. 2001;123:9974–9983. doi: 10.1021/ja016615t. [DOI] [PubMed] [Google Scholar]
- 4.(a) Horigome M, Motoyoshi H, Watanabe H, Kitahara T. Tetrahedron Lett. 2001;42:8207–8210. [Google Scholar]; (b) Motoyoshi H, Horigome M, Watanabe H, Kitahara T. Tetrahedron. 2006;62:1378–1389. [Google Scholar]
- 5.(a) Albert BJ, Koide K. Org Lett. 2004;6:3655–3658. doi: 10.1021/ol049160w. [DOI] [PubMed] [Google Scholar]; (b) Albert BJ, Sivaramakrishnan A, Naka T, Koide K. J Am Chem Soc. 2006;128:2792–2793. doi: 10.1021/ja058216u. [DOI] [PubMed] [Google Scholar]; (c) Albert BJ, Sivaramakrishnan A, Naka T, Czaicki NL, Koide K. J Am Chem Soc. 2007;129:2648–2659. doi: 10.1021/ja067870m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Ghosh AK, Anderson DD. Org Lett. 2012;14:4730–4733. doi: 10.1021/ol301886g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK, Li J. Org Lett. 2011;13:66–69. doi: 10.1021/ol102549a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]; (b) Prunet J. Curr Top Med Chem. 2005;5:1559–1577. doi: 10.2174/156802605775009801. [DOI] [PubMed] [Google Scholar]
- 8.(R)-isopropylidene glyceraldehyde is commercially available and also can be conveniently prepared from D-mannitol in gram-scale, see: Organic Syntheses, Coll. 1998;9:450.1995;72:6.
- 9.(a) Achmatowicz O, Bukowski P, Szechner B, Zwierzchowska Z, Zamojski A. Tetrahedron. 1971;27:1973–1996. [Google Scholar]; (b) Georgiadis MP, Albizati KF, Georgiadis TM. Org Prep Proc Int. 1992;24:95–118. [Google Scholar]
- 10.Trost BM, Quintard A. Org Lett. 2012;14:4698–4700. doi: 10.1021/ol302074h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The absolute stereochemistry of 12 and 13 were ultimately identified by comparing the 2D-NMR spectroscopy of advanced intermediate 16 and its C-4 epimer.
- 12.Chen ZH, Tu YQ, Zhang SY, Zhang FM. Org Lett. 2011;13:724–727. doi: 10.1021/ol102955e. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Rohanna J, Zhou J, Lyer K, Rainier JD. J Am Chem Soc. 2011;133:3208–3216. doi: 10.1021/ja200089f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams DR, Fultz MW. J Am Chem Soc. 2005;127:14550–14551. doi: 10.1021/ja054201k. [DOI] [PubMed] [Google Scholar]
- 15.Prasad KR, Gholap SL. J Org Chem. 2008;73:2–11. doi: 10.1021/jo0702342. [DOI] [PubMed] [Google Scholar]
- 16.For the details, please see the Supporting Information.
- 17.(a) Corey EJ, Chaykovsky M. J Am Chem Soc. 1965;87:1353–1364. [Google Scholar]; (b) Alcaraz L, Harnett JJ, Mioskowski C, Martel JP, Legall T, Shin D-S, Falck JR. Tetrahedron Lett. 1994;35:5449–5452. [Google Scholar]
- 18.Bode JW, Carreira EM. J Org Chem. 2001;66:6410–6424. doi: 10.1021/jo015791h. [DOI] [PubMed] [Google Scholar]
- 19.(a) Williams DR, Jass PA, Tse HLA, Gaston RD. J Am Chem Soc. 1990;112:4552–4554. [Google Scholar]; (b) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew Chem, Int Ed. 2001;40:196–199. [PubMed] [Google Scholar]
- 20.Horita K, Yoshioka T, Tanaka T, Oikawa Y, Yonemitsu O. Tetrahedron. 1986;42:3021–3028. [Google Scholar]
- 21.(a) Corey EJ, Roberts BE. J Am Chem Soc. 1997;119:12425–12431. [Google Scholar]; (b) Gazaille JA, Abramite JA, Sammakia T. Org Lett. 2012;14:178–181. doi: 10.1021/ol202966m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis MD, Cha JK, Kishi Y. J Am Chem Soc. 1982;104:4976–4978. [Google Scholar]
- 23.Woodward S. Chem Soc Rev. 2000;29:393–401. [Google Scholar]
- 24.Ghosh AK, Nicponski DR. Org Lett. 2011;13:4328–4331. doi: 10.1021/ol2016675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 26.Paquette LA, Gugelchuk M, McLaughlin ML. J Org Chem. 1987;52:4732–4740. [Google Scholar]
- 27.Rafferty RJ, Williams RM. J Org Chem. 2012;77:519–524. doi: 10.1021/jo202139k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeChristopher BA, Loy BA, Marsden MD, Schrier AJ, Zack JA, Wender PA. Nature Chem. 2012;4:705–710. doi: 10.1038/nchem.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.