

Abstract
Disorder in the position of the dioxygen ligand is a well-known problem in dioxygen complexes and in particular in those of picket fence porphyrin species. The dynamics of Fe–O2 rotation and tert-butyl motion in three different picket fence porphyrin derivatives has been studied by a combination of multi-temperature X-ray structural studies and Mössbauer spectroscopy. The structural studies show that the motions of the dioxygen ligand also require motions of the protecting pickets of the ligand binding pocket. The two motions appear to be correlated and the temperature-dependent change in the O2 occupancies can not be governed by a simple Boltzmann distribution. The three [Fe(TpivPP)(RIm)(O2)] derivatives studied have RIm = 1-methyl-, 1-ethyl-, or 2-methylimidazole. In all three species there is a preferred orientation of the Fe–O2 moiety with respect to the trans imidazole ligand and the population of this orientation increases with decreasing temperature. In the 1-MeIm and 1-EtIm species, the Fe–O2 unit is approximately perpendicular to the imidazole plane whereas in the 2-MeHIm species the Fe–O2 unit is approximately parallel. This reflects the low energy required for rotation of the Fe–O2 unit and the small energy differences in populating the possible pocket quadrants. All dioxygen complexes have a crystallographically required twofold axis of symmetry that limits the accuracy of the determined Fe–O2 geometry. However, the 80 K structure of the 2-MeHIm derivative allowed for the resolution of the two bonded oxygen atom positions and provided the best geometric description for the Fe–O2 unit. The values determined are: Fe–O = 1.811(5) Å, Fe–O–O = 118.2(9)°, O–O = 1.281(12) Å, and an off-axis tilt of 6.2°. The demonstration of the off-axis tilt is a first. We present detailed temperature-dependent simulations of the Mössbauer spectra that model the changing value of the quadrupole splitting and line widths. Residuals to fits are poorer at higher temperature. We believe that this is consistent with the idea that population of the two conformers are related to the concomitant motions of both Fe–O2 rotations and motions of the protecting tert-butyl pickets.
Introduction
The dynamics and interactions of the diatomic molecules, O2, NO, and CO that ligate the hemoproteins and related small molecules, are fundamental to understanding heme functions.1, 2 Oxyheme proteins continue to be the focus of much current research because of their important physiological roles. In spite of a large number of experimental investigations, substantial aspects of the electronic structure, biophysical properties and geometry of the oxyheme unit remain in doubt.3, 4, 5
The diamagnetism of oxyhemoglobin and the electronic configuration of FeII(S=0)–O2(S=0) was first assigned by Pauling and Coryell in 1936.6 Paramagnetic susceptibility with a thermally accessible triplet state S = 1 had once been claimed,7 but eventually the diamagnetism (over entire temperature range) was reconfirmed.8 The precise nature of the Fe–O2 bonding has been contentious. In addition to the Pauling model with its singlet dioxygen,6, 9 Weiss proposed the antiferromagnetically coupled model of FeIII(S=1/2)–O2−(S=1/2);10 and the ozone model FeII(S=1)–O2(S=1) of McClure,11 Harcourt,12, 13 and Goddard14 has also been put forward. Of these models, the Pauling model is closest to that supported by current data.
Some questions were resolved with the two available synthetic dioxygen structures of “picket fence” porphyrin derivatives: [Fe(TpivPP)(1-MeIm)(O2)]16, 17 and Fe(TpivPP)(2-MeHIm)(O2).18 The two structures were studied some time ago, when low-temperature studies were uncommon and precise structural details were hindered by the limited data and high thermal motion of the molecules.16b,19 Although the structures showed the dioxygen ligand bound to the iron in a bent, end-on Pauling fashion, the crystallographic data showed a large O2 disorder and consequent ambiguity in the position of the second oxygen atom. Information concerning subtle geometric effects, such as the off-axis tilts observed recently for NO complexes20, 21 were unavailable. Moreover, some of the structure-function relationships such as relative orientation of bound dioxygen and the proximal imidazole remained unexplored for lack of sufficient structural data.22 Only with low temperature structure determinations would such phenomena be observable, if then. Given the interesting ligand dynamics and other issues, we thought that a series of multiple-temperature X-ray studies could provide information on the O2 ligand rotations, much like those we were able to map out for several six-coordinate nitrosyl iron porphyrinates.23, 24 In these cases, the lower temperature structures resolved disorder, even though the rotation about the axial Fe–NO bond is a very low barrier process.
Mössbauer spectroscopy has been of great value in the study of electronic structure and Mössbauer studies have shown that the dynamics of the dioxygen complexes of hemes and heme proteins are especially rich. The oxyheme proteins and picket fence derivatives show similar but unusual Mössbauer spectra, which differ substantially from those of typical low-spin ferrous complexes. These distinctive features include (1) the anomalously large quadrupole splitting, ΔEQ ≥ 2 mm/s. Since in the low-spin ferrous state all three t2g orbitals would be doubly occupied, the electron shell should be almost spherical and the components of the electric field gradient (efg) tensor small. (2) An unexpected temperature dependence of the quadrupole splitting. The magnitude decreases with increasing temperature in oxymyoglobin and oxyhemoglobin as well as in the small molecule picket fence dioxygen derivatives. (3) Temperature-dependent line shape changes. The two lines of the quadrupole doublet broaden asymmetrically at intermediate temperatures. These features, all shown in spectra from the picket fence derivative [Fe(TpivPP)(1-MeIm)(O2)] were interpreted by Lang and co-workers as resulting from multiple configurations of the Fe–O2 unit because of rotation around the axial Fe–O bond.25 Later, Oldfield et al. proposed a different model to explain the anomalous temperature-dependent line width behavior for the Mössbauer spectra; the model also required rotations around the Fe–O bond.26
In this communication, we report multi-temperature structure determinations for [Fe(Tpiv-PP)(Im)(O2)] (Im = 1-MeIm, 1-EtIm, or 2-MeHIm) that shows that the population of the Fe–O2 conformers is temperature dependent. Further, for one system, we have been able to show that the Fe–O2 unit displays an off-axis tilt of the Fe–O2 bond. This structure provides the most accurate description of the geometry of the Fe–O2 unit currently available. Portions of the reported structure determinations were undertaken in support of Nuclear Resonance Vibrational Spectroscopy studies of the vibrational dynamics of the oxyiron systems. The results of those studies are now available.27 We have used recent enhancements in computing capabilities to better define the simulations of Mössbauer spectra of the three different dioxygen complexes. These new simulations demonstrate that the fits are poorest at higher temperatures consistent with the idea that the configurations are the result of concurrent motions of the O2 and the rotations of the protecting pickets. The multiple equilibrium processes lead to the failure of a simple Boltzmann distribution to completely model the experimental Mössbauer data.
Experimental Section
General Information
All reactions and manipulations were carried out under argon using a double-manifold vacuum line, Schlenkware and cannula techniques. Benzene and heptane were distilled over sodium/benzophenone. Chlorobenzene was washed with concentrated sulfuric acid, then with water until the aqueous layer was neutral, dried with MgSO4, and distilled twice over P2O5 under argon. Research grade oxygen (99.999%) was purchased from PRAXAIR and used as received. [H2(TpivPP)] and [Fe(TpivPP)Cl] was prepared according to a local modification of the reported synthesis.28 The 95% 57Fe enriched samples were prepared by a method similar to that of Landergren.29
Synthesis of [Fe(TpivPP)(1-EtIm)(O2)]
[Fe(TpivPP)(1-EtIm)2] was obtained from a local modification of the reported synthesis.16, 30 [Fe(TpivPP)(1-EtIm)2] (30 mg) was dried in vacuum for 30 mins. Drops of 1-ethylimidazole and benzene (~5 mL) were then transferred into the Schlenk by cannula. This mixture was gently heated with stirring and cooled to room temperature to give a clear red solution. Oxygen was then bubbled into this solution. X-ray quality crystals of [Fe(TpivPP)(1-EtIm)(O2)] were obtained in 8 mm × 250 mm sealed glass tubes by slow liquid diffusion using heptane as nonsolvent.
Synthesis of [Fe(TpivPP)(1-MeIm)(O2)]
Similar reaction procedures as above were performed with 1-MeIm in place of 1-EtIm. PhCl was used as solvent to give a similar red solution. After reaction with introduced oxygen, the solution was transferred into two glass tubes which had been placed inside a big Schlenk tube. 5 mL PhCl and 5 mL heptane were added outside of the two tubes. The system had been purged with argon, and then with oxygen. This big Schlenk tube was well-sealed, and crystals were obtained after 1 week.
Synthesis of [Fe(TpivPP)(2-MeHIm)(O2)]
Reduced [Fe(II)(TpivPP)]16d,30 was reacted with ~5 equiv of 2-methylimidazole in a mixed solution of benzene and ethanol. The mixture was stirred for 30 min and transferred into 8 mm × 250 mm glass tubes which were layered with heptane as nonsolvent. X-ray quality crystals of [Fe(TpivPP)(2-MeHIm)] were obtained after 2 weeks. Moderate-size single crystals were exposed at room temperature to pure dioxygen gas saturated with ethanol vapor. Crystals were obtained from exposure to (a) a 1 atm dioxygen atmosphere for 3 days or (b) to a 5 atm dioxygen atmosphere for 5 days in a high pressure reaction cylinder (Parr Instrument; Model 4635). After the oxygenation, the adducts were rapidly moved into the low temperature (100 K) nitrogen stream for structural and Mössbauer characterization.
Mössbauer Characterization
Mössbauer measurements were performed on a constant acceleration spectrometer from 15 to 298 K with optional small field (Knox College). Dark-red powder samples of [Fe(TpivPP)(1-RIm)(O2)] (R = Me or Et) were obtained from the reaction solution by slow addition of heptane. These oxygen complexes (160 mg, ~3 μmol 57Fe) were freshly filtered and immobilized with a minimum of Apiezon M grease. The sample was used immediately for the Mössbauer characterization. Crystals of [57Fe(TpivPP)(2-MeHIm)(O2)], 95% 57Fe, were prepared as noted above, crushed carefully to ensure its homogeneity and mulled with a minimum quantity of Apiezon M grease. The sample holder was sealed and used immediately for the Mössbauer characterization. This was the only sample that was measured with enriched iron; all other samples were based on natural abundance 57Fe. Replicate measurements on all three derivatives showed consistent T-dependent behavior. A sampling of spectra over the temperature range are given in the Supporting Information.
X-ray Structure Determinations
For [Fe(TpivPP)(1-MeIm)(O2)] structures, intensity data were collected on a D8 goniometer equipped with a Bruker APEXII CCD detector at Beamline 11.3.1 at the Advanced Light Source (Lawrence Berkeley National Laboratory) using synchrotron radiation tuned to λ = 0.7749 Å. 31 A series of 2-s data frames measured at 0.2° increments of ω were collected to calculate a unit cell. For data collection frames were measured for a duration of 2-s at 0.3° intervals of ω with a maximum 2θ value of ~60°. The data frames were collected using the program APEX2 and processed using the program SAINT routine within APEX2.32 For [Fe(TpivPP)(1-EtIm)(O2)], [Fe(TpivPP)(2-MeHIm)] and [Fe(TpivPP)(2-MeHIm)(O2)] structures, intensity data were collected on a Bruker Apex system with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å).
Absorption corrections were based on the multi-scan technique as implemented in SADABS.33 The structures were solved by direct methods using SHELXS-97 and refined against F2 using SHELXL-97;34, 35 subsequent difference Fourier syntheses led to the location of most of the remaining nonhydrogen atoms. For the structure refinement all data were used including negative intensities. All nonhydrogen atoms were refined anisotropically if not remarked otherwise below. Except as noted below, hydrogen atoms were idealized with the standard SHELXL-97 idealization methods. Solid-state analysis of crystal packing distances made use of the program MERCURY36 from the Cambridge Crystallographic Data Center. A brief summary of the crystallographic data is given in Table S1. Complete crystallographic details, atomic coordinates, anisotropic thermal parameters, and fixed hydrogen atom coordinates are given in the Supporting Information for the structures.
[Fe(TpivPP)(1-EtIm)(O2)]·C6H6 (100, 200, 300 K)
A black crystal with the dimensions 0.43 × 0.23 × 0.12 mm3, glued to the tip of a glass fiber, was used for the temperature-dependent structure determination. Measurement temperatures were 100, 200 and 300 K. In all structures, the asymmetric unit contains one-half picket fence porphyrin complex, and one-half benzene solvent molecule. There is a crystallographic twofold axis passing through the coordinated oxygen (O1) and the iron (Fe1) atoms. The 1-ethylimidazole ligand was found to disorder between two symmetry related sites. In rigid group refinements, an ideal 1-ethylimidazole group30 is applied to constrain this moiety. For each “picket”, the pivalamide methyl carbon atoms were found to disorder into two sets of positions. An ideal tetrahedral group37 is applied as rigid group to constrain each tert-butyl group. The occupancies of two pairs of tert-butyl groups are refined to be 0.555(6) (C19A, C20A, C21A and C22A) and 0.445(6) (C19B, C20B, C21B and C22B); 0.569(10) (C24A, C25A, C26A and C27A) and 0.431(10) (C24B, C25B, C26B and C27B). The terminal oxygen atom occupies two crystallographically independent positions and thus it exhibits fourfold positional disorder. The occupancy factors of two oxygen positions are refined by means of “free variables” and the final SOF are found to be 0.608(8) (O2A) and 0.392(8) (O2B). The fully occupied benzene solvent molecule lies on a twofold axis.
In the 200 and 300 K structures, the same crystal structure model and refinement procedures were applied. The terminal oxygen atom occupied two crystallographically independent positions and the final SOF are found to be 0.648(10) (O2A) and 0.352(10) (O2B) at 200K; 0.522(13) (O2A) and 0.478(13) (O2B) at 300K.
[Fe(TpivPP)(1-MeIm)(O2)]·0.48C6H5Cl·0.28C4H6N2 (100, 200, 300 K)
A black crystal with the dimensions 0.31 × 0.17 × 0.14 mm3 was used for structure determination. In the 100 K structure, the asymmetric unit contains one-half picket fence porphyrin complex, and disordered chlorobenzene and 1-methylimidazole solvent molecules. The porphyrin possessed a twofold axis, and the 1-methylimidazole disordered over two positions which were constrained by rigid group refinements. All the tert-butyl groups are constrained by rigid group refinements and the occupancy factors of disordered moieties are refined by means of “free variables”. The terminal oxygen atom occupies two crystallographically independent positions and thus it exhibits fourfold positional disorder. The occupancy factors of these two oxygen positions are refined to be 0.6740 and 0.326 by constraining the thermal parameter (Ueq) of the two positions to be the same. The partially occupied solvent molecules are heavily disordered at same position. The chlorobenzene is disordered between twofold symmetry related sites, while 1-methylimidazole appears as a half molecule with twofold symmetry. Their occupancies are refined to be 0.48 and 0.28 respectively.
In the 200 K structure, the terminal oxygen atom occupied two crystallographically independent positions and the final SOF are found to be 0.626 and 0.374. In the 300 K structure, the final SOF of terminal oxygen atom are refined to be 0.526 and 0.438.
[Fe(TpivPP)(2-MeHIm)(O2)]
[Fe(TpivPP)(2-MeHIm)(O2)] was prepared from the five-coordinate precursor [Fe(TpivPP)(2-MeHIm)], which had been structurally characterized before the oxygenation reaction. The first experiment used a crystal (A) with dimensions 0.30×0.26× 0.19 mm3 that had been reacted under a 1 atm dioxygen atmosphere. After the oxygenation, the crystal was rapidly moved to a diffractometer under a 100 K nitrogen stream, and X-ray data were collected. After the 100 K data collection, the crystal was slowly warmed to 300 K at 120 K/h under the N2 stream. Then the structural data were collected at 300 K.
A second crystal (B) with the dimensions 0.33 × 0.23 × 0.12 mm3 was oxygenated under a 5 atm dioxygen atmosphere. After oxygenation, the crystal was rapidly moved to the diffractometer under a 100 K nitrogen stream and structural data collected. After the 100 K data collection, the crystal was slowly warmed to 200 K at 120 K/h under the N2 stream, then the structural data were collected at 200 K. After this, the crystal was cooled to 100 K again and X-ray data were collected. Then the (same) crystal was further cooled to 80 K and structural data were collected. Finally the (same) crystal was warmed to 300 K to finish the final data collection at 300 K. Totally six X-ray data sets were collected for this crystal specimen including the precursor, five-coordinate [Fe(TpivPP)(2-MeHIm)]. During the whole procedure, the crystal was under the protection of N2 stream and the temperature was adjusted at a speed of 120 K/h.
The crystallographic data for all data sets were consistent with the monoclinic crystal system and space group C2/c; this is the same crystal system and space group as that of the five-coordinate precursor [Fe(TpivPP)(2-MeHIm)]. There is a crystallographic 2-fold axis passing through the iron (Fe1) and the coordinated oxygen (O1) atoms, except the 80 K structure of crystal (B) where the O1 tilt from the porphyrin normal is resolved and shows two oxygen positions. The 2-methylimidazole ligand is disordered between two symmetry related sites. The terminal oxygen atom (O2) occupies two crystallographically independent positions, and thus, it exhibits 4-fold positional disorder. The occupancy of the dioxygen ligand was carefully judged from the crystallographic results. First, the applied occupancy factor should give reasonable anisotropic displacement parameters (ADPs) compared to those of other atoms in the same molecule. Second, the command of FMAP-2 was applied to examine the resulting Fourier difference maps, especially the holes and peaks around the positions of dioxygen atoms. After careful refinement, the occupancy of dioxygen in the two 300 K structures were set at 0.65 (crystal A) and 0.90 (crystal B). The occupancy factors of the two terminal oxygen positions (O2A and O2B) are refined by means of “free variables” and the final SOF are given in the text. Two ethanol solvent molecules are found, and each is disordered around a 2-fold axis. In each structure, the first ethanol molecule is fully occupied; the second ethanol molecule occupancy was found to be variable.
Results
The synthesis, temperature-dependent molecular structures, and temperature-dependent Mössbauer characterization of three dioxygen iron picket fence porphyrin complexes are reported. The molecular structure determinations have been carried out over a range of temperatures between 80 and 300 K. The first two complexes, six-coordinate [Fe(TpivPP)(1-MeIm)(O2)] and [Fe(TpivPP)(1-EtIm)(O2)] were synthesized and crystallized from solution by means of vapor-liquid and liquid-liquid diffusion techniques, respectively. The third complex, [Fe(TpivPP)(2-MeHIm)(O2)], utilizing sterically hindered 2-methylimidazole, was synthesized from the heterogeneous reaction of gaseous dioxygen and crystalline, five-coordinate [Fe(TpivPP)(2-MeHIm)].18 Two crystal specimens of [Fe(TpivPP)(2-MeHIm)(O2)] (crystals A and B) were structurally characterized before and after the oxygenation reactions. The coordinated dioxygen ligand is disordered in all complexes with a total of four oxygen sites related in pairs by the required crystallographic twofold symmetry. The structure determinations show that the terminal oxygen atom occupancies between the two independent sites have a modest temperature dependence with one site pair becoming more populated with decreasing temperature. Attempts to achieve better ordering the coordinated dioxygen ligand with crystal annealing experiments, which were successful for a cobalt analog, were only partly successful for [Fe(TpivPP)(2-MeHIm)(O2)]. In that complex, the oxygen atom bonded to iron could be resolved into two positions and which provides the best description of the Fe–O–O geometry. This will be discussed subsequently. Selected parameters of the three oxyiron picket fence porphyrin structures are given in Tables 1 and 2.
Table 1.
Selected Structural Parameters for [Fe(TpivPP)(L)(O2)] (L = 1-EtIm and 1-MeIm)a
| Complex | T, K | Δ24b,c | (Fe–Np)av d | Fe–O1c | O1–O2Ac O1–O2Bc |
Fe–NImc | Fe–O1–O2Ae Fe–O1–O2Be |
γO2A, Be,f |
φO2Ae,g φO2Be,g |
φIme,g |
θO2A/Ime,h θO2B/Ime,h |
O2A/O2Bi | ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [Fe(TpivPP)-(1-MeIm)2)] | 100 | 0.04 | 1.992(3) | 1.9958(19) | 8.5 | 77.2 | 30 | ||||||
| 1.9921(18) | 21.1 | ||||||||||||
| [Fe(TpivPP)-(1-EtIm)2)] | 100 | 0.05 | 1.993(6) | 2.0244(18) | 6.6 | 62.4 | 30 | ||||||
| 1.9940(19) | 20.7 | ||||||||||||
| [Fe(TpivPP)-(1-EtIm)(O2)] | 100 | 0.008 | 1.9895(6) | 1.798(3) | 1.166(8) | 2.043(3) | 126.6(4) | 80.05 | 42.11 | 23.27 | 70.95 | 0.304(8) | tw |
| 1.111(12) | 132.4(7) | 37.93 | 29.22 | 0.196(8) | |||||||||
| 200 | 0.012 | 1.983(1) | 1.798(3) | 1.173(8) | 2.045(2) | 128.1(4) | 84.17 | 43.21 | 21.86 | 68.25 | 0.324(9) | ||
| 1.125(16) | 130.3(10) | 40.96 | 27.91 | 0.176(9) | |||||||||
| 300 | 0.009 | 1.9865(6) | 1.803(3) | 1.197(12) | 2.053(6) | 129.3(7) | 87.48 | 44.55 | 20.45 | 65.81 | 0.261(12) | ||
| 1.123(13) | 131.2(9) | 42.93 | 26.86 | 0.239(12) | |||||||||
| [Fe(TpivPP)-(1-MeIm)(O2)] | 100 | −0.004 | 1.991(3) | 1.864(4)j | 1.192(10) | 2.057(3) | 125.9(6) | 84.48 | 41.31 | 21.81 | 70.01 | 0.337 | tw |
| 1.143(19) | 127.4(13) | 43.17 | 26.01 | 0.163 | |||||||||
| 200 | 0.006 | 1.9995(17) | 1.856(4)j | 1.160(10) | 2.062(3) | 128.3(6) | 87.61 | 41.69 | 21.03 | 68.93 | 0.313 | ||
| 1.113(17) | 132.2(11) | 44.03 | 24.01 | 0.187 | |||||||||
| 300 | 0.008 | 1.9985(17) | 1.852(5)j | 1.106(13) | 2.085(4) | 135.4(9) | 87.58 | 44.18 | 21.43 | 66.20 | 0.281 | ||
| 1.093(17) | 131.9(12) | 43.40 | 27.25 | 0.219 | |||||||||
| [Fe(TpivPP)-(1-MeIm)(O2)] | 297 | 0.015 | 1.98(1) | 1.745(18) | 1.17(4) | 2.068(18) | 129(2) | 89.67 | 42.4 | 20 | 62.9 | 0.312 | 16b |
| 1.15(4) | 133(2) | 41.7 | 27.5 | 0.188 |
Estimated standard deviations are given in parentheses.
Displacement of iron atom from the 24-atom mean plane, a positive value is towards to the pocket porphyrin side.
Value in Å.
Average value in Å.
Value in degree.
Dihedral angle between the planes defined by Fe–O1–O2A and Fe–O1–O2B.
Dihedral angle between the planes defined by the closest Np–Fe–O1/Nax and the ligand plane (Fe–O1–O2A/B or imidazole plane).
Dihedral angle between proximate ligand plane (Fe–O1–O2A/B and imidazole plane) and unhindered side imidazole plane.
The occupancies of disordered terminal oxygen atoms is given; a second orientation with equal population is required by the twofold symmetry.
Value not reliable, see text.
Table 2.
Selected Structural Parameters for [Fe(TpivPP)(2-MeHIm)(O2)]a
| Complex | T, K | Δ24b,c | (Fe–Np)av d | Fe–O1c | O1–O2Ac O1–O2Bc |
Fe–NImc | Fe–O1–O2Ae Fe–O1–O2Be |
γO2A, Be,f |
φO2Ae,g φO2Be,g |
φIme,g |
θO2A/Ime,h θO2B/Ime,h |
O2A/O2Bi | ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [Fe(TpivPP)-(2-MeHIm)](A) | 100 | −0.391 | 2.066(8) | 2.112(5) | 23.44 | tw | |||||||
| [Fe(TpivPP)-(2-MeHIm)](B) | 100 | minus;0.384 | 2.070(6) | 2.113(3) | 23.32 | tw | |||||||
| [Fe(TpivPP)-(2-MeHIm)] | 294 | minus;0.399 | 2.070(6) | 2.095(6) | 22.82 | 18 | |||||||
| [Fe(TpivPP)-(2-MeHIm)(O2)](A) | 100 | minus;0.047 | 1.995(3) | 1.804(3) | 1.130(6) | 2.088(5) | 129.9(3) | 89.68 | 43.98 | 25.23 | 20.87 | 0.384(8) | tw |
| 1.204(19) | 129.5(12) | 44.60 | 69.86 | 0.116(8) | |||||||||
| [Fe(TpivPP-(2-MeHIm)0.65(O2)](A) | 300 | minus;0.139 | 2.007(3) | 1.909(8)j | 1.12(2) | 2.067(14) | 126.4(13) | 88.7 | 44.2 | 23.0 | 21.2 | 0.200(19) | tw |
| 1.13(4) | 131(2) | 44.5 | 67.6 | 0.124(19) | |||||||||
| [Fe(TpivPP)-(2-MeHIm)(O2)](B) | 80 | minus;0.044 | 1.991(3) | 1.811(5) | 1.281(12) | 2.082(5) | 118.2(9) | 87.9 | 44.4 | 22.5 | 23.1 | 0.398(8) | tw |
| 1.21(4) | 126(2) | 43.6 | 69.0 | 0.102(8) | |||||||||
| 100 | minus;0.043 | 1.994(3) | 1.793(3) | 1.128(6) | 2.091(5) | 130.3(3) | 89.27 | 44.73 | 21.38 | 23.89 | 0.392(7) | tw | |
| 1.19(2) | 128.2(13) | 44.55 | 66.83 | 0.108(7) | |||||||||
| 100 | minus;0.043 | 1.993(3) | 1.793(3) | 1.126(5) | 2.089(5) | 130.2(3) | 88.45 | 44.65 | 22.01 | 23.35 | 0.391(7) | tw | |
| 1.20(2) | 128.8(13) | 46.20 | 68.20 | 0.109(7) | |||||||||
| 200 | minus;0.038 | 1.9929(2) | 1.794(2) | 1.136(5) | 2.102(3) | 129.8(3) | 89.25 | 44.11 | 22.05 | 22.11 | 0.318(5) | tw | |
| 1.174(8) | 130.4(5) | 44.86 | 67.17 | 0.182(5) | |||||||||
| [Fe(TpivPP)-(2-MeHIm)0.9(O2)](B) | 300 | minus;0.065 | 1.9975(6) | 1.840(4)j | 1.164(9) | 2.097(6) | 127.7(6) | 89.0 | 44.9 | 20.3 | 25.0 | 0.296(10) | tw |
| 1.160(15) | 132.3(10) | 44.2 | 66.1 | 0.154(10) | |||||||||
| [Fe(TpivPP)-(2-MeHIm)(O2)] | 294 | minus;0.11 | 1.996(4) | 1.898(7)j | 1.205(16) | 2.107(4) | 129.0(12) | 86.18 | 43.16 | 22.2 | 24.43 | 0.3 | 18 |
| 1.232(22) | 128.5(18) | 43.90 | 69.40 | 0.2 |
Estimated standard deviations are given in parentheses.
Displacement of iron atom from the 24-atom mean plane, a positive value is towards to the pocket porphyrin side.
Value in Å.
Average value in Å.
Value in degree.
Dihedral angle between the planes defined by Fe–O1–O2A and Fe–O1–O2B.
Dihedral angle between the planes defined by the closest Np–Fe–O1/Nax and the ligand plane (Fe–O1–O2A/B or imidazole plane).
Dihedral angle between proximate ligand plane (Fe–O1–O2A/B and imidazole plane) and unhindered side imidazole plane.
The occupancies of disordered terminal oxygen atoms is given; a second orientation with equal population is required by the twofold symmetry.
Value not reliable, see text.
Solid-state Mössbauer spectra of the three species display similar, but not identical, temperature-dependent features. At low temperatures, the quadrupole doublet has a pair of narrow lines that broaden as the temperature is increased and then narrow again as the temperatures approaches room temperature. The value of the quadrupole splitting also shows substantial variation with temperature. Representative spectra for all three complexes are given in the Supporting Information and the temperature dependence will be discussed subsequently.
Discussion
The use of multiple-temperature structure determinations on the same single-crystal specimen can yield substantial insight into the nature of solid-state dynamic processes. Structure determinations for a series of iron(II) nitrosyl complexes at multiple temperatures showed that significant information into the pathways of NO ligand motion could be obtained.23, 24 The success of the NO studies led us to carry out studies of picket fence dioxygen complexes at a variety of temperatures from ambient down to 80 K. The picket fence dioxygen structures are known to have crystallographically imposed 2-fold symmetry leading to disorder of the bent dioxygen ligand and the trans imidazole ligand. In all known cases, a second crystallographically independent oxygen position is found at room temperature, and the coordinated dioxygen displays quasi 4-fold disorder. Only room temperature structures of these dioxygen complexes have been obtained prviously, despite Mössbauer spectroscopic evidence for dynamical processes that surely involve the coordinated O2. However, in the recent multiple-temperature study of a series of [Co(TpivPP)(R-Im)(O2)] (R-Im = 1-EtIm and 2-MeHIm) structures,28 we showed crystal annealing leads to an unusual reversible phase change for [Co(TpivPP)(1-EtIm)(O2)] that provides a completely ordered structure (P1̄ space group at 100 K) and excellent structural parameters. We concluded that the disorder of the tert-butyl groups of the protecting pickets is correlated with rotation of the O2 around the Co–O(O2) bond. Any rotation of O2 results in the picket motion and the correlated motion of the pickets and O2 is in a geared fashion which is required to avoid impossible atom · · · atom contacts. In this paper we report our studies on the dynamics of oxygen motion in iron picket fence derivatives that are based on multi-temperature Mössbauer spectroscopy and X-ray structure determinations.
Structures of [Fe(TpivPP)(1-MeIm)(O2)] and [Fe(TpivPP)(1-EtIm)(O2)]
The first crystals of dioxygen complexes obtained were those of the 1-methylimidazole derivative, [Fe(TpivPP)(1-MeIm)(O2)], and were quite small. This led to data having to be collected at a synchrotron line in order to obtain adequate data. Unfortunately, these crystals contained a small amount of an impurity ligand (probably adventitious water or chloride) in place of the O2. The presence of this small impurity is apparent in the electron density map (but not the difference Fourier map, see Figure S6). Although we have attempted to correct for this, there will be substantial uncertainty for the value of the Fe–O2 bond distances in the 1-methylimidazole derivative. Subsequently, larger and completely pure crystals with 1-ethylimidazole as the trans base were obtained and these results will be emphasized. Structures for both derivatives were obtained at three temperatures (100, 200 and 300 K), all on the same crystal specimen.
Basic structural features of [Fe(TpivPP)(1-MeIm)(O2)] and [Fe(TpivPP)(1-EtIm)(O2)] are depicted in Scheme 1 and are seen to be closely similar. There are strong similarities with the earlier reported structures of [Fe(TpivPP)(1-MeIm)(O2)]16b and [Co(TpivPP)(1-EtIm)(O2)]28 as shown in Figure S7 of the Supporting Information.
Scheme 1.
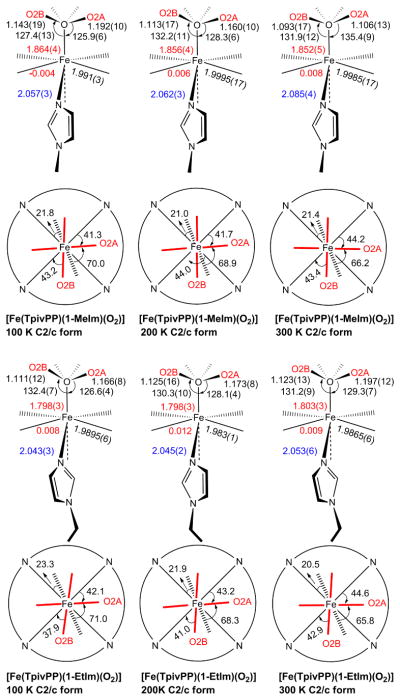
Formal diagrams of structures of [Fe(TpivPP)(1-MeIm)(O2)] and [Fe(TpivPP)(1-EtIm)(O2)] at different temperatures. The upper diagrams are edge-on views of the porphyrin plane and ligands with key parameters (distances in Å, angle in degrees) shown in sequence of O–O, O–O–M, M–O, Δ24 (positive to proximal side), M–Np and M–Nax. The lower diagrams are top-down views of the porphyrin plane showing the projections of dioxygen (red lines) and imidazole plane (dash line) and their angles between the closest M–Np vectors.
As expected, the bent Fe–O2 group shows a bisecting conformation; the terminal oxygen atom occupies two crystallographically independent positions and thus exhibits fourfold positional disorder. This can be seen in the right-hand side of Figure 1, a top-down perspective view for [Fe(TpivPP)(1-EtIm)(O2)]. All pickets are disordered, with each having two distinct orientations of the tert-butyl group; none of the picket disorder is a consequence of the crystallographic symmetry. A close examination of the right hand panel of Figure 1 shows details of the orientations of the tert-butyl groups. Each of the independent tert-butyl groups has two distinct but similar orientations, but the two independent pickets show different patterns. The disordered tert-butyl groups pointing toward O2A have two methyl groups oriented inward to the pocket cavity whereas the picket at the other site, pointing towards O2B, has only one methyl group inward to the pocket cavity. We will represent the orientation of the tert-butyl groups with the symbol “<o” for those with the two methyl groups inward and bracketing the oxygen and those with the single inward methyl group as “>o”.
Figure 1.

Thermal ellipsoid diagrams of [Fe(TpivPP)(1-EtIm)(O2)] at 100 K (edge-on view, left; top-down view, right) displaying selected atom labels. The terminal oxygen populations are also indicated. The axial imidazole is disordered over two positions related by the required symmetry axis. All picket orientations shown. Thermal ellipsoids are contoured at the 50% probability level. Hydrogens omitted for clarity.
The two sets of “bracketing” methyl groups (<o) interact with the terminal oxygen atom O2A, effectively on each side of the oxygen. The contacts are O2A· · ·C22A = 3.229 Å and O2A· · ·C20A = 4.323 Å for the first picket orientation and O2A· · ·C20B = 3.168 Å and O2A· · ·C22B = 4.008 Å for the second picket orientation. There is an equivalent set of such interactions 180° away, as required by the 2-fold symmetry. The short O· · ·C distances are very near the van der Waals radii sum of 3.22 Å. 39 The second set of disordered pickets have two inward methyl group orientation (>o), which interact with the terminal oxygen atom O2B. The contacts are O2B· · ·C27A = 2.443 Å for the first picket orientation and O2B· · ·C25B = 3.145 A for the second picket orientation. Similarly an equivalent set of interactions are found 180° away. It is noticed that the short contacts related to O2B are shorter than that of the O2A, which is consistent with its lower occupancy population (O2A:O2B = 0.60:0.40).
The <o orientation has been commonly observed when the pocket ligand is not linear, for example, N-coordinated nitrite. In a number of iron(II) and -(III) nitrite picket fence derivatives,38 the terminal nitrite oxygen atoms are bracketed by two methyl groups, but the pickets are completely ordered with a single orientation. For oxygen complexes, calculations suggested that picket behavior is intrinsically related to the oxygen ligand dynamics.40 Our recent high-resolution structural study of [Co(TpivPP)(1-EtIm)(O2)] demonstrated that the rotation of O2 around the Co–O bond correlates with picket motion, leading to the two picket orientations that bracket the terminal oxygen.28
It is interesting to make comparisons between the two iron and the cobalt dioxygen structures. Both “<o” and “>o” orientations are found for the tert-butyl groups. The “<o” orientations are always related with the major oxygen atom occupancy and the “>o” orientations with the minor oxygen positions. In the oxycobalt case, where there are only two oxygen atom positions, the “>o” orientations are still found as in the oxyiron analogues. This suggests that the short contacts with oxygen may not be the only factor in the picket orientations; the interactions between neighboring pickets may also be important. This is confirmed by the distances found between the carbon atoms of neighboring pickets in the structures ([Co(TpivPP)(1-EtIm)(O2)]: C22B· · ·C25B = 3.274 Å for the C2/c form and C44· · ·C38 = 3.632 Å for the P1̄ form; [Fe(TpivPP)(1-EtIm)(O2)]: C20B· · ·C25B = 3.181 Å), which are shorter than the C· · · ·C van der Waals radii sum of 3.4 Å.39 This is consistent with our previous conclusion of nondirectional influence of the oxygen ligand on the pickets.28
In these three structures, the major dioxygen orientation (M–O1–O2A plane) has a near perpendicular orientation with respect to the imidazole plane, suggesting that this orientation is the most favored one. The oxygen rotation around the M–O bond requires that the picket methyl groups must also move to lower the barrier. Thus, the dynamics and energetics of O2 rotation depend on two distinct molecular features: the barrier to rotation around the M–O bond and the energy required for the necessary movement of the pickets. It is likely that the picket disorder, which is seen as alternate orientations of the tert-butyl groups, is the “residue” from the M–O2 rotation. Correlated motion of the pickets and O2 in a geared fashion are required to avoid impossible atom· · ·atom (O· · ·C and C· · ·C) contacts between oxygen and picket, as well as picket to its neighboring picket; the differing positions of the tert-butyl groups must be nearly energetically equivalent. However, the energetics of the picket orientation will depend on whether or not there is an O2 ligand in proximity.
The effects of warming on the molecular structure was explored by collecting diffraction data between 100 and 300 K, which was accomplished without removing the crystal from the diffractometer. Top-down and side-on views of the molecule at the three temperatures are given in Figures 1 and S8–S10. The space group remains the same (C2/c, with required 2-fold symmetry) and in all cases, the dioxygen ligand is disordered over the four quadrants of the picket fence porphyrin. The disordered O2 occupancies (~0.3:~0.2:~0.3:~0.2 in the four quadrants) are similar, within experimental uncertainties at 100 and 200 K, with a modest preference for the Fe–O2 plane being near perpendicular to the imidazole plane. The occupancies become almost equal at 300 K. As will be discussed subsequently, the differences in the occupancy of the terminal oxygen over the explored temperature are smaller than anticipated from the Mössbauer measurements.
At 200 K, the pickets associated with the major occupancy terminal oxygen again display a pair of “<o” conformations with the closest O· · ·C contacts of 3.192 and 3.217 Å. Picket conformations associated with the minor occupancy oxygen atom are more complex, with both a “<o” and a “>o” conformation. The nonbonded contact with the minor terminal oxygen and the closest carbon of the “<o” conformation is 3.200 Å whereas the apparent O· · ·C contact with the “>o” picket conformation is a highly improbable 2.498 Å. These structural features are very comparable to the 200 K cobalt structure of [Co(TpivPP)(1-EtIm)(O2)].28
At 300 K, the oxygen atom occupancies are nearly equal (0.26:0.24:0.26:0.24). The orientation of the pickets is more diffuse, but shows a similar pattern to the 200 K structure. The picket associated with the (marginally) major occupancy terminal oxygen display a pair of “<o” conformations with the closest O· · ·C contacts of 3.033 and 3.188Å. The picket associated with the minor occupancy oxygen atom shows both “<o” and “>o” conformations. The nonbonded contact with the terminal oxygen and the closest carbon of the “<o” conformation is 3.219 Å whereas the apparent O· · ·C contact with the “>o” picket conformation is again an improbable 2.592 Å. Although the thermal parameters (or more precisely the anisotropic displacement parameters) for all atoms have increased over the temperature range, those of the picket methyl groups have especially increased. As can be seen in Figure S10, the appearance of these thermal parameters suggests a near continuum of methyl group positions about the CtBu–Camide bond and effectively appears close to free rotation at 300 K. The apparent easy movement of the pickets may be the reason that the oxygen atom occupancies are now near a statistical 0.25. Top down views of the O2-binding cavities are shown in Figure S11 with the terminal oxygen populations as a function of temperature indicated.
The current studies and the previous temperature-dependent study of [Co(TpivPP)(1-EtIm)(O2)]28 show that at temperatures of >~300 K the bent M–O–O group will equally populate the four quadrants of the porphyrin core. Molecular dynamic studies demonstrate accessible jumps between each quadrant with rates that are six times larger for cobalt than for iron.41 The crystal structure studies show that the location of the dioxygen ligand is the result of the constraints of the intramolecular interactions between dioxygen and picket atoms that relax as the temperature increases. Unequal populations in the four quadrants are clearly favored as the temperature decreases. As described below, there appears to be ligand orientation preferences, but in the iron system a single orientation is not found at the temperatures accessible for the crystal structure determinations.
The lower portions of Scheme 1 illustrate the relative orientations of the imidazole and the bent M–O2 ligand with respect to the M–Np directions of the porphyrin core. Bisecting oxygen orientations are found for both iron and cobalt. All M–O–O orientations have required twofold symmetry. The favored orientation of the axial O2 ligand appears to approach that of bisecting a bracketing pair of Fe–Np bonds, whereas the imidazole orientation tends toward eclipsing a Fe–Np bond. The orientation of the major Fe–O–O plane is approximately perpendicular to the imidazole plane with dihedral angles (θO2A) ranging between 65 and 71°. This favored orientation may reflect modest π donation from the trans imidazole because the imidazole ligand has been found to stabilize the oxygen adducts by acting as good π-donor.42 Some interesting temperature-dependent trends are noted. The θO2A angle decreases with the temperature warming up; whereas the angle between major Fe–O–O plane and its closest Fe–Np (φO2A) increases. The cobalt system shows a similar preference for the relative orientation of the Co–O–O plane and the imidazole plane.28 The θO2A angle of oxycobalt complexes also decrease with temperature increasing (ranging from 71 to 80°), but they are larger than that of the oxyiron complexes. This may reflect that there is stronger π-donation from imidazole to Co dπ and to O2 π*,43 which is consistent with the slightly shorter Co–Nim distances found in [Co(TpivPP)(1-EtIm)(O2)] at each temperature than in the iron analogue.
Values of the coordination group parameters of the 1-EtIm and 1-MeIm dioxygen derivatives are summarized in Table 1 along with values of related complexes for comparison. The iron shows very small displacements out of the 24-atom mean plane (Δ24⪅0.01 Å), compared to the ~04–0.05 Å values for [Fe(TpivPP)(RIm)2] (R= 1-Me, 1-Et and 1-Vinyl)30 and analogous cobalt dioxygen complexes,28 all of which are low-spin complexes. The range of Fe–Np bond distances, 1.983–2.000 Å, are consistent with the low-spin state.44 The Fe–O2 distances are found to be in the narrow range of 1.798(3)–1.803(3) Å for [Fe(TpivPP)(1-EtIm)(O2)]; the same range of values observed for the disordered forms of [Fe(TpivPP)(2-MeHIm)(O2)] (vide infra). The values for [Fe(TpivPP)(1-MeIm)(O2)] are seen to be modestly larger (1.852(5)–1.864(4) Å), but must be regarded as unreliable.45 As discussed subsequently, the value for the Fe–O2 bond is certainly limited to the range of 1.79 to 1.81 Å. The Fe–O2 bond distance is thus only slightly longer than the Fe–C bond (typically 1.75 Å) seen in a number of heme carbonyls, attesting to the Fe–O2 bond strength. Comparisons of the Fe–O2 geometry in the globins will be given subsequently.
Interestingly, no temperature dependence on the Fe–O2 bond distance is observed. All distances are longer than the reported value of 1.745(18) Å from the early room temperature structure of Collman et al.16b The Fe–O–O bond angles are in the range of 125–130°. The probable value, after correction for the disorder of the O2 that is likely to be slightly displaced from the twofold axis, would be a smaller value. Such a correction for the true location of the coordinated oxygen atom would also lead to slightly longer values for the O–O bond distance.
To our knowledge, there are no structures of iron dioxygen complexes other than those of the early work of Collman and co-workers with picket fence derivatives.16 Analogous cobalt complexes are known, based on both the picket fence scaffold as well as other cobalt chelates.28,46–53 In the cobalt systems, the M–O2 bond distance is ~0.15 Å longer than that in iron. The shorter distance must reflect a significantly stronger π bond in the iron systems. The Co–O2 bond has been described with the superoxide formalism of “ ” with the unpaired electron residing primarily in the π* antibonding orbital of oxygen,54 which is consistent with the EPR spectra. In contrast, the nature of the bonding in the Fe–O2 moiety has been very controversial (to this day). The nature of the complex bonding has been most recently studied by L-edge x-ray absorption spectroscopy by Solomon and co-workers5 and by theoretical studies of Shaik and co-workers.4
The axial Fe–NIm bond distances (range 2.043–2.085 Å) are all slightly longer (by 0.04 to 0.06 Å) than the values observed in the low spin bis(imidazole) complexes (Table 1).30 However, the observed distances are quite comparable to those observed in several six-coordinate carbonyl heme derivatives.55 Thus both CO and O2 are seen to have similar effects on the Fe–NIm bond trans to the diatomic ligand. This is probably the result of the strong π bonding between iron and the diatomic ligand.
In Situ Oxygenation of [Fe(TpivPP)(2-MeHIm)]
Single crystals of five-coordinate [Fe(TpivPP)(2-MeHIm)] are oxygenated by exposure to an atmosphere of dioxygen saturated with ethanol at either 1 or 5 atm O2 pressure. The solid-state reaction occurs with minimal change in cell volume (as noted by the cell volumes at 100 K, Table S3) and the space group of the crystalline materials does not change. However, the studies show that the solid-state oxygenation reaction yields not only the dioxygen complex but also initiates molecular dynamics involving motions of the coordinated dioxygen and motions of the protecting pickets.
In the solid state, five-coordinate [Fe(TpivPP)(2-MeHIm)] has completely ordered pickets as do the analogous cobalt species.56 Both have the imidazole on the side opposite the protected ligand binding pocket. The axial imidazole ligand is disordered in accord with the required twofold symmetry of the observed space group. The solid-state addition of the dioxygen ligand takes place without change in crystallinity or space group. However, the crystal structures show that the coordinated dioxygen is disordered. More surprisingly, the protecting pickets are now also disordered. Clearly there are strong interactions between dioxygen and the pickets. Moreover, the temperature dependent details of the Mössbauer spectra show that this disorder is a dynamic process. This is demonstrated by Mössbauer for all iron derivatives and is likely true for the cobalt species as well. The disorder in the coordinated imidazole in both the five- and the six-coordinate complexes is possibly dynamic as well, the ordering observed in one cobalt derivative shows that crystal packing can allow for the movement (rotation) of the imidazole.56
Two oxygenation experiments/structure determination sequences were done. For crystal A, after structure determination of the precursor at 100 K, oxygenation at 1 atm O2 was followed by determinations at 100 and 300 K. For crystal B, after structure determination of the precursor at 100 K, oxygenation at 5 atm O2 was followed by a number of structure determinations at 80, 100, 200 and 300 K. Strong agreements are found for the precursor complex and the 100 K structures, but the 300 K structures showed some differences to be discussed subsequently.
Structure of [Fe(TpivPP)(2-MeHIm)] and [Fe(TpivPP)(2-MeHIm)(O2)]
A thermal ellipsoid plot view of the [Fe(TpivPP)(2-MeHIm)] molecule is given in Figure S13 of the Supporting Information and, as noted above, is completely ordered save for the axial imidazole ligand. Details of the coordination group parameters for the two structure determinations are given in Table 2 and are seen to be closely similar. The parameters are also in general agreement with an earlier structure determination at 294 K,18 with the major significant difference in the value of the Fe–NIm bond that is shorter in the 294 K structure. This may be the result of the 2-MeHIm group being refined as a rigid group in that study.
The structure of the five-coordinate precursor is typical of previously characterized five-coordinate imidazole-ligated iron porphyrinates. (range of Fe–NIm = 2.130 to 2.171 Å).57, 58, 59, 60 The sterically hindered 2-methylimidazole ligand causes a modest increase in the value of the Fe–NIm bond (2.113 Å), compared to 1.9985 Å found in nonhindered imidazoles derivatives of [Fe(TpivPP)(1-RIm)2] (R = Me, Et and Vinyl).30 However such an elongation is not observed for the cobalt picket fence derivatives as noted previously.28 The out-of-plane iron displacement is within the range previously seen.
The temperature dependent behavior of the pickets in [Fe(TpivPP)(2-MeHIm)(O2)] is more complex than that observed in any of the other iron or cobalt dioxygen species. The 100 K structure of crystal A showed disordered terminal oxygen atoms with a site occupancy factor of 0.384(8) for the major position with the disordered pickets showing two distinct “>o” orientations. The pickets associated with the minor terminal oxygen atom (SOF = 0.116(8)) are completely ordered. Warming the crystals to 300 led to both loss of some coordinated dioxygen and picket order at the minor oxygen position. Both of the minor disorder pickets have the “<o” orientation.
These observations led us to the more detailed temperature-dependent measurements (Crystal B) that are outlined in Figure 2. As already noted, the five-coordinate species is ordered. Oxygenation at 5 atm O2, followed by an immediate structure determination at 100 K gave similar oxygen occupancy factors and picket order/disorder as that in Crystal A. Results from the two different crystals can be compared in Table 2. Warming the crystal to 200 K led changes in oxygen occupancy and to disorder in both independent pickets. Cooling the crystal back to 100 K reestablished the same pattern of picket ordering with the pickets associated with the major oxygen displaying disorder and the minor oxygen position showing completely ordered pickets (Table 2). The complete reversibility of the process can be seen in the virtually identical bonding parameters and oxygen atom occupancies.
Figure 2.

Thermal ellipsoid diagrams of [Fe(TpivPP)(2-MeHIm)] and its product [Fe(TpivPP)(2-MeHIm)(O2)] (crystal B), showing reaction conditions and routes. The temperatures are given in highlighted boxes. In all species, the axial imidazole is disordered over two positions related by the required 2-fold symmetry axis, but only one orientation is shown. Thermal ellipsoids are contoured at the 50% (80 and 100 K), 40% (200 K) and 30% (300 K) probability levels. The reaction and structure characterization were carried out for the same crystalline specimen. Hydrogens are omitted for clarity.
Given the earlier success in obtaining molecular ordering in [Co(TpivPP)(1-EtIm)(O2)] by crystal annealing,28 we attempted annealing for the iron system as well. Further cooling to 80 K did not lead to a substantial increase in ordering, with only a small increase in the SOF of the major oxygen position relative to the 100 K results. But the lowered temperature did allow us to resolve the position of the coordinated oxygen atom. Diagrams of the electron density in the 80 K difference Fourier maps are shown in Figures S4 and S5 of the Supporting Information. The off-axis position of the oxygen ligand is shown in Figure 3. The off-axis resolution of the coordinated oxygen atom gives the best available description of coordinated oxygen in an iron complex (vide infra). Top down views of the structure at 80 K and at 200 K are given in Figure 4. The terminal oxygen (O2A and O2B) contacts with the pickets are closer than those observed in the analogous 1-ethyl- and 1-methylimidazole derivatives. The two “>o” type O2A· · ·C contacts are 2.491 and 3.063 Å at 80 K and 2.612 and 3.029 Å at 200 K. Clearly only one of the orientation of the pickets is compatible with the presence of the terminal oxygen and the picket atom populations, determined independently of the oxygen atom populations, are consistent with this possibility. Although the pickets associated with the minor terminal oxygen atom have a single orientation at 80 K with O2B· · ·C contacts of 2.798 and 4.223 Å, a second set of picket orientations are seen at 200 K. Both are bracketing orientations (“<o”) with (paired) contact distances of 2.809 and 3.451 Å or 3.324 and 4.307 Å.
Figure 3.
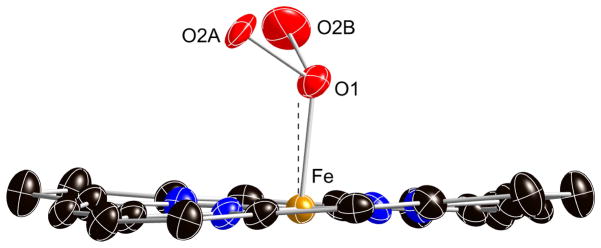
Diagram illustrating the off-axis tilt of the coordinated dioxygen ligand at 80 K. Both the major (O2A) and minor (O2B) oxygen positions are shown. The dashed line is the position of the twofold axis and the normal to the porphyrin plane.
Figure 4.
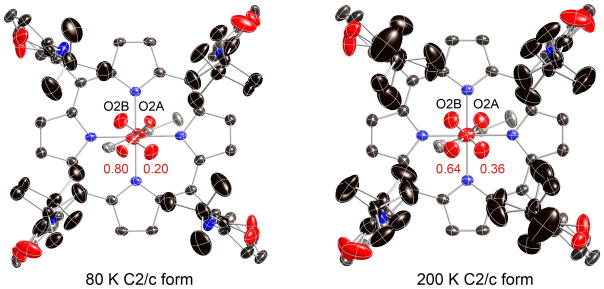
Top-down thermal ellipsoid diagrams of [Fe(TpivPP)(2-MeHIm)(O2)] at 80 and 200 K showing the resolution of the bonded oxygen at 80 K and the increasing order of one Fe–O2 configuration. Thermal ellipsoids are contoured at the 50% (80 K) and 40% (200 K) probability level. Hydrogens are omitted for clarity.
Warming the crystal (B) to 300 K leads to more disorder. There appears to be a modest loss of the dioxygen ligand (~10%). A larger loss of dioxygen was observed for the crystal A sample (~35%). A comparison of the room temperature structural values with those obtained at lower temperatures suggests that the loss of oxygen leads to errors in the geometric parameters (Table 2). It is likely that the room temperature structural parameters previously reported18 were affected by an unrecognized loss of dioxygen from the crystal. We believe that the low temperature values represent the most appropriate set; the congruence in those values can be seen in Table 2. It is important to note that similar oxygen loss was not observed for nonhindered imidazole (1-RIm, R= Et or Me) derivatives.
Crystal B was aged for an additional 3.5 days and the structure then redetermined (at 300 K). The crystal has clearly lost additional dioxygen (to about 60% total) and the structure is clearly a mixture of the five-coordinate precursor and the oxygen complex. Unexpectedly, the dioxygen loss appears to be preferentially from the minor site that now shows very small electron density. The dioxygen ligand is thus primarily disordered over the two sites related by the crystallographic twofold axis. Both sets of pickets are still disordered.
The changes in structure upon oxygenation are shown in Table 2; Scheme 2 gives values for selected parameters of the coordination group of [Fe(TpivPP)(2-MeHIm)(O2)] at 80 and 100 K. Values for the cobalt analogue are also given for comparison. The out-of-plane iron displacement decreases but does not decrease to zero; the iron remains on the imidazole side of the porphyrin plane just as in the cobalt analogue. The axial Fe–NIm bond decreases to 2.089 Å, but it does not decrease to the 2.043 Å value seen in the 1-EtIm derivative (Table 1). A similar smaller decrease in the Co–NIm bond distance is also seen for the cobalt species.28 Thus comparable behavior is observed in both the cobalt and iron species with the hindered 2-methylimidazole trans to O2, relative to the derivatives with an unhindered imidazole. Both effects are seen to be consistent with a modest amount of strain or “tension” in the M–NIm bond of the O2 complexes owing to the increased contact between the 2-methylimidazole and the porphyrin ligand. It should be noted that there is no hindered imidazole effect on the Fe–O bond strength, e.g., the Fe–O bond trans to 2-MeHIm is the same as the bond trans to 1-EtIm, despite earlier claims based on both experiment18 and theory.61
Scheme 2.

Formal diagrams of structures of five-coordinate [M(TpivPP)(2-MeHIm] and their oxygen adducts [M(TpivPP)(2-MeHIm)(O2)] (M = Fe (crystal B), Co). The left two columns of schemes are edge-on view of the porphyrin plane and ligands with the key parameters (distances in Å, angle in degrees) of O–O–M, M–O, Δ24, M–Npav and M–Nax. The right column of schemes are top view of the porphyrin plane showing the projections of dioxygen (red line) and imidazole plane (dash line) and their angles between the closet M–Np vectors.
Although the picket fence derivatives remain the only characterized iron dioxygen complexes, obtaining precise values of the Fe–O and Fe–O–O bond distances and angles has been compromised by crystalline disorder. It is clear, especially from the lowest temperature structure determinations, that the position of the coordinated oxygen is unlikely to be on the crystallographic twofold axis found for all the dioxygen complexes62 but displaced off this axis. Resolution and refinement of the off-axis position was not possible until the 80 K structure determination of [Fe(TpivPP)(2-MeHIm)(O2)]. In this structure we find that the two oxygen positions are separated by 0.39 Å. The off-axis position leads to these new values: Fe–O = 1.811(5) Å, Fe–O–O = 118.2(9)°, O–O = 1.281 (12) Å, and an off-axis tilt of 6.2°. The above values are more consonant with values that might have been expected. The off-axis correction leads to slightly longer Fe–O bond distance, a smaller Fe–O–O angle, and a larger O–O bond distance. In our opinion, these are substantially improved values relative to those determined based on the on-axis position of the coordinated oxygen. The observed O–O bond length of 1.281 Å is now in the range of values expected from the O–O stretching frequency of 1163 cm−1.63, 64
An off-axis tilt for the Fe–O2 group had been predicted in earlier DFT calculations by Rovira and Parrinello41 where the tilt was predicted to be in the range of 3–5°. In DFT calculations carried out in conjunction with NRVS vibrational studies of the dioxygen complexes,27 we found that the predicted off-axis tilt was between 2.9–3.6°.66 In the ordered form of [Co(TpivPP)(1-EtIm)(O2) ], the Co–O2 bond is tilted off the normal to the porphyrin plane by 2.7°. Off-axis tilts of the diatomic NO ligand had been observed in both five-coordinate20, 67 and six-coordinate iron nitrosyls23, 24 and five-coordinate cobalt nitrosyls,68 but in the NO systems, the sense of the off-axis tilt is the opposite of the dioxygen complexes. What is not resolved in this iron study is the correct value of the trans O–Fe–N bond angle. It is clear that both axial bonds are off-axis; the O–Fe–N value must either be 168.6 or 177.4°. The corresponding value in the ordered cobalt dioxygen complex is 178.2°.
Finally, there is now satisfying agreement between the small molecule results and the reported values for myoglobin. The Fe–O distances range upward from 1.81 Å, with Fe–NAx = 2.06–2.07 Å, and an Fe–O–O angle of 115–125°.69, 70
Temperature-Dependent Mössbauer Spectra
The Mössbauer spectra of all three dioxygen complexes studied herein show similar, unusual temperature dependent behavior. Representative spectral data are shown in Figures S1–S3 of the Supporting Information. At very low temperature in zero field, a quadrupole split doublet with very narrow lines is observed. As temperature increases, the line widths become broader and value of the quadrupole splitting decreases. With further increases in temperature, the lines narrow but the quadrupole splitting value remains much smaller than that observed at the lowest temperature. This behavior was first seen by Lang and co-workers for [Fe(TpivPP)(1-MeIm)(O2)].25 The temperature dependent data we observe for [Fe(TpivPP)(1-MeIm)(O2)] agree well with those obtained previously by Lang and co-workers,25 although their measured set of temperatures were more limited than those that we have obtained. This same characteristic behavior is also seen for oxymyoglobin and oxyhemoglobin although in these cases the highest temperature accessible was ~240 K.71 It is considered likely that similar physical phenomena (motion of the coordinated O2) lead to this comparable behavior in the small molecule and protein dioxygen complexes.
There are several important features to be recognized. In addition to the temperature dependence of the quadrupole splitting, the value of the low-temperature quadrupole splitting is much larger than that for most other low-spin iron(II) porphyrinates. One other porphyrinate to display such a large QS is low-spin [Fe(TpivPP)(NO2)]−.72 Huynh, Scheidt and co-workers suggested that the large QS value in this species was the result of a large in-plane rhombicity caused by strong π-bonding to one dπ Fe orbital. Such a rhombicity would also be appropriate for the strongly bonded and bent Fe–O2 units of the oxy complexes. Such strong π-bonding between Fe and O2 is also consistent with the small (for the iron(II) oxidation state) value of the isomer shift, which is comparable to those of heme carbonyls.73
Lang and coworkers have presented a model for the temperature-dependent Mössbauer that results from different interconverting configurations of the Fe–O2 unit.25 The model postulates two distinct conformations of the iron coordination group with two electric field gradients (EFG’s) corresponding to the two conformations that differ in energy. This dynamic model successfully reproduced the experimental data. In the model, one conformation is favored at low temperature with both conformers populated at higher temperatures according to a Boltzmann distribution. The low-temperature quadrupole splitting was −2.07 mm/s, with an asymmetry parameter η of 0.23. The higher energy configuration, that starts becoming populated at higher temperatures, had a quadrupole splitting of −0.79 mm/s and η = 0.80. It was assumed that both configurations had a common fixed component of the EFG, presumably perpendicular to the heme plane. Given the similarity of the two conformers, the large differences between the two sets of EFG tensors is unexpected and even unsatisfying.
Oldfield and co-workers revisited this question of understanding the expected value of the EFG tensor with rotation of the O2 orientation.26 Using DFT calculations based on a truncated molecular model, they found that the EFG changes very little with a 90° rotation of the O2 and that the principle axes of the EFG follow the O2, with the largest component of the EFG being in the heme plane. This result suggests a simpler version of the Lang model in which the EFG principle axis components are constant, but jump in orientation around the z axis by some angle. However, Oldfield et al. did not attempt to fit their model to the range of temperatures that had been measured.
With the substantially increased computing power that can now be brought to model these systems, we have carried out detailed temperature-dependent fittings of the spectra. We have used both the assumptions made in the Lang and the Oldfield models to obtain fits to the spectra. [Fe(TpivPP)(1-EtIm)(O2)] was the system most thoroughly studied and results of the fittings are shown in Figure 5 that displays the Oldfield model results and Figure S14 that show results based on the Lang assumptions. Fit parameters are (temperature independent): quadrupole splitting, asymmetry parameter, Lorentzian linewidth, excited site energy, and (temperature dependent): isomer shift, jump rate. The rotation angle between the two EFG orientations is assumed to be 90°. The fit residuals are shown above each spectrum. Equally similar fits were obtained for [Fe(TpivPP)(2-MeHIm)(O2)] and the Lang model fits are shown in Figure 6. Oldfield model simulations for [Fe(TpivPP)(1-MeIm)(O2)] are give in Figure S15 of the Supporting Information. The simulations shown are simultaneous least-squares fits of up to eight spectra. Fit parameters are (temperature independent): quadrupole splitting, asymmetry parameter, lorentzian linewidth, excited site energy, and (temperature dependent): isomer shift, jump rate. The rotation angle between the two EFG orientations is assumed to be 90°.
Figure 5.

Diagram illustrating the simulation of the Mössbauer spectra of [Fe(TpivPP)(1-EtIm)(O2)] using the Oldfield approach. Parameters: QS= −2.09 mm/s, η= 0.12, energy difference = 123 cm−1, line width = 0.24 mm/s, rate of interconversion = 2.55 × 106 sec−1 (50 K), 6.36 × 108 sec−1 (250 K). Fit residuals between the experimental data and the simulation are shown as the top line.
Figure 6.

Diagram illustrating the Lang model simulation for [Fe(TpivPP)(2-MeHim)(O2)]. Parameters: QS(I)= −2.05 mm/s, QS(II)= −2.09 mm/s, η(I and II) = 0.15, energy difference = 157 cm−1, line width = 0.26 mm/s, rate of interconversion = 7.30 × 105 sec−1 (30 K), 4.82 × 109 sec−1 (275 K), jump angle 80°. Fit residuals between the experimental data and the simulation are shown as the top line.
A second independent set of temperature-dependent measurements and simulations were performed for [Fe(TpivPP)(1-EtIm)(O2)]. The data and simulations are shown in Figures S16 and S17 (Supporting Information). Satisfactory agreement for both the experimental data and the simulation parameters were observed.
We note that while the residuals of the fits up to 125 K are relatively good, they become poorer at higher temperatures. In particular, neither the Lang nor the Oldfield approach yielded a sufficiently small quadrupole splitting above 250 K. In various attempts to rectify this misfit, we tried without significant success a number of variations on the Oldfield model, including rotations of the EFG by an angle other than 90°. Another concern is that the temperature dependence of the site populations predicted by the energy parameter from our fits was hard to square with the temperature dependence of O2 site populations observed in X-ray structures.
In our initial approach to an understanding of the dynamics of the [Fe(TpivPP)((R-Im)(O2)] systems, we expected that the temperature-dependent X-ray structural and Mössbauer analyses would lead to a convergent estimate of the energy differences between the two conformers. This expected result was not forthcoming. We believe that this is the result of an important factor, namely the structural results for the iron and cobalt28 are strongly supportive of the idea that the conformational states of oxygen are not simply an equilibrium governed by a rotation of O2 around the the Fe–O bond but rather is the result of concomitant motions of both the O2 and the protecting pickets, and perhaps vibronic contributions to a reduced quadrupole splitting at higher temperature.74, 75 With these complex motions of the system, a simple Boltzmann distribution cannot be expected to adequately model the data and the complete temperature dependence of the Mössbauer data is not perfectly simulated. Nonetheless, the original two state model of Lang et al.25 captures the essence of the dynamics of these picket fence dioxygen systems.
Summary
The dynamics of O2 motion in three different dioxygen complexes of iron(II) picket fence porphyrins have been studied by multi-temperature structure determinations and Mössbauer spectroscopy. Populations of the disordered Fe–O2 groups related by apparent rotation around the axial Fe–O2 bond are temperature dependent with no orientational preference at or near room temperature. All studies show that the distinct Fe–O2 conformations interconvert by correlated motion of the dioxygen and rotation of the protecting pickets. Evidence is presented for an off-axis tilt of the axial Fe–O2 bond.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by a CAS Hundred Talent Program to JFL. We also acknowledge support of the National Institutes of Health under award number GM-038401 to WRS.
Footnotes
Supporting Information Available. Figures S1–S3 display experimental Mössbauer data over the complete temperature range, Figures S4 to S6 show electron density plots, Figure S7 gives formal diagrams comparing [Fe(TpivPP)(1-MeIm)(O2)] and [Co(TpivPP)(1-EtIm)(O2)], Figures S8 to S10 give thermal ellipsoid plots of [Fe(TpivPP)(1-EtIm)(O2)] at 100–300 K, Figure S12 shows an edge-on view of [Fe(TpivPP)(1-MeIm)(O2)], Figure S13 shows the thermal ellipsoid plot of five-coordinate [Fe(TpivPP)(2-MeHIm)] and Figures S14 to S17 presents simulations and comparison of the temperature dependent Mössbauer spectra. Complete crystallographic details for all structures determined are given Tables S1 to S3. Crystallographic information files (CIF) for the 15 structures determinations are also available. Ordering information is given on any current masthead page. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Ghosh A, editor. The Smallest Biomolecules: Diatomics and Their Interactions with Heme Proteins. Elsevier; Amsterdam: 2008. [Google Scholar]
- 2.(a) Stryer L. Biochemistry. 3. Freeman; New York: 1988. [Google Scholar]; (b) Fermi G. In: Molecular Structures in Biology. Diamond R, Koetzle TF, Prout K, Richardson J, editors. Chap 6. Oxford University Press; New York: 1993. pp. 164–191. [Google Scholar]
- 3.Kirchner RF, Loew GH. J Am Chem Soc. 1977;99:4639. doi: 10.1021/ja00456a020. [DOI] [PubMed] [Google Scholar]
- 4.Chen H, Ikeda-Saito M, Shaik S. J Am Chem Soc. 2008;130:14778. doi: 10.1021/ja805434m. [DOI] [PubMed] [Google Scholar]
- 5.Wilson SA, Kroll T, Decreau RA, Hocking RK, Lundberg M, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2013;135:1124. doi: 10.1021/ja3103583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pauling L, Coryell CD. Proc Natl Acad Sci USA. 1936;22:210. doi: 10.1073/pnas.22.4.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Cerdonio M, Congiu-Castellano A, Mogno F, Pispisa B, Romani GL, Vitale S. Proc Natl Acad Sci USA. 1977;74:398. doi: 10.1073/pnas.74.2.398. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cerdonio M, Congiu-Castellano A, Calabrese L, Morante SB, Pispisa B, Vitale S. Proc Natl Acad Sci USA. 1978;75:4916. doi: 10.1073/pnas.75.10.4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Savicki JP, Lang G, Ikeda-Saito M. Proc Natl Acad Sci USA. 1984;81:5417. doi: 10.1073/pnas.81.17.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boso B, Debrunner PG, Wagner GC, Inubushi T. Biochim Biophys Acta. 1984;791:244. doi: 10.1016/0167-4838(84)90015-3. [DOI] [PubMed] [Google Scholar]
- 9.Pauling L. Nature. 1964;203:182. [Google Scholar]
- 10.Weiss JJ. Nature. 1964;202:83. doi: 10.1038/202083b0. [DOI] [PubMed] [Google Scholar]
- 11.McClure DS. Radiation Res Suppl. 1960;2:218. [Google Scholar]
- 12.Harcourt RD. Int J Quantum Chem. 1971;5:479. [Google Scholar]
- 13.Harcourt RD. Chem Phys Lett. 1990;167 [Google Scholar]
- 14.Goddard WA, Olafson BD. Proc Natl Acad Sci USA. 1975;72:2335. doi: 10.1073/pnas.72.6.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paulsen H, Schunemann V, Trautwein AX, Winkler H. Coord Chem Rev. 2005;249:255. [Google Scholar]
- 16.(a) Collman JP, Gagne RR, Reed CA, Robinson WT, Rodley GA. Proc Natl Acad Sci USA. 1974;71:1326. doi: 10.1073/pnas.71.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jameson GB, Rodley GA, Robinson WT, Gagne RR, Reed CA, Collman JP. Inorg Chem. 1978;17:850. [Google Scholar]; (c) Collman JP, Gagne RR, Halbert TR, Marchon JC, Reed CA. J Am Chem Soc. 1973;95:7868. doi: 10.1021/ja00804a054. [DOI] [PubMed] [Google Scholar]; (d) Collman JP, Gagne RR, Reed C, Halbert TR, Lang G, Robinson WT. J Am Chem Soc. 1975;97:1427. doi: 10.1021/ja00839a026. [DOI] [PubMed] [Google Scholar]
- 17.Abbreviations: TpivPP, dianion of α,α,α,α-tetrakis(o-pivalamidophenyl)porphyrin; 1-MeIm, 1-methylimidazole; 1-EtIm, 1-ethylimidazole; 2-MeHIm, 2-methylimidazole; RIm, generalized imidazole; Np, porphinato nitrogen atom; Mb, Myoglobin; Hb, Hemoglobin.
- 18.(a) Jameson GB, Molinaro FS, Ibers JA, Collman JP, Brauman JI, Rose E, Suslick KS. J Am Chem Soc. 1980;102:3224. [Google Scholar]; (b) Jameson GB, Molinaro FS, Ibers JA, Collman JP, Brauman JI, Rose E, Suslick KS. J Am Chem Soc. 1978;100:6769. [Google Scholar]
- 19.(a) Collman JP. Acc Chem Res. 1977;10:265. [Google Scholar]; (b) Jene PG, Ibers JA. Inorg Chem. 2000;39:3823. doi: 10.1021/ic000127p. [DOI] [PubMed] [Google Scholar]
- 20.Scheidt WR, Duval HF, Neal TJ, Ellison MK. J Am Chem Soc. 2000;122:4651. [Google Scholar]
- 21.Wyllie GRA, Scheidt WR. Chem Rev. 2002;102:1067. doi: 10.1021/cr000080p. [DOI] [PubMed] [Google Scholar]
- 22.Momenteau M, Reed CA. Chem Rev. 1994;94:659. [Google Scholar]
- 23.Silvernail NJ, Pavlik JW, Noll BC, Schulz CE, Scheidt WR. Inorg Chem. 2008;47:912. doi: 10.1021/ic701700p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silvernail NJ, Barabanschikov A, Sage JT, Noll BC, Scheidt WR. J Am Chem Soc. 2009;131:2131. doi: 10.1021/ja8055613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spartalian K, Lang G, Collman JP, Gagne RR, Reed CA. J Chem Phys. 1975;63:5375. [Google Scholar]
- 26.Godbout N, Sanders LK, Salzmann R, Havlin RH, Wojdelski M, Oldfield E. J Am Chem Soc. 1999;121:3829. [Google Scholar]
- 27.Li JF, Peng Q, Barabanchikov A, Pavlik JW, Alp EE, Sturhahn W, Zhao J, Schulz CE, Sage JT, Scheidt WR. Chemistry Eur J. 2011;17:11178. doi: 10.1002/chem.201101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li JF, Noll BC, Oliver AG, Scheidt WR. J Am Chem Soc. 2012;134:10595. doi: 10.1021/ja303475a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landergren M, Baltzer L. Inorg Chem. 1990;29:556. [Google Scholar]
- 30.Li J, Nair SM, Noll BC, Schulz CE, Scheidt WR. Inorg Chem. 2008;47:3841. doi: 10.1021/ic702498c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crystallographic data were collected through the SCrALS (Service Crystallography at Advanced Light Source) program at the Small-Crystal Crystallography Beamline 11.3.1 (developed by the Experimental Systems Group) at the Advanced Light Source (ALS). The ALS is supported by the U.S. Department of Energy, Office of Energy Sciences Materials Sciences Division, under contract DE-AC02-05CH11231 at Lawrence Berkeley National Laboratory.
- 32.APEX2 v2.1-4 and SAINT v7.34A data collection and data processing programs, respectively, Bruker Analytical X-ray Instruments, Inc., Madison, WI.
- 33.Sheldrick GM. Program for empirical Absorption Correction of Area Detector Data. Universität Göttingen; Germany: 1996. [Google Scholar]
- 34.Sheldrick GM. Acta Crystallogr, Sect A. 2008;A64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 35.R1 = Σ||Fo| − |Fc ||/Σ|Fo| and wR2 ={Σ[w Fo2 − Fc2)2]/Σ[wFo4]}1/2. The conventional R-factors R1 are based on F, with F set to zero for negative F2. The criterion of F2 > 2σ(F2) was used only for calculating R1. R-factors based on F2 (wR2) are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger.
- 36.(a) Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J. J Appl Cryst. 2006;39:453. [Google Scholar]; (b) Bruno IJ, Cole JC, Edgington PR, Kessler MK, Macrae CF, McCabe P, Pearson J, Taylor R. Acta Crystallogr. 2002;B58:389. doi: 10.1107/s0108768102003324. [DOI] [PubMed] [Google Scholar]; (c) Taylor R, Macrae CF. Acta Crystallogr. 2001;B57:815. doi: 10.1107/S010876810101360X. [DOI] [PubMed] [Google Scholar]
- 37.Kitazawa T, Nishikiori S-I, Kuroda R, Iwamoto T. J Chem Soc, Dalton Trans. 1994:1029. [Google Scholar]
- 38.(a) Nasri H, Goodwin JA, Scheidt WR. Inorg Chem. 1990;29:185. [Google Scholar]; (b) Nasri H, Wang Y, Huynh BH, Scheidt WR. J Am Chem Soc. 1991;113:717. [Google Scholar]; (c) Nasri H, Wang Y, Huynh BH, Walker FA, Scheidt WR. Inorg Chem. 1991;30:1483. [Google Scholar]; (d) Nasri H, Haller KJ, Wang Y, Huynh BH, Scheidt WR. Inorg Chem. 1992;31:3459. [Google Scholar]; (e) Nasri H, Ellison MK, Chen S, Huynh BH, Scheidt WR. J Am Chem Soc. 1997;119:6274. [Google Scholar]; (f) Nasri H, Ellison MK, Krebs C, Huynh BH, Scheidt WR. J Am Chem Soc. 2000;122:10795. [Google Scholar]; (g) Nasri H, Ellison MK, Shang M, Schulz CE, Scheidt WR. Inorg Chem. 2004;43:2932. doi: 10.1021/ic035119y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bondi A. J Phys Chem. 1964;68:441. [Google Scholar]
- 40.Rovira C, Parrinello M. Chem Eur J. 1999;5:250. [Google Scholar]
- 41.(a) Degtyarenko I, Nieminen RM, Rovira C. Biophys J. 2006;91:2024. doi: 10.1529/biophysj.106.083048. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rovira C, Parrinello M. Biophys J. 2000;78:93. doi: 10.1016/S0006-3495(00)76575-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gall RS, Rogers JF, Schaefer WP, Christoph GG. J Am Chem Soc. 1976;98:5135. [Google Scholar]
- 43.(a) Tsubaki M, Yu NT. Proc Natl Acad Sci USA. 1981;78:3581. doi: 10.1073/pnas.78.6.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mackin HC, Tsubaki M, Yu NT. Biophys J. 1983;41:349. doi: 10.1016/S0006-3495(83)84446-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheidt WR, Reed CA. Chem Rev. 1981;81:543. [Google Scholar]
- 45.As described earlier, these crystals have a very small amount of another ligand in place of O2. Thermal ellipsoid plots show a somewhat elongated ellipsoid for the bonded oxygen along the twofold axis consistent with additional electron density along the twofold (See Figure S12. Attempts to model the impurity ligand showed that the resulting Fe–O2 bond length varies remarkably with the model. Given the uncertainty of the impurity ligand content, extraction of a precise Fe–O2 distance seems unwarranted. Edge-on ORTEP plots at all three temperatures are given in the Supporting Information.
- 46.Rodley GA, Robinson WT. Nature. 1972;235:438. doi: 10.1038/235438a0. [DOI] [PubMed] [Google Scholar]
- 47.Calligaris M, Nardin G, Randaccio L. J Chem Soc, Dalton Trans. 1973:419. [Google Scholar]
- 48.Jameson GB, Robinson WT, Rodley GA. J Chem Soc, Dalton Trans. 1978:191. [Google Scholar]
- 49.Gall RS, Rogers JF, Schaefer WP, Christoph GG. J Am Chem Soc. 1976;98:5135. [Google Scholar]
- 50.Gall RS, Rogers JF, Schaefer WP, Christoph GG. Inorg Chem. 1980;19:340. [Google Scholar]
- 51.Avdeef A, Schaefer WP. J Am Chem Soc. 1976;98:5153. [Google Scholar]
- 52.Gall RS, Schaefer WP. Inorg Chem. 1976;15:2758. [Google Scholar]
- 53.Doppelt P, Fischer J, Richard L, Weiss R. Nouv J Chim. 1987;11:357. [Google Scholar]
- 54.(a) Walker FA. J Am Chem Soc. 1970;92:4235. [Google Scholar]; (b) Hoffman BM, Diemente DL, Basolo F. J Am Chem Soc. 1970;92:61. [Google Scholar]; (c) Getz D, Melemud E, Silver BC, Dori Z. J Am Chem Soc. 1975;97:3846. [Google Scholar]
- 55.Silvernail NJ, Roth A, Schulz CE, Noll BC, Scheidt WR. J Am Chem Soc. 2005;127:14422. doi: 10.1021/ja053148x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J, Noll BC, Oliver AG, Ferraudi G, Lappin AG, Scheidt WR. Inorg Chem. 2010;49:2398. doi: 10.1021/ic902309f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ellison MK, Schulz CE, Scheidt WR. Inorg Chem. 2002;41:2173. doi: 10.1021/ic020012g. [DOI] [PubMed] [Google Scholar]
- 58.Hu C, Roth A, Ellison MK, An J, Ellis CM, Schulz CE, Scheidt WR. J Am Chem Soc. 2005;127:5675. doi: 10.1021/ja044077p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu C, An J, Noll BC, Schulz CE, Scheidt WR. Inorg Chem. 2006;45:4177. doi: 10.1021/ic052194v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu C, Noll BC, Piccoli PMB, Schultz AJ, Schulz CE, Scheidt WR. J Am Chem Soc. 2008;130:3127. doi: 10.1021/ja078222l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liao MS, Huang MJ, Watts JD. J Phys Chem A. 2010;114:9554. doi: 10.1021/jp1052216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.The one exception is that of the annealed crystalline form of [Co(TpivPP)(1-EtIm)(O2)].28 In that complex, the Co–O2 bond is tilted off the normal to the porphyrin plane by 2.7°.
- 63.Collman JP, Brauman JI, Halbert TR, Suslick KS. Proc Natl Acad Sci USA. 1976;73:3333. doi: 10.1073/pnas.73.10.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.This argument follows that of the well-known superoxo, peroxo formalism. See for example.22, 65
- 65.Reed CA, Cheung SK. Proc Natl Acad Sci USA. 1977;74:1780. doi: 10.1073/pnas.74.5.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peng Q, Scheidt WR. unpublished results [Google Scholar]
- 67.Ellison MK, Scheidt WR. J Am Chem Soc. 1997;119:7404. [Google Scholar]
- 68.Ellison MK, Scheidt WR. Inorg Chem. 1998;37:382. doi: 10.1021/ic971109j. [DOI] [PubMed] [Google Scholar]
- 69.Phillips SEV. J Mol Biol. 1980;142:531. doi: 10.1016/0022-2836(80)90262-4. [DOI] [PubMed] [Google Scholar]
- 70.(a) Vojtechovsky J, Chu K, Berendzen J, Sweet RM, Schlichting I. Biophys J. 1999;77:2153. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Unno M, Chen H, Kusama S, Shaik S, Ikeda-Saito M. J Am Chem Soc. 2007;129:13394. doi: 10.1021/ja076108x. [DOI] [PubMed] [Google Scholar]; (c) Quillin ML, Arduini RM, Olson JS, Phillips GN., Jr J Mol Biol. 1993;234:140. doi: 10.1006/jmbi.1993.1569. [DOI] [PubMed] [Google Scholar]
- 71.Spartalian K, Lang G. J Phys. 1976;37:C6–195. [Google Scholar]
- 72.Nasri H, Wang Y, Huynh BH, Scheidt WR. J Am Chem Soc. 1991;113:717. [Google Scholar]
- 73.Debrunner PG. In: Iron Porphyrins part 3. Lever ABP, Gray HB, editors. VCH Publishers; New York: 1989. pp. 177–181. [Google Scholar]
- 74.Price DC. Aust J Phys. 1978;31:397. [Google Scholar]
- 75.Srivastava KKP, Sinha TP. J Phys France. 1987;48:2119. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.