

Abstract
Background
Pain and renal dysfunction occur in sickle cell disease. Morphine used to treat pain also co-activates platelet-derived growth factor receptor-β (PDGFR-β), which can adversely affect renal disease. We examined the influence of morphine in mesangial cells in vitro and in mouse kidneys in vivo.
Methods
Mouse mesangial cells treated with 1 μM morphine in vitro or kidneys of transgenic homozygous or hemizygous sickle or control mice (n=3 for each), treated with morphine (0.75, 1.4, 2.14, 2.8, 3.6, and 4.3 mg kg−1 day−1 in two divided doses during the first, second, third, fourth, fifth, and sixth weeks, respectively), were used. Western blotting, bromylated deoxy uridine incorporation-based cell proliferation assay, reverse transcriptase-polymerase chain reaction, immunofluorescent microscopy, and blood/urine chemistry were used to analyse signalling, cell proliferation, opioid receptor (OP) expression, and renal function.
Results
Morphine stimulated phosphorylation of PDGFR-β and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) to the same extent as induced by platelet-derived growth factor-BB (PDGF-BB) and promoted a two-fold increase in mesangial cell proliferation. The PDGFR-β inhibitor, AG1296, OP antagonists, and silencing of μ- and κ-OP abrogated morphine-induced MAPK/ERK phosphorylation and proliferation by ∼100%. Morphine treatment of transgenic mice resulted in phosphorylation of PDGFR-β, MAPK/ERK, and signal transducer and activator of transcription 3 (Stat3) in the kidneys. Morphine inhibited micturition and blood urea nitrogen (BUN) clearance and increased BUN and urinary protein in sickle mice.
Conclusion
Morphine stimulates mitogenic signalling leading to mesangial cell proliferation and promotes renal dysfunction in sickle mice.
Keywords: morphine, nephropathy, pain, platelet-derived growth factor, sickle cell disease
Editor's key points.
Morphine can worsen kidney disease in sickle cell disease (SCD).
This study investigated the effects of morphine on mesangial cells in vitro and mouse kidneys in vivo.
Morphine activated several key mitogenic signalling pathways both in vitro and in vivo.
Morphine worsened renal function in sickle cell mice.
Inhibition of the identified mitogenic pathways may prevent morphine-induced renal damage in patients with SCD.
Pain and end-stage renal disease are associated with reduced survival and poor quality of life in sickle cell disease (SCD).1–4 Morphine and its congeners are used to treat severe pain after vasoocclusive crises and ongoing chronic pain in SCD.5,6 As a possible complication of this therapeutic approach, we observed previously that clinically relevant doses of morphine incite kidney pathology, glomerular enlargement, and albuminuria in wild-type and transgenic sickle mice.7–9 Here, we investigate the mechanism(s) by which morphine stimulates renal disease.
In addition to their induction of analgesia, opioids activate growth, survival, and cytoprotection via opioid receptors (OPs) on multiple cell types including glomerular mesangial cells.7,10–15 Mitogenic signalling induced by morphine involves co-activation of receptor tyrosine kinases (RTKs) for vascular endothelial growth factor receptor-2 and platelet-derived growth factor receptor-β (PDGFR-β) and downstream mitogenic signalling of mitogen-activated protein kinase/extracellular signal regulated kinase (MAPK/ERK) and signal transducer and activator of transcription 3 (Stat3) pathways via μ-opioid receptor (MOP) on endothelium and pericytes.16–19 PDGFR-β signalling is critical to mesangial function, because overexpression or deletion of PDGF-B or PDGFR-β influences mesangial proliferation and glomerular pathology in the kidney.20
We investigated the mitogenic signalling of morphine in vitro in the mesangial cells and in vivo in the kidneys of transgenic mice expressing sickle haemoglobin (HbS) or normal human haemoglobin (HbA). Furthermore, we examined whether treatment with morphine influenced renal function in HbS mice.
Methods
All mouse studies were performed after receiving approval from our Institutional Animal Care and Use Committee (#0806A37663).
Mice
Homozygous (BERK), hemizygous (hBERK1), and control (HbA-BERK) transgenic mice were used. BERK, the homozygous sickle mice, have knockout of both murine α- and β-globins and carry the linked transgenes for human α- and βS-globins and express >99% human HbS.21 BERK mice present the sickle phenotype of human disease including renal disease, haemolysis, reticulocytosis, anaemia, organ damage, pain, and reduced survival.21–23 hBERK1 mice are hemizygous for knockout of murine β-globin, but are homozygous for knockout of murine α-globin and carry a single copy of the linked transgenes for human α- and βS-globins. They express ∼25% human HbS and show kidney pathology.9,21,22 HbA-BERK (control) mice exclusively express human α- and βA-globins (thus, normal human HbA) but no murine α- or β-globins.21 All three types of mice are littermates and hence have similar genetic background. Mice were bred <12 h light-to-dark cycle, in our pathogen-free facility and phenotyped by isoelectric focusing for Hb and by real-time polymerase chain reaction for transgene number.
Cell culture
Mouse mesangial cells (American Type Culture Collection, Manassas, VA, USA) were cultured using a 3:1 mixture of Dulbecco’s modification of Eagle's medium and F12-Hams medium, respectively, supplemented with 10% heat-inactivated fetal calf serum, 100 U ml−1 penicillin, 100 U ml−1 streptomycin, and 10 mM HEPES (all from Invitrogen, Carlsbad, CA, USA). Cells were stimulated with 1 μM morphine or 20 ng ml−1 PDGF-BB (Invitrogen) in the presence or absence of the PDGFR tyrosine kinase inhibitor, AG-1296 (10 μM; Cayman Chemicals, Ann Arbor, MI, USA), naloxone (1 μM), methylnaltrexone (MNTX) 0.1 μM (Wyeth Pharmaceuticals, Philadelphia, PA, USA) and norbinaltorphimine (nor-BNI) 1 μM (Sigma-Aldrich, St Louis, MO, USA).
Drugs and treatments
Six each of HbA-BERK and hBERK1 and 12 BERK mice were divided into two equal groups and s.c. injected with either phosphate-buffered saline (PBS) or morphine sulphate (Baxter Esilerderle Healthcare, Cherry Hill, NJ, USA) daily (0.75, 1.4, 2.14, 2.8, 3.6, and 4.3 mg kg−1 divided doses in Weeks 1–6, respectively; equivalent to ∼50–301 mg in a 70 kg human per day). At the end-point, mice were killed with compressed carbon dioxide and the kidneys were removed immediately for analysis.
MOP-1 and KOP-1 gene silencing
A cocktail of sequences of siRNA specifically targeting the mouse MOP-1 and KOP-1 genes (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were used (for further information see Supplementary material).
Each siRNA consisted of a pool of three-target-specific 20–25 nucleotide siRNAs. On Day 1, mouse mesangial cells were plated in a six-well plate. On Day 2, 100 nM siRNA and 4 μl of transfection agent si-PORT Lipid (Ambion, Inc., Austin, TX, USA) were separately diluted with OPTI-MEM I medium (Invitrogen) and then mixed rapidly and incubated at room temperature for 15 min to form a complex. The siRNA/transfection agent complex was overlaid on the mesangial cells drop wise. Control, scramble-siRNA (Santa Cruz Biotechnology, Inc.) was used as a negative control. After fourth incubation, an equal volume of mesangial cell growth medium was added to the cells followed by overnight incubation. Cells were then used to determine gene expression by reverse transcription–polymerase chain reaction (RT–PCR) or were used for other experiments.
Western immunoblotting
Kidney and mesangial cell lysates containing 100 μg of protein were resolved on a 3–15% gradient sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Immobilon, Millipore, Bedford, MA, USA) as described previously.7 For immunoblotting, we used antibodies to phospho-p44/42 MAPK/ERK Thr 202/Tyr 204 (1:500), total p44/42 MAPK/ERK (1:500), phospho-Stat3 Tyr 705 (1:500), phospho-Stat3 Ser 727 (1:500), and total-Stat3 (1:500) from Cell Signalling Technology (Beverly, MA, USA); phospho-PDGFR-β Tyr 716 (1:250) from Upstate (Lake Placid, NY, USA); total PDGFR-β (1:500) from Cell Signalling Technology (Beverly); Thy-1(1:200). Immunoreactive proteins were seen using a species-specific antibody linked to alkaline phosphate followed by development of chemiluminescent signals using the ECF western blotting system (Amersham Life Sciences, Buckinghamshire, UK). Chemiluminescent signals were acquired using a Storm 860 PhosphorImager (Molecular Dynamics, Sunnyvale, CA, USA) and densitometric analysis was performed using Image J software (NIH, Bethesda, MD, USA).
Proliferation assay
Proliferation of mesangial cells was measured using bromylated deoxyuridine (BrdU) cell proliferation enzyme-linked immunosorbent assay (Roche, Indianapolis, IN, USA) according to the manufacturer's instructions.7 Briefly, 2000 cells per well were seeded on a 96-well plate in 10% fetal calf serum containing mesangial cell medium. Cells were allowed to adhere to the plate overnight and then incubated overnight with 0.1% fetal calf serum containing medium. Cells were pre-incubated with AG-1296, naloxone, nor-BNI, MNTX, or sunitinib (2 μM, Pfizer, New York, NY, USA) for 60 min; then morphine or PDGF-BB was added for an additional 48 h. Appropriate control wells were used for each experiment.
Reverse transcription–polymerase chain reaction
Total RNA was isolated using Trizol reagent from the kidneys of 5 HbA-BERK, 7 hBERK1, and 6 BERK. Five micrograms of total RNA were reverse transcribed using the first-strand synthesis system (Invitrogen). Taq DNA polymerase (Continental Lab Products, San Diego, CA, USA) was used for performing PCRs. Primer sequences are available in the Supplementary material. Amplification was performed for 30 cycles at 94°C for 50 s, 56°C for 50 s, and 72°C for 50 s with a final extension cycle for 10 min at 72°C in a PTC-100 thermocycler (MJ Research, Waltham, MA, USA) as described earlier.7 DNA samples were seen using ethidium bromide by 2% agarose gel electrophoresis and sequenced (Microchemical Facility, University of Minnesota) to verify that they matched the expected DNA sequences. Densitometric analysis of the bands was performed using Image J (NIH).
Immunofluorescent staining
Fresh kidneys were embedded in optimum cutting temperature compound (Sakura Finetek USA, Inc., Torrance, CA, USA) and flash frozen in isopentane/liquid nitrogen. Cryo-sections were cut at 4 µm. Sections were blocked with 3% donkey serum in PBS for 30 min at 37°C and incubated with primary antibodies diluted in 3% donkey serum in PBS for 60 min at room temperature.
Immunostaining was performed as described by us earlier for kidney sections with slight modifications to accommodate different antibodies used for co-staining in addition to those used by us earlier (see Supplementary material).23 Primary antibodies for MOP and KOP did not show any immunoreactivity on the sections of kidneys from MOP- and KOP-knockout mice, respectively (Supplementary Fig. S1). In parallel, negative controls were incubated with an isotype-matched immunoglobulin G and secondary antibodies.
Immunofluorescent images were acquired using an Olympus 1X70 microscope with a ×1 eyepiece and a ×100 objective fitted with an Olympus DP70 digital camera (Olympus, Tokyo, Japan).
Urine and serum chemistry
Six each (total, 12) of BERK mice were treated with PBS or morphine as described in ‘Drugs and Treatments’ above. Urine was collected for a 24 h period using metabolic cages. BUN and urine protein and creatinine were estimated using urea and protein assay kits (Bio-assay systems, Hayward, CA, USA) and a creatinine assay kit (Cayman Chemicals). BUN clearance (C) was calculated using the equation, C=(U×V)/P, where U and P are urinary and BUN concentration, and V is urine flow rate in ml min−1. Urine volume and BUN were analysed in six PBS- and six morphine-treated mice, whereas urine protein and BUN clearance were analysed in six PBS- and three morphine-treated mice.
Data and statistical analysis
Data are expressed as the mean (sd). Comparisons were made using Mann–Whitney U-test when n=3 and using analysis of variance with Bonferroni's post hoc test when n≥5. Statistical significance was defined as P<0.05.
Results
Morphine stimulates mitogenic signalling by co-activating PDGFR-β via OPs in kidney mesangial cells
Mesangial cells stimulated with morphine in vitro showed a time-dependent increase in the phosphorylation of PDGFR-β and MAPK/ERK (Fig. 1a). Morphine-induced phosphorylation of MAPK/ERK was abrogated by naloxone and by AG1296, a pharmacological inhibitor of PDGFR, suggesting that morphine stimulates mitogenic signalling via OPs and PDGFR-β (Fig. 1b). PDGF-BB also stimulated MAPK/ERK phosphorylation, which was inhibited by AG1296 or naloxone, indicative of a cross-talk between PDGFR-β and OPs. Silencing of MOP and KOP on mesangial cells inhibited morphine-induced phosphorylation of MAPK/ERK, suggesting that morphine stimulates MAPK/ERK signalling via MOP and KOP (Fig. 1c). Interestingly, silencing of MOP and KOP also inhibited PDGF-BB-induced MAPK/ERK phosphorylation, indicating that OPs may contribute to PDGFR-β signalling (Fig. 1d). MAPK/ERK phosphorylation by morphine or PDGF-BB was inhibited by OP antagonists, MNTX and nor-BNI (Fig. 1e and f). These data show that morphine acts via OPs to stimulate mitogenic signalling by co-activating PDGFR-β.
Fig 1.

Morphine signalling in mouse mesangial cells in vitro. (a) Stimulation of mesangial cells with 1 μM morphine showing a time-dependent increase in phosphorylation of PDGFR-β and MAPK/ERK. (b) Morphine- and 20 ng ml−1 PDGF-BB-induced PDGFR-β and MAPK/ERK phosphorylation are inhibited by 10 μM AG1296, a PDGFR-β inhibitor and naloxone, a non-selective OP antagonist. (c and d) Silencing of MOP and KOP on mesangial cells inhibits morphine- and PDGF-BB-induced MAPK/ERK phosphorylation. (e and f) OP antagonist, MNTX (0.1 μM) and nor-BNI (1 μM), inhibit morphine- and PDGF-BB-induced MAPK/ERK phosphorylation. Mean (sd), n=3.
Morphine-induced mesangial cell proliferation depends upon OPs, and PDGFR-β
Morphine-induced proliferation was prevented by PDGFR inhibition using AG1296 or sunitinib (Sutent), a clinically used inhibitor of PDGFR and other growth factor receptors (Fig. 2). Similarly, morphine-induced proliferation was abrogated by OP antagonism using naloxone, MNTX, or nor-BNI. Sutent and nor-BNI also inhibited the basal proliferation of mesangial cells, while naloxone and MNTX did not have a significant effect when compared with PBS.
Fig 2.
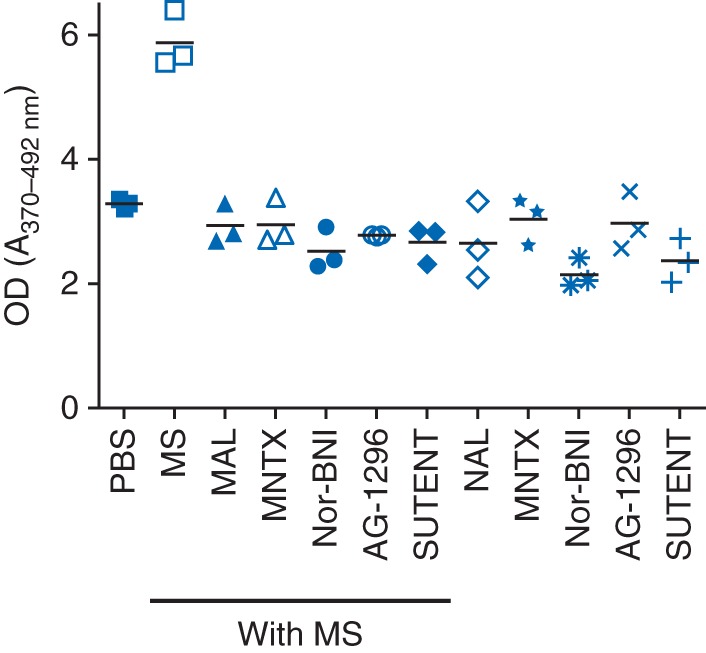
Mesangial cell proliferation shown as optical density (OD) in response to morphine and PDGFR inhibitors or OP antagonists. Mesangial cells were incubated for 48 h with 1 μM morphine in the presence or absence of 1 μM naloxone, 0.1 μM MNTX, 1 μM nor-BNI, 10 μM AG1296, and 2 μM sunitinib. Mean (sd), n=3.
MOP and KOP expression is increased in the sickle kidney
Kidneys of all three types of mice showed the expression of MOP and KOP, but not that of DOP (Fig. 3a and b). However, the expression of both MOP and KOP was significantly higher in BERK kidneys when compared with HbA-BERK. MOP and KOP expression was increased throughout the kidney in the BERK sickle mouse kidney including the glomerular epithelium and mesangium (Fig. 3c). In HbA-BERK, MOP was weakly expressed throughout the kidney, whereas KOP appeared to be strongly expressed on the vasculature when compared with other cells of the HbA-BERK kidney. More than one band for MOP was seen in HbA-BERK kidney when compared with a single band in sickle kidneys (Fig. 3a).
Fig 3.

Expression of MOPs and KOPs in the kidney of sickle mice. (a and b) RT–PCR analysis showing increased expression of MOP and KOP in BERK kidneys when compared with HbA-BERK. (c) Immunofluorescent staining of kidney sections showing MOP- and KOP-specific, green and red fluorescence, and 4′, 6-Diamidino-2-phenyl indole-positive blue nuclei, respectively. Each column shows the respective co-staining in the same field of view. The first column shows strong immunoreactivity (ir) of MOP and KOP on the large vessels in HbA-BERK mice kidney. Glomerular and epithelial regions of the HbA-BERK kidney do not show appreciable MOP-ir or KOP-ir (yellow arrows, middle column). BERK kidneys exhibit strong MOP-ir and KOP-ir in glomerular and epithelial cells (yellow arrows). n=5, 7, and 6 for HbA-BERK, hBERK1, and BERK, respectively. bv, blood vessels.
Morphine treatment of mice promotes mitogenic signalling in the kidney
Treatment of HbA-BERK, hBERK1, and BERK mice for 6 weeks with morphine resulted in increased phosphorylation of PDGFR-β, Stat3, and MAPK/ERK, when compared with PBS-treated mice (Fig. 4). Expression of the mesangial cell-specific protein, Thy1, also increased in morphine-treated kidneys. Kidneys of sickle BERK mice, which have the highest level of HbS expression, showed higher constitutive phosphorylation of PDGFR-β, Stat3, and MAPK/ERK and Thy1 expression when compared with HbA-BERK or hBERK1. Morphine further activated these signalling pathways in BERK.
Fig 4.

Morphine signalling in kidneys of mice treated with morphine (MS) or PBS for 6 weeks. Density of phospho-protein bands was determined relative to respective total protein bands or with respect to glyceraldehyde-3-phosphate dehydrogenase. Each band is representative of three separate mice kidneys. Mean (sd), n=3.
Morphine treatment influences renal function in sickle mice
Morphine treatment of BERK mice for 6 weeks led to complete inhibition of micturition in three of six mice, while the other three mice showed 60% decreased urinary volume when compared with PBS-treated mice over a 24 h period (Fig. 5a). BUN levels increased while BUN clearance decreased significantly upon morphine treatment when compared with PBS (P<0.01; Fig. 5b and c). A ∼2.5-fold increase occurred in urinary protein in morphine when compared with PBS-treated BERK mice (P<0.01, vs PBS) (Fig. 5d).
Fig 5.
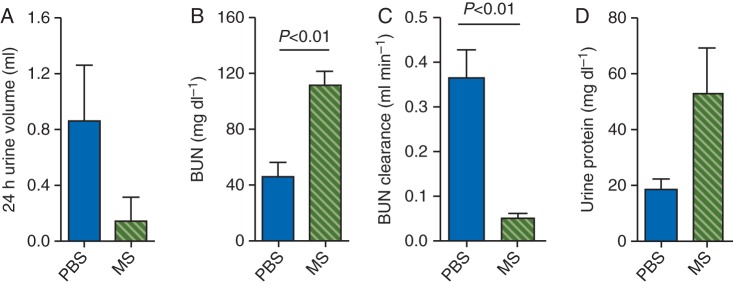
Renal function in BERK mice treated with morphine (MS) or PBS for 6 weeks. Three of six mice did not urinate in the 24 h period after 6 weeks of MS treatment, but all PBS-treated mice urinated. Therefore, n=6 for each data point except for MS in (c and d) where n=3. *P<0.01 vs PBS.
Discussion
We demonstrate that chronic exposure to morphine stimulates mitogenic signalling via co-activation of PDGFR-β in the kidney of control and sickle mice. Notably, morphine also impaired renal function in BERK sickle mice. Morphine stimulates mesangial cell proliferation through OP-mediated mitogenic signalling, which may in part contribute to renal disease. As morphine is often used as an analgesic for chronic pain, its use may exacerbate existing renal disease in SCD. We identify several molecular components critical to morphine's mitogenic activity in the kidney, including OPs and PDGFR-β, which could potentially be targeted with FDA-approved drugs, such as MNTX and PDGFR-β inhibitors, to prevent morphine-induced renal injury without compromising analgesia.
Opioids and their receptors expressed in the central nervous system have also been demonstrated in the kidney.7,24,25 We observed an increase in expression of MOP and KOP in transgenic sickle mouse kidneys when compared with control mice. While it is unclear whether increased MOP and KOP expression is a cause or consequence of renal disease, our OP antagonism and silencing studies clearly show that morphine-induced renal effects are mediated by OPs. Previous studies suggest that morphine contributes to mesangial expansion by increasing glomerular mesangial cell proliferation, matrix synthesis, and immune complex formation.12,26 In the current study, using both in vitro mesangial cells and in vivo mouse models, we find that morphine stimulates mesangial cell proliferation, via phosphorylation of PDGFR-β and MAPK/ERK signalling, suggesting that morphine has a mesangial cell-specific effect. Notably, our finding that BERK kidneys express higher levels of MOP and KOP compared with control mouse kidneys is consistent with the higher levels of constitutive phosphorylation of growth signalling pathways and Thy1 protein expression seen in BERK kidneys when compared with HbA-BERK kidneys. Thus, our study provides clear in vivo evidence that morphine promotes mitogenic signalling in the kidney.
PDGFR-β, Stat3, and MAPK/ERK signalling are key regulatory pathways in cellular proliferation and kidney disease.27–29 OPs activate MAPK/ERK and Stat3 phosphorylation7,13,16,30 and co-activate RTKs for growth factors including PDGFR-β.7,13,16,17,19,27,30–32 Deletion of PDGF-B or PDGFR-β results in the loss of mesangium in the glomerulus, while systemic overexpression of PDGF-B leads to mesangioproliferative alterations and renal fibrosis.20 Morphine stimulates PDGF-BB expression in endothelial cells.19,33 Therefore, it is likely that co-activation of PDGFR-β may amplify its mitogenic signalling in the kidney, which is consistent with our earlier observations on morphine-induced kidney growth, mesangial proliferation, and glomerular volume expansion in wild-type and sickle mice.7–9
Glomerulomegaly may be a precursor to glomerular sclerosis and result in increased urinary protein excretion.34 Morphine-induced mesangial signalling may contribute to impairment in renal function in sickle mice. However, morphine also induces vacuolization of tubular epithelium and tubular damage in mice and rats.9 It is likely that morphine-induced tubular pathology in part contributes to proteinuria observed in BERK mice treated with morphine for 6 weeks. Our observations are consistent with the significantly increased BUN in Wistar rats treated with morphine for 30 days.35 Morphine also inhibited micturition in sickle mice similar to that observed clinically because of bladder retention caused by lowering of perfusion pressure and possibly by inducing renal pathology in these mice observed by us earlier.9,36,37 Therefore, chronic morphine treatment leads to renal dysfunction in sickle mice.
To prevent the inadvertent effect of morphine on renal disease, we identified OPs and PDGFR-β, as targets of its mitogenic activity. Earlier, we identified KOP as one of the targets of morphine activity in the kidney using OP-specific knockout mice.7 In the present study, we used pharmacological and genetic approaches to identify the receptors associated with the activity of morphine in the mesangial cells. Pharmacologically, both nor-BNI and MNTX inhibited morphine-induced signalling and proliferation, suggestive of the OP-mediated effect. The pKi of nor-BNI for KOP was shown to be 0.1 nM, and for MOP of 102 nM.38 MNTX has a pKi of 8.0, 7.5, and 6.2 for MOP, KOP, and DOP, respectively.39 Therefore, based on pharmacological inhibition, it is likely that morphine acts via MOP, KOP, or both agonism on mesangial cells. This is similar to our observations showing inhibition of morphine signalling after silencing of either MOP or KOP. It is therefore likely that morphine acts via more than one OP.
Splice variants of MOP have been suggested to play a physiologically important role through heterodimerization.40 Multiple bands of MOP observed by us are suggestive of the presence of splice variants and perhaps heterodimerization of MOP with KOP. Although not known in the kidney, OPs are known to act as heterodimers in some cell types41 and therefore silencing or antagonizing one receptor may abrogate the activity of the other. An advantage of targeting KOP is that it may not antagonize morphine-analgesia, while MOP antagonists may block morphine-analgesia. It is possible that chronic treatment with morphine may lead to tolerance and therefore we administered escalating doses of morphine in our study. While activity of morphine via OPs may be reduced because of desensitization of OPs, it may act via non-opioid family of receptors. Indeed, morphine has been shown to bind to toll-like receptor 4 and promote neuroinflammation.42 Therefore, targeting RTKs with Sunitinib-like clinically used drugs may prevent the inadvertent effect of morphine in the kidney. However, Sunitinib significantly inhibited mesangial cell proliferation, supporting the notion that RTKs are critical to the maintenance of normal mesangium. Therefore, doses of RTK inhibitors must be carefully determined or it may be safer to use OP antagonists, such as MNTX, to block morphine's mitogenic effect. The mechanistic understanding herein provides insights into ameliorating the inadvertent mitogenic activity of morphine in the kidney. As these studies are performed in mice, any alteration in clinical decision-making should not be made until proved in human studies.
Supplementary material
Supplementary material is available at British Journal of Anaesthesia online.
Authors’ contributions
M.L.W. performed morphine treatments and tissue processing, analysed data, and assisted with experimental design and writing; C.C. performed western immunoblotting and proliferation assay; Y.L. did western immunoblotting and RT–PCR; J.N. and T.P. performed immunofluorescence microscopy; and M.F. performed RT–PCR; R.P.H. genotyped and provided mice and advice on editing the manuscript; K.G. conceived, designed, and supervised the entire study, analysed and interpreted data, and wrote and edited the manuscript.
Declaration of interest
None declared.
Funding
This work was supported by National Institutes of Health—National Heart, Lung and Blood Institute grants (grant numbers RO1 HL68802, HL103773 to K.G., PO1 HL55552 to R.P.H.).
Supplementary Material
Acknowledgements
We thank Stefan Kren for technical assistance, Derek Vang for statistical analysis, and Professor John E. Connett for advice on statistical analysis; and Carol Taubert for assistance with manuscript preparation.
References
- 1.McClish DK, Penberthy LT, Bovbjerg VE, et al. Health related quality of life in sickle cell patients: the PiSCES project. Health Qual Life Outcomes. 2005;3:50. doi: 10.1186/1477-7525-3-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anie KA, Grocott H, White L, Dzingina M, Rogers G, Cho G. Patient self-assessment of hospital pain, mood and health-related quality of life in adults with sickle cell disease. BMJ Open. 2012;2 doi: 10.1136/bmjopen-2012-001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–44. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 4.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–31. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- 5.Ballas SK. Update on pain management in sickle cell disease. Hemoglobin. 2011;35:520–9. doi: 10.3109/03630269.2011.610478. [DOI] [PubMed] [Google Scholar]
- 6.Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120:3647–56. doi: 10.1182/blood-2012-04-383430. [DOI] [PubMed] [Google Scholar]
- 7.Weber ML, Farooqui M, Nguyen J, et al. Morphine induces mesangial cell proliferation and glomerulopathy via kappa-opioid receptors. Am J Physiol Renal Physiol. 2008;294:F1388–97. doi: 10.1152/ajprenal.00389.2007. [DOI] [PubMed] [Google Scholar]
- 8.Arerangaiah R, Chalasani N, Udager AM, et al. Opioids induce renal abnormalities in tumor-bearing mice. Nephron Exp Nephrol. 2007;105:e80–9. doi: 10.1159/000098564. [DOI] [PubMed] [Google Scholar]
- 9.Weber ML, Vang D, Velho PE, et al. Morphine promotes renal pathology in sickle mice. Int J Nephrol Renovasc Dis. 2012;5:109–18. doi: 10.2147/IJNRD.S33813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singhal PC, Sharma P, Gibbons N, Franki N, Kapasi A, Wagner JD. Effect of morphine on renomedullary interstitial cell proliferation and matrix accumulation. Nephron. 1997;77:225–34. doi: 10.1159/000190277. [DOI] [PubMed] [Google Scholar]
- 11.Singhal PC, Sharma P, Sanwal V, et al. Morphine modulates proliferation of kidney fibroblasts. Kidney Int. 1998;53:350–7. doi: 10.1046/j.1523-1755.1998.00758.x. [DOI] [PubMed] [Google Scholar]
- 12.Singhal PC, Gibbons N, Abramovici M. Long term effects of morphine on mesangial cell proliferation and matrix synthesis. Kidney Int. 1992;41:1560–70. doi: 10.1038/ki.1992.226. [DOI] [PubMed] [Google Scholar]
- 13.Gupta M, Yunfang L, Gupta K. Opioids as promoters and regulators of angiogenesis. In: Maragoudakis ME, Papadimitriou E, editors. Angiogenesis: Basic Science and Clinical Applications. Kerala, India: Transworld Research Network; 2007. pp. 303–18. [Google Scholar]
- 14.Gupta K, Stephenson EJ. Existence and modus operandii of opioid receptors in endothelium. In: Aird WC, editor. The Endothelium: A Comprehensive Reference. New York: Cambridge University Press; 2007. pp. 451–60. [Google Scholar]
- 15.Tegeder I, Geisslinger G. Opioids as modulators of cell death and survival—unraveling mechanisms and revealing new indications. Pharmacol Rev. 2004;56:351–69. doi: 10.1124/pr.56.3.2. [DOI] [PubMed] [Google Scholar]
- 16.Gupta K, Kshirsagar S, Chang L, et al. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signalling and promotes breast tumor growth. Cancer Res. 2002;62:4491–8. [PubMed] [Google Scholar]
- 17.Chen C, Farooqui M, Gupta K. Morphine stimulates vascular endothelial growth factor-like signalling in mouse retinal endothelial cells. Curr Neurovasc Res. 2006;3:171–80. doi: 10.2174/156720206778018767. [DOI] [PubMed] [Google Scholar]
- 18.Singleton PA, Lingen MW, Fekete MJ, Garcia JG, Moss J. Methylnaltrexone inhibits opiate and VEGF-induced angiogenesis: role of receptor transactivation. Microvasc Res. 2006;72:3–11. doi: 10.1016/j.mvr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Luk K, Boatman S, Johnson KN, et al. Influence of morphine on pericyte-endothelial interaction: implications for antiangiogenic therapy. J Oncol. 2012;2012:458385. doi: 10.1155/2012/458385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floege J, Ostendorf T, Wolf G. Growth factors and cytokines. In: Neilsen EG, Couser WG, editors. Immunologic Renal Disease. 2nd Edn. Baltimore: Lippincott, Williams and Wilkins; 2001. [Google Scholar]
- 21.Paszty C, Brion CM, Manci E, et al. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278:876–78. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 22.Diwan BA, Gladwin MT, Noguchi CT, Ward JM, Fitzhugh AL, Buzard GS. Renal pathology in hemizygous sickle cell mice. Toxicol Pathol. 2002;30:254–62. doi: 10.1080/019262302753559597. [DOI] [PubMed] [Google Scholar]
- 23.Kohli DR, Li Y, Khasabov SG, et al. Pain-related behaviors and neurochemical alterations in mice expressing sickle hemoglobin: modulation by cannabinoids. Blood. 2010;116:456–65. doi: 10.1182/blood-2010-01-260372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quirion R, Finkel MS, Mendelsohn FA, Zamir N. Localization of opiate binding sites in kidney and adrenal gland of the rat. Life Sci. 1983;33(Suppl. 1):299–302. doi: 10.1016/0024-3205(83)90502-7. [DOI] [PubMed] [Google Scholar]
- 25.Neidle A, Manigault I, Wajda IJ. Distribution of opiate-like substances in rat tissues. Neurochem Res. 1979;4:399–410. doi: 10.1007/BF00963809. [DOI] [PubMed] [Google Scholar]
- 26.Singhal PC, Pan CQ, Sagar S, Gibbons N, Valderrama E. Morphine enhances deposition of ferritin-antiferritin complexes in the glomerular mesangium. Nephron. 1995;70:229–34. doi: 10.1159/000188589. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi T, Abe H, Arai H, et al. Activation of STAT3/Smad1 is a key signalling pathway for progression to glomerulosclerosis in experimental glomerulonephritis. J Biol Chem. 2005;280:7100–6. doi: 10.1074/jbc.M411064200. [DOI] [PubMed] [Google Scholar]
- 28.Grande MT, Lopez-Novoa JM. Therapeutical relevance of MAP-kinase inhibitors in renal diseases: current knowledge and future clinical perspectives. Curr Med Chem. 2008;15:2054–70. doi: 10.2174/092986708785132889. [DOI] [PubMed] [Google Scholar]
- 29.Floege J, Eitner F, Alpers CE. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008;19:12–23. doi: 10.1681/ASN.2007050532. [DOI] [PubMed] [Google Scholar]
- 30.Fujioka N, Nguyen J, Chen C, et al. Morphine-induced epidermal growth factor pathway activation in non-small cell lung cancer. Anesth Analg. 2011;113:1353–64. doi: 10.1213/ANE.0b013e318232b35a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singleton PA, Garcia JG, Moss J. Synergistic effects of methylnaltrexone with 5-fluorouracil and bevacizumab on inhibition of vascular endothelial growth factor-induced angiogenesis. Mol Cancer Ther. 2008;7:1669–79. doi: 10.1158/1535-7163.MCT-07-2217. [DOI] [PubMed] [Google Scholar]
- 32.Farooqui M, Li Y, Rogers T, et al. COX-2 inhibitor celecoxib prevents chronic morphine-induced promotion of angiogenesis, tumour growth, metastasis and mortality, without compromising analgesia. Br J Cancer. 2007;97:1523–31. doi: 10.1038/sj.bjc.6604057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wen H, Lu Y, Yao H, Buch S. Morphine induces expression of platelet-derived growth factor in human brain microvascular endothelial cells: implication for vascular permeability. PLoS One. 2011;6:e21707. doi: 10.1371/journal.pone.0021707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hostetter TH. Hyperfiltration and glomerulosclerosis. Semin Nephrol. 2003;23:194–9. doi: 10.1053/anep.2003.50017. [DOI] [PubMed] [Google Scholar]
- 35.Atici S, Cinel I, Cinel L, Doruk N, Eskandari G, Oral U. Liver and kidney toxicity in chronic use of opioids: an experimental long term treatment model. J Biosci. 2005;30:245–52. doi: 10.1007/BF02703705. [DOI] [PubMed] [Google Scholar]
- 36.Gentili M, Bonnet F. Spinal clonidine produces less urinary retention than spinal morphine. Br J Anaesth. 1996;76:872–3. doi: 10.1093/bja/76.6.872. [DOI] [PubMed] [Google Scholar]
- 37.Mahinda TB, Lovell BM, Taylor BK. Morphine-induced analgesia, hypotension, and bradycardia are enhanced in hypertensive rats. Anesth Analg. 2004;98:1698–704. doi: 10.1213/01.ANE.0000115148.03515.56. table of contents. [DOI] [PubMed] [Google Scholar]
- 38.Hjorth SA, Thirstrup K, Grandy DK, Schwartz TW. Analysis of selective binding epitopes for the kappa-opioid receptor antagonist nor-binaltorphimine. Mol Pharmacol. 1995;47:1089–94. [PubMed] [Google Scholar]
- 39.Beattie DT, Cheruvu M, Mai N, et al. The in vitro pharmacology of the peripherally restricted opioid receptor antagonists, alvimopan, ADL 08–0011 and methylnaltrexone. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:205–20. doi: 10.1007/s00210-007-0146-x. [DOI] [PubMed] [Google Scholar]
- 40.Majumdar S, Grinnell S, Le Rouzic V, et al. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc Natl Acad Sci USA. 2011;108:19778–83. doi: 10.1073/pnas.1115231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Loram LC, Ramos K, et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci USA. 2012;109:6325–30. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.