

Abstract
It is well known that electrons below 15 eV induce strand breaks in DNA essentially via the formation of transient anions which decay by dissociative electron attachment (DEA) or into dissociative electronics states. The present article reports the results of a study on the influence of organic ions on this mechanism. tris and EDTA are incorporated at various concentrations within DNA films of different thicknesses. The amino group of tris molecules and the carboxylic acid function of ethylenediamine tetra-acetic acid (EDTA) molecules together can be taken as simple model for the amino acids components of proteins, such as histones, which are intimately associated with the DNA of eukaryotic cells. The yield of single strand breaks induced by 10 eV electrons is found to decrease dramatically as a function of the number of organic ions/nucleotide. As few as 2 organic ions/nucleotide are sufficient to decrease the yield of single strand breaks by 70%. This effect is partly explained by an increase in multiple inelastic electrons scattering with film thickness but changes in the resonance parameters can also contribute to DNA protection. This can occur if the electron captures cross section and the lifetime of the transient anions (i.e., core-excited resonances) formed at 10 eV are reduced by the presence of organic ions within the grooves of DNA. Moreover, it is proposed that the tris molecules may participate in the repair of DNA anions [such as G(-H)−]induced by DEA on DNA bases.
I. INTRODUCTION
It is generally accepted that when high energy particles interact with living organisms, cellular transformations are predominantly the result of DNA damage.1,2 Radiation damage to DNA can be classified into base and sugar modifications, base release, single strand breaks (SSBs), and clustered lesions, which include a combination of two or mores single modifications, e.g., double strand breaks (DSB), tandem lesions, and crosslinks.1 This damage is produced principally by secondary species generated along the radiation track. Secondary electrons, whose distribution lies mostly below 20 eV, are the most abundant of these secondary species.3 Whether created by ionization of DNA or surrounding molecules, such as H2O, secondary electrons can interact with DNA, break chemical bonds, and create harmful radicals and ions. Thus, they account for a large portion of the direct and indirect effects of ionizing radiation on DNA. At low energies (0–20 eV)within condensed matter, electrons disrupt chemical bonds by either producing an electronic excited state that dissociates or a transient anion which decays via dissociative electron attachment (DEA), or to the creation of a dissociative electronic excited state.4 Thus, due to the potentially crucial role of low energy electrons (LEEs)in radiobiology and its applications (i.e., radiation protection and radiotherapy), considerable research has been devoted to understanding their interactions with the DNA molecule.
In recent years, our knowledge of LEE-DNA interactions has grown considerably and we now understand some of the fundamental mechanisms leading to DNA damage. This damage occurs at the molecular scale5 where the fragmentation is localized on the individual DNA subunits (i.e., DNA bases,6–9 deoxyribose,7,10 phosphate,11,12 or the surrounding water molecules).13,14 Below 15 eV, fragmentation occurs mainly via the formation of transient anions which decay into the DEA channel or autoionization leaving a subunit in a repulsive state. At higher energy such states result principally from direct scattering.5 All of these processes can irreversibly modify plasmid and linear DNA producing the previously mentioned lesions.15,16 According to several experimental17–19 and theoretical20–24 studies, LEE breaks DNA strands by cleavage of the CO bond of the backbone at the 3′and 5′ positions,25 not only via direct capture by a phosphate group26 but also via electron transfer.19–23,27 In brief, an incoming electron captured by one of the lowest π*-resonance states of the bases transfers to the sugar-phosphate group of the molecule where it resides for a sufficiently long time to cause C–O σ bond rupture via DEA. Scission of the C–O bond may also be induced by proton transfer to a negatively charged base during the lifetime of a resonance.24 Such a proton transfer from sugar to base anion radical forms a sugar carbanion and this is followed by a barrier-free sugar-phosphate C–O bond cleavage. Both mechanisms have been shown to be effective below 3 eV. However, as recently suggested, electronic excitation of a base by 5–15 eV electrons could lower their energies below 3 eV and trigger this mechanism. LEE-induced N-glycosidic bond cleavage (i.e., base release in DNA) was also found to occur primarily via initial electron capture by a base for LEE with energies below 15 eV.19
At the experimental level, this knowledge of LEE-DNA interactions was derived from a multitude of experiments of increasing target complexity, ranging from gaseous DNA subunits or their analogs to plasmid DNA. This range was necessary to systematically understand how known fundamental principles of LEE scattering from simple molecules and attachment to them would apply to larger ones up to plasmid DNA containing over 6×103 nucleotides. In the solid phase experiments, the targets were investigated in the form of thin films of the highest possible purity, so as to keep the system as simple as possible, despite the complexity of DNA. Although the dry film experiments of pure DNA in vacuum were essential to unveil basic mechanisms of damage, they clearly do not correspond to cellular conditions. We therefore initiated a series of systematic investigations to probe DNA with LEE under conditions which are closer to those found in living cells.28,29 Recently, we showed that the presence of additional water around DNA (1) modifies the transient negative ion manifold and corresponding decay channels29 and (2) the indirect effect of LEE irradiation contributes to 60% of the damage.28
Ions are an important constituent of cells; they play a role in the stabilization of the B-DNA conformation in vitro by their interaction with the major and minor grooves as well as the negatively charged phosphate. In the present study, we investigate the role of organic ions, in the form of tris and EDTA, on LEE-induced DNA damage. tris and EDTA were chosen for two reasons. First, 10 mM tris with 1 mM EDTA constitute the standard buffer solution for many in vitro experiments with DNA. Furthermore, as shown in Fig. 1 and the next section, the tris molecule possesses an amine group (NH2), while the EDTA molecule contains four carboxylic acid groups (COOH). Because of their functional groups, these organic ions can mimic some aspects of the protein structure. In addition, the histone proteins, which are intimately associated with the DNA of eukaryotic cells, are rich in basic amino acids such as lysine and arginine which have additional positively charged amino groups. Experiments with these molecules can therefore provide some insight into the effect of proteins on LEE-induced DNA damage. Results on a series of experiments on the influence of the number of these organic ions on the yield of SSB induced by LEE in plasmid DNA films is reported herein. Our investigations were performed on DNA films of different thicknesses placed in an ultrahigh vacuum (UHV) system and irradiated by 10 eV electrons. At this energy, DNA damage is dominated by the formation of transient anions which can decay via two pathways of fragmentation: DEA or into electronically dissociative states. Briefly, our results show that the yield of SSB decrease as a function of the number of organic ions/nucleotide in DNA films of variable thickness as well as in films of constant thickness. This protective effect can not only be explained by a thickness dependence but also by modification of the resonance parameters and/or the repair of DNA anionic fragments induced by DEA on DNA bases.
FIG. 1.
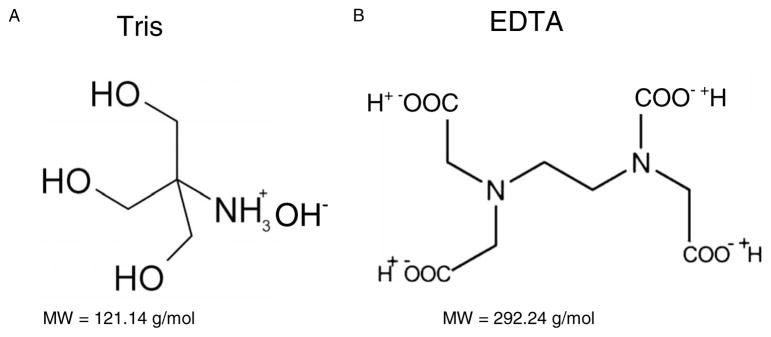
(a) Structure and molecular weight of tris molecule. (b) Structure and molecular weight of EDTA molecule.
II. EXPERIMENTAL
A. Preparation of plasmid DNA
pGEM-3Zf (-) plasmid DNA (3197 bp, Promega) was extracted from E. Coli JM109 and purified with QIAfilter plasmid Giga kit (QIAGEN). Agarose gel electrophoresis analysis showed that 95% of the purified plasmid DNA was in the supercoiled form and the rest was in the concatemeric (1%) and nicked circular form (4%). The relative amount of proteins in the plasmid was determined by measuring the ratio of the UV absorption of DNA at 260 nm and the UV absorption of protein at 280 nm, giving a ratio of 1.98 corresponding to less than 15% of proteins.30–34 The UV absorption at 260 nm also provided the absolute amount of plasmid DNA, based on the common factor of 1.0 optical density unit for a solution of double stranded DNA with a concentration of 50 ug/ml.35 TE buffer (10 mM tris and 1 mM EDTA) was removed by homemade columns filled with Sephadex G50 (Pharmacia) resin on a glass bead bed. After equilibration and washes with ddH2O, the pure DNA was eluted by centrifugation.
Unless they become saturated, Sephadex G-50 columns are highly efficient for removing salt from a solution.36 Given that we applied only 150 μl of DNA solution containing 0.24 mg of tris and EDTA ions to purify, we can affirm that we operated under conditions far from saturation. The high efficiency of the columns to remove small molecules such as salt was verified previously by Cecchini et al., who purified a P-32 labeled oligonucleotides solution from unreacted free 32P [gamma-ATP (adenosine-5′-triphosphate)], by passing the solution through the column. Comparison of the amount of radioactivity before and after purification determined that the column removed more than 99.9% of unreacted free 32P (gamma-ATP).37 We therefore estimate that the amount of remaining tris and EDTA in our DNA solution after the purification protocol is about 1 organic ion/nucleotide, in addition to counterions. In any case, some residual tris and EDTA molecules are needed to stabilize DNA in solution and to produce minimal damage by the manipulation of samples. In the presentation of the results, we refer to the quantities of organic ions added to the purified samples. Thus, DNA films with 0 organic ions/nucleotide refers to purified DNA samples that contain a minimal amount of tris and EDTA. The other samples were prepared by adding different amount of tris and EDTA varying between 1 and 32 organic ions/nucleotide to the purified DNA. The structures of tris and EDTA, as well as their respective molecular weights are given in Fig. 1. The positive charge of tris molecule is expected to be attracted by the negatively charged phosphate of DNA backbone. A few others of these molecules will reside into minor and major groove of DNA helix. Because of its interaction with DNA, it is the amine group of the tris molecules that is the most likely to play a role in protecting DNA against LEE-induced damage. The interactions of EDTA with DNA should be much less important, owing to the four negative charges on the molecule. However, both tris and EDTA contribute to the film thickness, keeping in mind the 10 to 1 ratio, respectively.
B. LEE irradiation
To obtain the results presented in Figs. 2–4, different amount of tris and EDTA varying between 0 and 32 organic ions/nucleotide were added to a constant amount (260 ng) of DNA and deposited on a chemically clean tantalum foil (Goodfellow, 99.9% purity, 25 μm thickness). Each sample was frozen at −70 °C for 5 min and then lyophilized with a hydrocarbon-free sorption pump under a pressure of 1 mTorr for 2 h. The lyophilized DNA formed films of different average thicknesses between 5 ML (for samples without organic ions) and 60 ML (for samples with 32 organic ions/nucleotide). The correspondence between film thickness and amount of ion is given in Table I. The results presented in Fig. 5 were recorded with DNA films containing between 0 and 8 organic ions/nucleotide. They were given the same treatment as described above, except that they were deposited on a tantalum film of 450±50 nm thickness evaporated on a 0.4 mm thickness silicon substrate. In order to obtain constant film thickness of approximately 5 ML, the amount of DNA was deceased while the number of organic ions/nucleotide increased, as described in Table II. After lyophilisation, each sample of (3.5±0.2) mm diameter was directly transferred to a UHV chamber. After 24 h evacuation, the DNA was exposed to a fixed incident electron current of 1.5 nA (current density of 1.2×1010 electrons s−1 cm−2) under a background pressure of 1×10−8 Torr at room temperature. The incident electron energy was set at 10±0.5 eV for irradiation times of seconds up to 3 min. The area of the electron beam was adjusted to be slightly smaller than that of the sample.
FIG. 2.

Dose response curve for plasmid DNA irradiated with 10 eV electrons in presence (■) or absence (●) of 12 organic ions/nucleotide. Panel A represents the formation of SSB while panel B represents the loss of super-coiled DNA. The current density was kept constant at 1.2 × 1010 electrons/s cm2. Each data points correspond to the mean value of four samples ± standard deviation. The solid line is a guide for the eyes. The initial slope was determines by the first three data points and are given for each curves (s). S1 and S2 correspond to the initial slope for the formation of SSB in plasmid DNA without organic ions and plasmid DNA in presence of 12 organic ions/nucleotide, respectively. S3 and S4 correspond to the initial slope for the loss of supercoiled DNA in plasmid DNA in presence of 12 organic ions/nucleotide and plasmid DNA without organic ions, respectively.
FIG. 4.

(a) Experimental yield of SSBs induced by 10 eV electrons as a function of the number of organic ions/nucleotide in a film of constant DNA content. (b) Comparison of the experimental yield (■) with the expected yield (dashed line) for the formation of SSBs induced by 10 eV electrons as a function of DNA film thickness. Each data point corresponds to the mean value of 12 samples irradiated in 15 s (in the linear portion of the dose-response curve) ± standard deviation. The solid line corresponds to the smoothing of experimental data generated with the method of percentile filter. The current density was kept constant at 1.2×1010 electrons/s cm2.
TABLE I.
Estimated averaged film thicknesses in nanometers and monolayers as a function of the number of organic ions/nucleotide for samples with a constant amount of DNA (260 ng).
| Film thickness (nm) | Film thickness (ML) | |
|---|---|---|
| 0 organic ion/nucleotide | 10 | 5 |
| 1 organic ion/nucleotide | 14 | 7 |
| 2 organic ions/nucleotide | 16 | 8 |
| 8 organic ions/nucleotide | 34 | 17 |
| 32 organic ions/nucleotide | 120 | 60 |
FIG. 5.

Experimental (■) and theoretical (- - - -) yields of SSBs at constant DNA film thickness as a function of the number of organic ions/nucleotide. The dashed line is the expected yield if the interaction between DNA and organic ions had no effect. Each data points correspond to the mean value of six samples irradiated in 15 s (in the linear portion of the dose-response curve) ± standard deviation.
TABLE II.
Amount of DNA needed in nanograms to obtain at constant thickness while varying the number of organic ions/nucleotide present in a 5 ML film.
| Number of organic ions/nucleotide | DNA (ng) |
|---|---|
| 0 | 260 |
| 1 | 168 |
| 2 | 126 |
| 5 | 78 |
| 8 | 56 |
C. Analysis of plasmid DNA by Agarose Gel Electrophoresis
After irradiation, the tantalum foil was removed from the UHV chamber and DNA was immediately dissolved in 10 μl of TE buffer and pH 8.0. The recovery of DNA was approximately 98% and was calculated by measuring the absolute amount of DNA. The different forms of DNA were separated by 1% neutral agarose gel electrophoresis run in TAE buffer (40 mM tris acetate, 1 mM EDTA, and pH 8.0) at 100 V for 7 min and 75 V for 90 min. Both the gel and the DNA samples were prestained by SYBR Green I (Molecular Probes), 10 000× for gel and 100× for samples, respectively. After electrophoresis, gels were scanned with the STORM860 using the blue fluorescence mode (Molecular Dynamics) at an excitation wavelength of 430 nm. The percentage of each form was obtained from Image Quant analysis. These values were corrected for the weaker binding of SYBR Green I to the supercoiled form of DNA compared to nicked circular and linear configurations.38 In order to find this correction factor, 100 ng of supercoiled DNA and 100 ng of linear DNA were deposited in two different wells of an electrophoresis gel. The linear fragments of DNA were obtained by cutting the plasmid with the restriction enzyme EcoRI which linearized the plasmid. After determining the intensity of each band, the area under the peak of linear DNA was divided by the area under the peak of supercoiled DNA to give a correction factor of 1.2.
For each experiment, four control samples were deposited on tantalum, lyophilized, kept under UHV conditions, and recovered, but were never irradiated. The comparison of these controls with the controls in the solution shows that reduction of supercoiled DNA due to manipulation depended on the amount of organic ions. A loss of 8 percentage points was observed with no addition of ions, whereas supercoiled DNA was reduced by only 2.5 percentage points in the presence of 32 organic ions/nucleotide.
The quantum yields [(Y) in number of damages per incident electron per DNA molecule] for the induction of SSB and loss of supercoiled DNA were derived from the initial linear slopes (S in % electron−1) of the respective exposure-response curves given by Y =S/f where f is the percentage of supercoiled DNA in the control.
III. RESULTS AND DISCUSSION
A. DNA damage as function of electron exposure
Figure 2 shows exposure-response curves obtained by bombarding two different types of DNA films with 10 eV electrons: plasmids in the presence of 12 organic ions/nucleotide added (■) and plasmids without any added organic ions (●). The percentage loss of supercoiled DNA as well as the formation of SSB were plotted as a function of the total number of incident electrons. Each point on the figure corresponds to the mean value of the four measurements and the error bars denote the standard deviation of these measurements. As expected, the amount of supercoiled DNA decreases continuously with irradiation whereas the number of SSB increases. As seen from Fig. 2, the values of the initial slopes of the curves recorded without organic ions compared to those recorded in presence of tris-EDTA show that the presence of organic ions protects DNA. DNA is twice as sensitive to the formation of SSB in the absence of organic ions than in the presence of 12 organic ions/nucleotide.
B. Protection of DNA by organic ions against the damage induced by the tantalum surface
Figure 3 shows the stability of nonirradiated DNA (i.e., plasmid DNA deposited on tantalum, lyophilized, kept under UHV, and recovered) as a function of the number of organic ions/nucleotide present in the film. Tantalum was chosen as the standard substrate for plasmid DNA films because it induces minimal DNA damage5 in the presence of organic ions. Lyophilization on other metal substrates produces considerably more damage; e.g., a gold surface induces up to 25% SSB to 5 ML nonirradiated buffered DNA films.28 According to Fig. 3, in the absence of added organic ions, non-irradiated samples have a low percentage of supercoiled DNA (86%). The amount of intact DNA increases up to two organic ions/nucleotide; beyond this number, the additional ions do not have any significant effect.
FIG. 3.

Percentage of supercoiled DNA in nonirradiated samples deposited on a tantalum surface as a function of the number of organic ions/nucleotide. Each data point corresponds to the mean value of four samples ± standard deviation.
Assuming that the tantalum surface is responsible for the DNA damage, the latter should arise from the first layer in direct contact with the substrate. If all the DNA molecules were uniformly distributed and those in contact with the metal necessarily destroyed, while the others in the film were kept intact, 20% of the DNA film would be damaged by tantalum, leaving 80% of the DNA in the supercoiled form. The percentage of supercoiled DNA in our nonirradiated samples in absence of organic ion is close to this value (86%). Since there are irregularities in the DNA film, this finding supports the hypothesis that tantalum plays a major role in the induction of damage to nonirradiated DNA films in the absence of organic ions. This hypothesis can be further verified by adding organic ion to purified samples. For example, adding a single organic ion/nucleotide increases the film thickness from 5 to 7 ML. In this case, if the layer in contact with tantalum is completely damaged and the others are intact, the proportion of DNA damage in uniformly distributed DNA films should change from 20% to 14%. This is close to what we observe experimentally; there is 5% less damaged plasmids with one added organic ion/nucleotide compared to films without added organic ion. Indeed, Fig. 3 shows that the percentage of supercoiled DNA increases from 86% to 91% by adding 1 organic ion/nucleotide. The protective effect of organic ions against damage induced by the tantalum surface is thus simply explained by the increase in film thickness which decreases the amount of DNA in direct contact with tantalum, thus leaving a larger proportion of intact DNA in the sample.
C. Damage as a function of film thickness and the number of organic ions in the DNA film
The yield of SSB induced by 10 eV electrons as a function of the number of organic ions/nucleotide present in films of constant DNA content (260 ng) is shown in Fig. 4(a). In the absence of organic ions, the yield of SSB is high, followed by a dramatic decrease as a function of the number of organic ions/nucleotide, with saturation beyond 8 organic ions/nucleotide. The apparent slight increase in the yield observed between 0 and 1 organic ion/nucleotide is not statistically significant. It is important to note that all the yields of SSB measured in this study are expressed per DNA molecule. In other words, the measured yields are independent of the number of DNA molecules in the film and also independent of DNA dilution by the presence of tris and EDTA molecules. Thus, Fig. 4(a) clearly shows that the plasmids are protected by the tris molecules. Since the decrease in the yield of SSB cannot be explained by the effect of dilution, it must arise from the effect of film thickness on electron beam intensity and electron energy, and/or modification of the damage by the presence of the organic cations in DNA.
The experimental yields measured in Fig. 4(a) are expressed as a function of film thicknesses in Fig. 4(b) (■). The solid line corresponds to the smoothing of experimental data generated with the method of percentile filter. These thicknesses were calculated taking the density of the organic ions to be the same as that of DNA (1.7 g/cm3) (Ref. 39) and considering that each sample possess a constant amount of DNA (260 ng) which accounts for 5 ML of the total film thickness. Calculated film thicknesses are given in Table I as a function of the number of organic ions present in the film. Addition of as few as eight organic ions/nucleotide increases film thickness by 3.4 fold. It is thus essential to know the quantity of ions present during the preparation of the samples in order to include their contribution in the calculation of film thickness. Not considering the contribution of non-DNA components to total film thickness would have the effect of giving incorrect values for cross sections and effective electron ranges. Alternatively, effective cross sections for the production of strand breaks by LEEs can be measured in highly purified plasmid DNA, as recently done by Panajotovic et al.,40 i.e., with samples having the minimal amount of tris and EDTA ions.
When electrons penetrate a DNA film, they continuously lose energy by inelastic collisions. Thus the cross section for the induction of strand breaks changes continuously as a function of the penetration depth. At a certain distance, some electrons do not have sufficient energy to cause further DNA strand breaks. The number of electrons having sufficient energy to cause SSB then diminishes exponentially41 as the film thickness increases and so does the yield of SSB. The dashed line in Fig. 4(b) corresponds to the best exponential decay fit to our experimental yields of SSB. Obviously, it does not fit our results, indicating that the yields of SSB cannot be explained uniquely by a thickness effect. The measured yields decrease more rapidly as a function of film thickness than expected. This discrepancy supports the hypothesis that the ion-DNA interaction plays a role in protecting DNA.
D. Damage as a function of the number of organic ions per nucleotide induced by LEEs in DNA films of constant thickness
In order to test the hypothesis that the decrease in the yield of SSB in Fig. 4(b) is not only the result of increasing thickness, the experiment was repeated with a constant film thickness of 5 ML; i.e., with the amount of DNA in the samples decreasing while the number of organic ions per nucleotide increased. Figure 5 shows the experimental yield of SSB measured at 5 ML thickness. It decreases dramatically as a function of the number of organic ions per nucleotide. The presence of 2 organic ions/nucleotide is sufficient to decrease the yield of SSB by approximately 70% while the presence of 8 organic ions/nucleotide decreases the yield of SSB by 88%. In these experiments, the maximum number of organic ions/nucleotide was limited to 8 because additional organic ions resulted in insufficient amounts of DNA available for precise analysis of SSB. The expected SSB yield at constant film thickness, if the presence of organic ions had no effect, is given by the dashed line in Fig. 5. The comparison with the experimental yields clearly shows that the presence of positively charged organic ions in the film protects the DNA from SSBs induced by 10 eV electrons.
Many radioprotective molecules are known to act within fast radiochemistry time-scales (10−13–10–10 s). For example, thiol compounds are excellent free radical scavengers for species such as hydroxyl radicals.42,43 But there are no examples of molecules that protect DNA from damage induced by LEEs. At 10 eV, electrons break DNA strands essentially via core-excited resonances; i.e., the formation of a transient anion consisting of the incoming electron trapped by the positive electron affinity of an electronically excited state of a basic unit of DNA. When created on the phosphate unit of DNA, some of these two-electron one-hole states are dissociative in the Franck–Condon region, leading to rupture of the C–O bond in the backbone.11,25 Alternatively, the incident electron can be captured by a base and subsequently transfer to the dissociative anion states of the phosphate group to form the 10 eV core-excited anion leading to a SSB.25 Even at 10 eV, the transfer of electrons from a base to the phosphate group appears to be the major pathway of strand break formation in DNA.27 The different transfer pathways following electron capture by a base are shown schematically in Fig. 6. At an initial energy Eo =10 eV, the electron can temporarily attach to a base, creating a core-excited transient anion [base*]−. Via pathway 1, the electron can be released with its initial energy into the continuum (ec−) (i.e., the surrounding medium or vacuum) or it can be transferred (et−) to another site, within DNA, where it can form another transient anion. As shown by the arrow on the left, this site can be the phosphate unit. Pathway 3 is similar except that before transferring the electron loses a considerable amount of energy to electronically excited the base (E≪E). The base transient anion can also dissociate (DEA, pathway 2). In this case, the additional electron on the anionic fragment could also transfer to the PO4 unit (pathway 4) leading to a SSB. In the case of pathways 2 and 4, repair by proton transfer from the tris molecule may at least partially explain protection of DNA by organic ions. Furthermore, when the core-excited transient base anion [base*]− is formed (i.e., a one-hole-two-electron state), DEA could leave the neutral moiety in a dissociative electron excited state or the hole could be associated with the sugar-phosphate group with two electrons in the π* orbital of the base. Such configuration may lead to a DSB with a single 10 eV electron (Z. Li, J. R. Wagner, and L. Sanche, unpublished results).
FIG. 6.

Decay channels of a transient anion of a DNA base formed at electron energy Eo. Pathways 1 to 3 represent the elastic, DEA, and electronically inelastic channels, respectively. represents the transfer of an electron emitted from the base or an anionic fragment (pathway 4) to the phosphate unit.
The following equations show an example of the capture of an electron by guanine which has the lowest oxidation potential of the four bases. The transient negative ion (G−) can dissociate into an anionic dehydrogenated base [i.e., G +e−→G−→G(-H)−+H•] and a radical or it can dissociate into H− and a dehydrogenated base G(-H).
Thus, the formation of a dehydrogenated anionic base can lead to the formation of SSB via pathway 4. However, tris molecules in our DNA films could repair dehydrogenated guanine anions by donating an H atom to the base and accepting the unpaired electron from it. This two-electron-coupled-proton transfer or one-step-hydrogen atom and electron transfer mechanism results in a normal guanine and an organic anion. This process prevents the transfer of electrons to the phosphate and thus inhibits pathway 4. As the amount of organic ions/nucleotide increases, we expect the probability of hydrogen transfer to DNA to increase preventing the formation of SSB. Jena et al.44 showed from density functional theory calculations that by this mechanism certain amino acids, such as cysteine and tyrosine, can repair DNA damage in the form of guanine anion induced by LEE in both the gaseous and aqueous phases and prevent the formation of SSB. Our experiments were performed in the solid phase but in the absence of water molecules. These conditions may therefore be close to those of the gaseous phase studied by Jena et al.44 Because the EDTA molecules tend to accept hydrogen atoms, they are not expected to participate in the repair process.
It is also possible that the resonance parameters of the transient anions involved in the formation of SSB, via the pathways described in Fig. 6, are modified by the presence of organic ions within the groves of DNA. The cross section for DEA to the base which can lead to pathway 4, is given by
| (1) |
where Ps represents the survival probability of the anion against autodetachment of the electron and σcap the cross section for electron capture by the base. σcap can be written
| (2) |
where λ is the de Broglie wavelength of the incident electron, g is a statistical factor, and χv is the normalized vibrational nuclear wave function. Γa is energy width of the resonance and Γd corresponds to the width of Frank–Condon region. The width of the transient anion state in the autodetaching region defines the lifetime τa toward autodetachment, τa(R) = ħ/Γa(R), such that the survival probability of the temporary anion, after electron capture, can be expressed as
| (3) |
where RE is the bond length of the anion at energy E (in our case Eo =10 eV) and Rc is that internuclear separation, along the dissociation coordinate, beyond which autodetachment is no longer possible. If we define an average lifetime τ̄a and let K ≡ λ2g|χv|2, then Eq. (1) becomes
| (4) |
Here τ̄c(E) ≡ |Rc -RE|/v where v is the average velocity of separation of the dissociating fragments. Hence, the DEA cross section depends exponentially on ratio of the lifetime of the transient anion and the velocities of the fragments.45
The same equation can be derived for DEA to the phosphate group. However, in this case, the capture probability depends on the direct capture cross section by PO4 at 10 eV and the electron transfer probability via pathways 1 at 10 eV and that via pathway 3 at lower energy. Furthermore, different transient anion states have to be considered at the two different energies.
Despite the complexity of the problem Eqs. (1)–(4) clearly indicate that any change in the environment of a base or a phosphate unit is bound to influence the magnitude of the DEA cross section to the basic unit, most effectively, by modifying the lifetime of the transient anion (i.e., τa). It is known that DEA to molecular solids and biomolecules including DNA is highly dependent on environment.46,47 In particular, modification of an electric field close to the target molecule can considerably alter the DEA cross section.46 This has been verified by changing the distance between a molecule undergoing DEA and its metal substrate by intercalating an inert dielectric layer between them. At large distances (≥1 nm), the electric field created by the polarization field in the dielectric and in the metal (induced by the transient anion) increases the DEA cross section principally by reducing Rc in Eq. (3).46 At shorter distances from the metal surface (≤1 nm), the electric field is sufficiently high to decrease the capture cross section and reduce the average lifetime τ̄a by increasing electron transfer to the metal.46,48 This latter mechanism has been found to decrease σDEA to condensed O2 by two orders of magnitude when the target molecule goes from lying on a thick dielectric layer to the bare substrate.48 Considering the sensitivity of DEA to the modification of a near electric field, it is very likely that the modification of electric fields within DNA caused by the addition of cations at specific sites would modify σDEA leading to base damage and rupture of the C–O bond of the backbone. It is therefore highly probable that changes in the internal charge distribution by cations within DNA are involved in its protection. In order to confirm this possibility, a study involving plasmid DNA in presence/absence of 2 organic ions/nucleotide will soon be performed as a function of electron energy. This experiment will allow to verify if the presence of positively charged molecules change resonance parameter as the energy width of the resonance and its position.
IV. SUMMARY AND CONCLUSIONS
We have investigated the damage induced to plasmid DNA by lyophilization on a tantalum substrate followed by exposure to UHV and that induced by 10 eV electrons. In these experiments, the amount of added organic cations to the DNA was varied from 0 to 32 per nucleotide. Damage to the DNA in nonirradiated samples appears to be induced by the tantalum surface in contact with the first layer of the lyophilized DNA films. Additional damage is induced by irradiation with 10 eV electrons. Both types of damage decrease with increasing cation concentration. For example, the addition of a single organic ion/nucleotide in a film increases the percentage of supercoiled DNA from 86% to 91% of the original amount. This type of protection of DNA in unirradiated samples is easily explained by the decrease in the number of DNA molecules in contact with the metal substrate as the number of organic ions increases. This effect of dilution of DNA within the film also provides protection from LEE-induced damage because organic ions absorb some of the energy of electrons which would otherwise be deposited within DNA. However, this phenomenon can only partially explain the damage dependence on cation concentration. The DNA-cation interaction also protects DNA from the damage induced by 10 eV electrons. This interaction may modify the capture cross section and lifetime of the transient anions formed on the bases and the phosphate group of DNA. Since bond dissociation is highly sensitive to these resonance parameters, damage to DNA is reduced by the presence of additional organic cations. Furthermore, the damage induced to the bases by DEA can be restored by the amine function of the tris organic cation and thus also reduces DNA damage. As demonstrated theoretically by Jena et al.44 in the case of amino acids, a dehydrogenated anionic base can be restored to the original molecular moiety by a two-electron-coupled-proton transfer or one-step-hydrogen-electron transfer. From this result, we may conjecture that the histone proteins associated with eukaryotic DNA also serve to protect DNA via a similar repair mechanism, but this hypothesis remains to be tested. In order to complete this study, it would be interesting to repeat the experiment by using spermine which allows the incorporation of a poly cationic amine without increasing the film thickness. A second experiment studying the yield of SSB as a function of electron energy with plasmid DNA in presence of organic cations and organic anions, independently, would be useful to more clearly understand how the presence of ions modifies resonance parameters.
Acknowledgments
The authors would like to thank P. Cloutier, S. Girouard, and Professor William Bernhard for helpful comments and suggestions. Financial support for this work was provided by the Canadian Institutes of Health Research.
References
- 1.von Sonntag C. The Chemical Basis of Radiation Biology. Taylor & Francis; London: 1987. [Google Scholar]
- 2.DeVita VT, Jr, Hellman S, Rosenberg SA. Cancer: Principle and Practice of Oncology. Lippincott; New York: 2001. [Google Scholar]
- 3.Pimblott SM, LaVerne JA, Mozumber A. J Phys Chem. 1996;100:8595. [Google Scholar]; Radiat Phys Chem. 2007;76:1244. [Google Scholar]
- 4.Sanche L. Scanning Microsc. 1995;9:619. [Google Scholar]
- 5.Sanche L. Mass Spectrom Rev. 2002;21:349. doi: 10.1002/mas.10034. [DOI] [PubMed] [Google Scholar]
- 6.Abdoul-Carime H, Cloutier P, Sanche L. Radiat Res. 2001;155:625. doi: 10.1667/0033-7587(2001)155[0625:leeesd]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Sevilla M, Sanche L. J Am Chem Soc. 2003;125:8916. doi: 10.1021/ja034286u. [DOI] [PubMed] [Google Scholar]
- 8.Huels MA, Hahndorf I, Illenberger E, Sanche L. J Chem Phys. 1998;108:1309. [Google Scholar]
- 9.Abdoul-Carime H, Gohlke S, Illenberger E. Phys Rev Lett. 2004;92:168103. doi: 10.1103/PhysRevLett.92.168103. [DOI] [PubMed] [Google Scholar]
- 10.Ptasińska S, Denifl S, Scheier P, Märk TD. J Chem Phys. 2004;120:8505. doi: 10.1063/1.1690231. [DOI] [PubMed] [Google Scholar]
- 11.Pan X, Sanche L. Chem Phys Lett. 2006;421:404. [Google Scholar]
- 12.Pan X, Cloutier P, Hunting D, Sanche L. Phys Rev Lett. 2003;90:208102. doi: 10.1103/PhysRevLett.90.208102. [DOI] [PubMed] [Google Scholar]
- 13.Simpson WC, Parenteau L, Smith RS, Sanche L, Orlando TM. Surf Sci. 1997;390:86. [Google Scholar]
- 14.Pan X, Abdoul-Carime H, Cloutier P, Bass AD, Sanche L. Radiat Phys Chem. 2005;72:193. [Google Scholar]
- 15.Boudaïffa B, Cloutier P, Hunting D, Huels MA, Sanche L. Science. 2000;287:1658. doi: 10.1126/science.287.5458.1658. [DOI] [PubMed] [Google Scholar]
- 16.Huels MA, Boudaiffa B, Cloutier P, Hunting D, Sanche L. J Am Chem Soc. 2003;125:4467. doi: 10.1021/ja029527x. [DOI] [PubMed] [Google Scholar]
- 17.Martin F, Burrow PD, Cai Z, Cloutier P, Hunting DJ, Sanche L. Phys Rev Lett. 2004;93:068101. doi: 10.1103/PhysRevLett.93.068101. [DOI] [PubMed] [Google Scholar]
- 18.Ptasińska S, Sanche L. Chem Phys. 2007;9:1730. doi: 10.1039/b616619a. [DOI] [PubMed] [Google Scholar]
- 19.Zheng Y, Wagner R, Sanche L. Phys Rev Lett. 2006;96:208101. doi: 10.1103/PhysRevLett.96.208101. [DOI] [PubMed] [Google Scholar]
- 20.Barrios R, Skurski P, Simons J. J Phys Chem. 2002;106:7991. [Google Scholar]
- 21.Berdys J, Anusiewicz I, Skurski P, Simons J. J Am Chem Soc. 2004;126:6441. doi: 10.1021/ja049876m. [DOI] [PubMed] [Google Scholar]
- 22.Berdys J, Skurski P, Simons J. J Phys Chem B. 2004;108:5800. [Google Scholar]
- 23.Berdys J, Anusiewicz I, Skurski P, Simons J. J Phys Chem A. 2004;108:2999. [Google Scholar]
- 24.Dąbkowska I, Rak J, Gutowski M. Eur Phys J D. 2005;35:429. [Google Scholar]
- 25.Zheng Y, Cloutier P, Hunting DJ, Sanche L, Wagner JR. J Am Chem Soc. 2005;127:16592. doi: 10.1021/ja054129q. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Sevilla MD, Sanche L. J Am Chem Soc. 2003;125:13668. doi: 10.1021/ja036509m. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Zheng Y, Cloutier P, Sanche L, Wagner RJ. J Am Chem Soc. 2008;130:5612. doi: 10.1021/ja077601b. [DOI] [PubMed] [Google Scholar]
- 28.Brun E, Cloutier P, Sicard-Roselli C, Fromm M, Sanche L. J Phys Chem B. 2009;113:10008. doi: 10.1021/jp902540k. [DOI] [PubMed] [Google Scholar]
- 29.Ptasinska S, Sanche L. Phys Rev. 2007;75:030915. doi: 10.1103/PhysRevE.75.031915. [DOI] [PubMed] [Google Scholar]
- 30.Warberg O, Christian W. Biochem Z. 1942;310:384. [Google Scholar]
- 31.Glasel JA. BioTechniques. 1995;18:62. [PubMed] [Google Scholar]
- 32.Manchester KL. BioTechniques. 1996;20:968. doi: 10.2144/96206bm05. [DOI] [PubMed] [Google Scholar]
- 33.Manchester KL. BioTechniques. 1995;19:208. [PubMed] [Google Scholar]
- 34.Wilfinger WW. BioTechniques. 1997;22:474. doi: 10.2144/97223st01. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Vol. 3. Cold Spring Harbor Laboratory Press; New York: 1989. p. C.1. [Google Scholar]
- 36.Gel Filtration Principles and Method Handbook. GE Healthcare Life Science; Sweden: 2007. [Google Scholar]
- 37.Cecchini S, Girouard S, Huels MA, Sanche L, Hunting DJ. Radiat Res. 2004;162:604. doi: 10.1667/rr3267. [DOI] [PubMed] [Google Scholar]
- 38.Cai Z, Cloutier P, Hunting D, Sanche L. J Phys Chem B. 2005;109:4796. doi: 10.1021/jp0459458. [DOI] [PubMed] [Google Scholar]
- 39.Fasman GD. Handbook of Biochemistry and Molecular Biology. 3. Vol. 1 CRC; Cleveland: 1975. [Google Scholar]
- 40.Panajotovic R, Martin F, Cloutier P, Hunting DJ, Sanche L. Radiat Res. 2006;165:452. doi: 10.1667/rr3521.1. [DOI] [PubMed] [Google Scholar]
- 41.Bouchiha D, Gorfinkiel JD, Caron LG, Sanche L. J Phys B. 2006;39:975. [Google Scholar]
- 42.Cadet J, Douki T, Gasparutto D, Gromova M, Pouget JP, Ravanat JL, Romieu A, Sauvaigo S. Nucl Instrum Methods Phys Res B. 1999;151:1. [Google Scholar]
- 43.Cadet J, Douki T, Gasparutto D, Ravanat JL. Radiat Phys Chem. 2005;72:293. [Google Scholar]
- 44.Jena NR, Misra PC, Suhai S. J Phys Chem B. 2009;113:5633. doi: 10.1021/jp810468m. [DOI] [PubMed] [Google Scholar]
- 45.O’Malley TF. Phys Rev. 1966;150:14. [Google Scholar]
- 46.Bass AD, Sanche L. Radiat Environ Biophys. 1998;37:243. doi: 10.1007/s004110050125. [DOI] [PubMed] [Google Scholar]
- 47.Sanche L. Eur Phys J D. 2005;35:367. [Google Scholar]
- 48.Sanche L, Parenteau L. J Chem Phys. 1990;93:7476. [Google Scholar]