

Abstract
Bacterial small RNAs (sRNAs) are short transcripts that typically do not encode proteins and often act as regulators of gene expression through a variety of mechanisms. Regulatory sRNAs have been identified in many species, including Mycobacterium tuberculosis, the causative agent of tuberculosis. Here, we use a computational algorithm to predict sRNA candidates in the mycobacterial species M. smegmatis and M. bovis BCG and confirmed the expression of many sRNAs using Northern blotting. Thus, we have identified 17 and 23 novel sRNAs in M. smegmatis and M. bovis BCG, respectively. We have also applied a high-throughput technique (Deep-RACE) to map the 5′ and 3′ ends of many of these sRNAs and identified potential regulators of sRNAs by analysis of existing ChIP-seq datasets. The sRNAs identified in this work likely contribute to the unique biology of mycobacteria.
Introduction
The genus Mycobacterium contains many clinically relevant pathogens, including Mycobacterium tuberculosis and Mycobacterium leprae, the etiologic agents of tuberculosis (TB) and leprosy, respectively. M. tuberculosis alone was responsible for 8.7 million incident cases and 1.4 million deaths globally in 2011 [1]. The treatment of TB has become increasingly difficult due to its high drug resistance and adaptability; hence, the development of new and more effective treatments for TB is imperative.
Bacterial “small RNAs” (sRNAs) are small (50–400 nt), typically untranslated transcripts. Many sRNAs play important roles in gene regulation in response to environmental changes [2]. sRNAs can originate from their own independent genes or through the processing of larger transcripts [3]. To exert their function, sRNAs typically base-pair with target messenger RNAs (mRNAs), resulting in altered transcription, mRNA stability, or translation [4]. sRNAs are key regulators of pathogenesis in many bacterial species [5]. Recently, RNA-seq has been widely applied to identify novel sRNA candidates in many bacterial species [6]–[10], including M. tuberculosis [11]. sRNAs have also been identified/predicted in M. tuberculosis using other experimental approaches [12]–[14] and computational analysis of DNA sequence [13], [15], [16]. In total, 63 sRNAs have been experimentally validated in M. tuberculosis. sRNAs have also been identified in other mycobacterial species: 34 and 15 sRNAs have been experimentally validated in Mycobcaterium bovis BCG and Mycobacterium smegmatis, respectively [13].
In a previous study, we used computational predictions from the SIPHT (sRNA Identification Protocol using High-throughput Technologies) [17] algorithm to identify 144 sRNA candidates in M. bovis BCG. We selected 34 conserved sRNA candidates which we experimentally confirmed by Northern blot [13]. In the current study, we expanded our search to include all the remaining SIPHT predictions for M. bovis BCG as well as all SIPHT predictions for M. smegmatis (these were not explored in the previous study). By combining SIPHT predictions with large-scale Northern blot validation, we have identified 23 and 17 novel sRNAs in M. bovis BCG and M. smegmatis, respectively. Thus, we have substantially increased the number of experimentally validated sRNAs in mycobacterial species. We also analyzed existing ChIP-seq datasets to identify possible regulators of sRNA expression, and used Deep-RACE, a technique that combines high throughput RNA-seq with Rapid Amplification of cDNA Reads (RACE) [18], to identify sRNA 5′ and 3′ ends. Lastly, it is worth noting that this work is one of the first efforts to better coordinate genome annotation; all sRNA candidates identified in this study have been renamed with a new nomenclature [19].
Materials and Methods
Strains and Plasmids
M. bovis BCG (Pasteur strain, Trudeau Institute), and M. tuberculosis H37Rv were grown in mycomedium (as previously reported, [13]). M. bovis BCG and M. tuberculosis cultures were grown for 7 days, with shaking, to late-log phase. Cultures of M. smegmatis MC2155 were grown shaking at 37°C, in trypticase soy media supplemented with 0.05% Tween 80 for 18 hours with shaking (late-log phase).
Phylogenetic Selection of Computationally Predicted sRNA Candidates
Small RNA candidates of M. smegmatis were predicted using the SIPHT program with the same parameters as described previously [17], [20]. SIPHT identifies potential sRNA candidates based on the presence of intergenic sequence conservation upstream of putative Rho-independent terminators. SIPHT has been widely applied in sRNA studies [21]–[23], and its reliability has been tested and compared with other algorithms [24].
RNA Isolation and Northern Blot Analysis
RNA was isolated as previously reported [13]. Northern blot analysis was performed as previously reported [13]; probes were designed according to SIPHT predicted sequences and tested in M. bovis BCG, M. smegmatis and M. tuberculosis [13]. All the oligonucleotides that were used in this study are listed in Table S1.
ChIP-seq Analysis
We analyzed existing ChIP-seq datasets for 55 M. tuberculosis transcription factors extracted from a previous study [25]. ChIP-seq peak positions were compared to the 5′ end positions of M. bovis BCG and M. tuberculosis sRNAs from the current study and two previous studies [12], [15]. For M. bovis BCG sRNAs, we first identified the equivalent region of the M. tuberculosis H37Rv genome. Possible sRNA regulators were selected if the ChIP-seq peak was located within 100 bp upstream and 20 bp downstream of an sRNA 5′ end.
Deep 5′ and 3′ RACE
Deep 5′ RACE and Deep 3′ RACE were performed as previously described [18] with the following exceptions. Deep 5′ RACE libraries and Deep 3′ RACE libraries were pooled and sequenced together using an Ion Torrent 316 chip (Wadsworth Center Applied Genomic Technologies Core Facility). For Deep 5′ RACE, sequence reads were identified by the presence of the expected adapter sequence at the read 5′ end. Adapter sequences were removed and reads of >40 nt were mapped to the reference genomes using BWA [26]. For Deep 3′ RACE, sequence reads were identified by the presence of the expected adapter sequence. Adapter sequences were removed. The oligo-dT stretch was removed by identifying the first consecutive pair of bases not including a “T” and removing all sequence upstream of this. Sequences of >40 nt were mapped to the reference genomes using BWA [26]. For both Deep 5′ RACE and Deep 3′ RACE, 5′ and 3′ ends were identified as the position with the most sequence reads, and with a minimum of 5 reads. Sequences of all primers used for Deep RACE are listed in Table S2.
Results and Discussion
Prediction of sRNAs in silico using SIPHT
Using SIPHT, we identified 93 candidate sRNAs in M. smegmatis (refseq: NC_008596) (Table S3) and 144 candidate sRNAs in M. bovis BCG (refseq: NC_008769) (Table S4). Tables S3 and S4 include a detailed description of the predicted coordinates, orientations, sizes and neighboring upstream and downstream genes. Northern probes were designed according to SIPHT prediction. Figure 1 summarizes the overall approach that was employed in this work for sRNA identification and confirmation.
Figure 1. Schematic for sRNA identification.
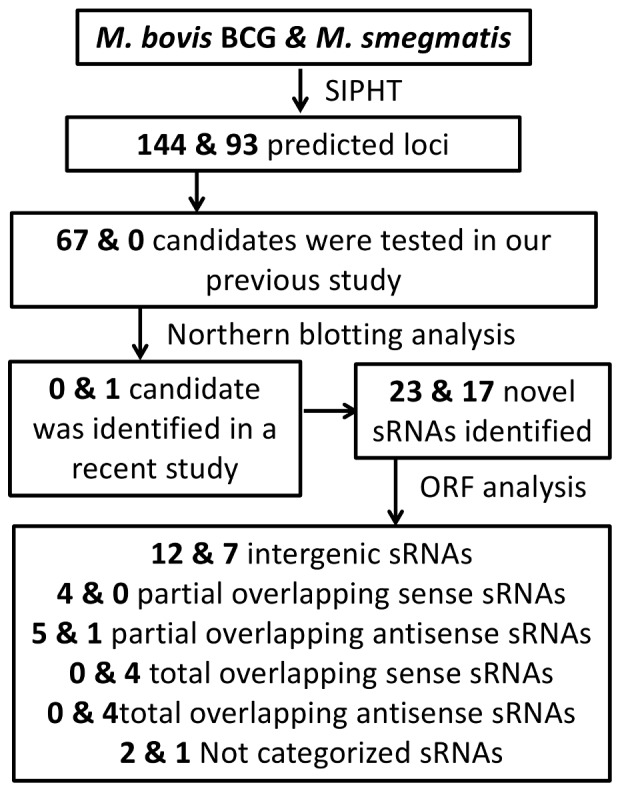
This schematic shows the combination of computational approaches and Northern blotting analysis used to identify the reported novel sRNAs in M. bovis BCG and M. smegmatis.
17 Novel sRNAs Identified in Mycobacterium Smegmatis
All 93 M. smegmatis sRNA candidates were tested by Northern blot using oligonucleotides in both orientations; expression was confirmed for 18 sRNA (listed in Table 1; see blot pictures in Figure 2 and Figure S1). One of them (Sm32/33) was identified in recent work as IGR-1 with similar size, coordinates and same orientation [27]. Thus, 17 M. smegmatis sRNAs identified here have not been experimentally demonstrated in any previous studies. In our previous study [13], we reported homologs of 6 M. smegmatis sRNA candidates (Sm32/Sm33, Sm35, Sm46, Sm47, and Sm74) in M. bovis BCG (Mpr13/Mcr14, Mpr20, Mpr3, Mpr4, and Mpr5, respectively). These were confirmed directly in M. smegmatis by Northern blotting in current study and listed in the 17 novel confirmed sRNAs. A homologue of Sm76 was previously identified in M. tuberculosis by RNA-seq [15] and microarray analysis [14] but not otherwise experimentally confirmed. All of the validated sRNAs were in the same orientation to that predicted by SIPHT. This suggests that the sequence specificity of SIPHT for this prediction is higher than in our previous work, in which 9 out of 37 of the validated sRNAs were in the opposite orientation to the prediction [13]. All confirmed sRNAs were assigned gene names according to a recently-proposed nomenclature [19].
Table 1. sRNAs confirmed by Northern blotting analysis in M. smegmatis.
| Homology confirmed by Northern analysis in: | ||||||
| 5′ end* | 3′ end* | M. bovis BCG | M. tuberculosis | New nomenclature | Length Confirm by Northern blotting analysis(nt) | |
| Intergenic sRNAs | ||||||
| Sm19 | 5029661 | 5029530 | ncMSMEG14931Ac | 100–118 | ||
| Sm32/33# | 417709 | 417796 | ✓ | ncMSMEG10373A | 44 | |
| Sm35 | 1458488 | 1458562 | ✓ | ncMSMEG11363A | 118 | |
| Sm46 | 5864890 | 5864989 | ✓ | ncMSMEG15796A | 82 | |
| Sm49 | 1086797 | 1087035 | ncMSMEG11016A | 48–66 | ||
| Sm64 | 2523008 | 2522888 | ✓ | ✓ | ncMSMEG12439Ac | 100–118 |
| Sm76 | 3690377 | 3690280 | ✓ | ✓ | ncMSMEG13628Ac | 100–118 |
| Sm82 | 4392939/4392970 | 4393039 | ✓ | ncMSMEG14302A | 66/82–100 | |
| Total overlapping sense sRNAs | ||||||
| Sm38 | 2236980 | 2237466 | ✓ | ncMSMEG2161A | 48–66/66–82 | |
| Sm41 | 3815700/3815647 | 3815581 | ✓ | ncMSMEG3749Ac | 48–66/100–118 | |
| Sm90 | 6845964 | 6846035 | ✓ | ✓ | ncMSMEG6799A | 48–66 |
| Sm93 | 858482 | 858588 | ✓ | ncMSMEG0774A | 66–82/100–118 | |
| Total overlapping antisense sRNAs | ||||||
| Sm42 | 4290417/4290487 | 4290537 | ✓ | ✓ | ncMSMEG4206A | 48–66/100–118 |
| Sm67 | 2600405/2600425 | 2600485 | ncMSMEG2514A | 48–66/66–82 | ||
| Sm68 | 2600389 | 2600701 | ncMSMEG2514B | 82–100 | ||
| Sm74 | 3111233 | 3111268 | ✓ | ncMSMEG3037A | 66–82 | |
| Partial overlapping antisense sRNAs | ||||||
| Sm11 | 2835860 | 2835984/2835999 | ✓ | ncMSMEG12771A | 100–118/140–151 | |
| Not categorized sRNAs | ||||||
| Sm47 | 6242319 | 6242668 | ✓ | ncMSMEG16173A | 311 | |
The coordinates in bold were determined by 5′ or 3′ Deep-RACE. Where only one end was determined by Deep RACE, the other end was estimated based on the size determined by Northern blot. Where neither end was determined by Deep RACE, SIPHT-predicted coordinates are listed (underlined text).
Experimentally demonstrated in previous study [27].
Figure 2. Northern blotting confirmation of sRNA candidates in M. smegmatis.

Selected images of Northern blotting analysis for validated M. smegmatis sRNAs; the remaining images are included in Figure S1. Lane 1 and 2 indicate total RNA samples extracted from M. smegmatis and M. bovis BCG, respectively. We used Phi-X174/Hae III Marker for the size prediction. The probes we used for this analysis are listed in Table S1.
Given the practical convenience of testing RNA from both species simultaneously to search for novel sRNA candidates, we used the designed probes for sRNA detection in M. smegmatis to also probe expression of these candidates in M. bovis BCG and M. tuberculosis. Although our focus was to validate M. smegmatis predictions, we fortuitously discovered homologues of 9 candidates in M. bovis BCG and 4 candidates in M. tuberculosis (Table 1, Figure S2). Since these probes were not specifically designed for the other two species, lack of detection could be due to either the absence of sRNA expression or to non-optimization of the probe sequence that was used for hybridization to the targeted region in the M. bovis BCG and M. tuberculosis genome. Also, differences in culture medium might contribute to the low number of expressed homologous sRNAs of M. smegmatis in M. tuberculosis as expression of these sRNAs could be specific to different conditions in M. tuberculosis. Given our focus in sRNA identification, specific conditions that could lead to differences in sRNA expression will be explored in future work.
23 Novel sRNAs Identified in Mycobacterium Bovis BCG
Twenty-one of the sRNA candidates for M. bovis BCG (Bo12, Bo15, Bo41, Bo52, Bo58, Bo67, Bo68, Bo75, Bo80, Bo85, Bo99, Bo100, Bo111, Bo113, Bo115, Bo117, Bo122, Bo125, Bo126, Bo137, and Bo139) were previously identified, under the nomenclature Mpr 1–21, respectively [13]. Forty-six other candidates were also tested previously but showed no signal; therefore, only the remaining 77 candidates were tested using Northern blotting analysis in this study, and we confirmed expression of 23 new sRNA candidates (Figure 3 and Figure S3). A homologue of Bo46 was previously identified in M. tuberculosis by RNA-seq [15] but not otherwise experimentally validated. All of the validated sRNAs were in the same orientation as that predicted by SIPHT. We also applied the probes to M. smegmatis and M. tuberculosis and identified 20 and 5 sRNA homologues, respectively (Table 2; Figure 3; Figure S3). All the confirmed sRNAs in M. bovis BCG and M. tuberculosis are listed in Table 2, along with the new nomenclature for sRNAs.
Figure 3. Northern blotting confirmation of sRNA candidates in M. bovis BCG.

Selected images of Northern blotting analysis for validated M. bovis BCG sRNAs; the remaining images are included in Figure S2. Lane 1 and 2 indicate total RNA samples extracted from M. bovis BCG and M. smegmatis, respectively. We used Phi-X174/Hae III Marker for the size prediction. The probes we used for this analysis are listed in Table S1.
Table 2. sRNAs confirmed by Northern blotting analysis in M. Bovis BCG.
| Homology confirmed by Northern analysis in: | ||||||
| 5′ end* | 3′ end* | M. smegmatis | M. tuberculosis | New nomenclature | Length Confirm by Northern blotting analysis(nt) | |
| Intergenic sRNAs | ||||||
| Bo35 | 576179 | 576067/576104 | ✓ | ncBCG10493Ac | 48–66/82–100 | |
| Bo48 | 3028936 | 3028876 | ✓ | ✓ | ncBCG12782Ac | 48–66 |
| Bo53 | 1044606 | 1044706/1044720/1044727 | ✓ | ncBCG10960A | 82–100/100–118 | |
| Bo60 | 1247638 | 1247538 | ✓ | ncBCG11150Ac | 66/100 | |
| Bo71 | 1588853 | 1588693 | ✓ | ✓ | ncBCG11448Ac | 66 |
| Bo73 | 1647817 | 1647853 | ✓ | ncBCG11504A | 66–82 | |
| Bo78 | 207337 | 207179 | ✓ | ✓ | ncBCG10186Ac | 66/100 |
| Bo86 | 2325795 | 2325960 | ✓ | ncBCG12107A | 66–82 | |
| Bo101 | 2919337 | 2919277 | ✓ | ncBCG2654Ac | 48–66 | |
| Bo118 | 3765977 | 3765917 | ✓ | ncBCG13438Ac | 48–66 | |
| Bo132 | 4260533 | 4260610 | ✓ | ncBCG13885A | 66–82 | |
| Bo105 | 3073445 | 3073541 | ncBCG12831A | 66–82 | ||
| Partial overlapping sense sRNAs | ||||||
| Bo27 | 2157804 | 2157704 | ✓ | ncBCG11948Ac | 82–100 | |
| Bo46 | 2603016 | 2602916 | ✓ | ✓ | ncBCG12368Ac | 100 |
| Bo82 | 2235286 | 2235196 | ✓ | ncBCG12024Ac | 82–100 | |
| Bo87 | 2351000/2351046 | 2350915/2350874 | ✓ | ✓ | ncBCG12128Ac | 48–66/66–82/82 |
| Partial overlapping antisense sRNAs | ||||||
| Bo32 | 817571 | 817483 | ✓ | ncBCG10734Ac | 66–82/82 | |
| Bo47 | 2705925/2705838 | 2705735 | ncBCG12462Ac | 82–100 | ||
| Bo81 | 218700 | 2187796 | ✓ | ncBCG10195A | 66/100 | |
| Bo96 | 2686849 | 2686909/2686989 | ✓ | ncBCG12441A | 66–82/118–142 | |
| Bo130 | 413416 | 413288 | ✓ | ncBCG0352Ac | 66–82/118–142 | |
| Not categorized sRNAs | ||||||
| Bo13 | 3126934 | 3127070 | ncBCG12882A | 48–66/82–100 | ||
| Bo29 | 1770012 | 1769806 | ✓ | ncBCG11603Ac | 66–82 | |
The coordinates in bold were determined by 5′ or 3′ Deep-RACE. Where only one end was determined by Deep RACE, the other end was estimated based on the size determined by Northern blot. Where neither end was determined by Deep RACE, SIPHT-predicted coordinates are listed (underlined text).
Deep-RACE Identifies sRNA 5′ and 3′ Ends
We used Deep-RACE, a previously described approach that combines conventional RACE and deep sequencing to identify 5′ and 3′ ends of selected RNAs [18], [28]. In total, we identified 5′ ends for 9 sRNAs and 3′ ends for 21 sRNAs. Examples are shown in Figure 4. For some sRNAs we identified multiple 5′/3′ ends. Multiple 5′ ends could be due to multiple transcription start sites or RNA processing. Multiple 3′ ends could be due to RNA processing or may indicate imprecise Rho-dependent termination of transcription.
Figure 4. Identification of sRNA 5′ and 3′ ends by Deep RACE.

Blue lines show the number of 5′ RACE reads mapped to respective genome, while red lines show the number of 3′ RACE reads. The coordinates with the highest number of mapped reads (the peak) indicate the likely 5′ and 3′ ends of sRNAs and are labeled in the figure. The orange arrow under the chart shows where the Northern probes base-paired and the blue arrows are the adjacent annotated coding regions. Results for other sRNAs can be found in Figure S4.
Size Comparisons between Experimental and Prediction Analysis
As noted in our earlier study [13], the predicted size of the candidate sRNAs correlates only weakly with experimental observations. Only about 17% of the confirmed sRNAs were within 10% of their predicted sizes. Additionally, in many cases, multiple bands were detected by Northern analysis, suggesting the presence of multiple start sites, multiple termination sites, and/or sRNA processing. This is consistent with the Deep RACE data (Figure 4; Figure S4). Deep RACE identified both 5′ and 3′ ends for seven sRNAs. In these cases, the sizes determined by Deep RACE are similar to those confirmed by Northern blotting.
Location of sRNAs with Respect to Genes
To investigate the potential roles of the novel sRNAs, we mapped them all to the latest annotated genome (National Center for Biotechnology Information, NCBI). Although we aimed to find intergenic sRNAs, half of the candidates we identified in this study overlap partially or entirely protein-coding genes in either the sense or antisense orientation (Table 1, Table 2). We categorized sRNAs into different classes according to their position relative to adjacent coding regions. Where possible, we used 5′/3′ end information from Deep-RACE data. For sRNAs that have only one end mapped by Deep-RACE, the other end was estimated according to the length confirmed by Northern blotting analysis (Figure 4). For sRNAs that have neither end mapped by Deep-RACE, the farthest possible ends were estimated according to Northern blotting analysis and the sRNAs would be categorized as “not determined” if multiple class options exist.
Nine sRNAs in M. smegmatis (Sm19, Sm32/33, Sm35, Sm46, Sm49, Sm64, Sm76, Sm82) and twelve sRNAs in M. bovis BCG (Bo35, Bo48, Bo53, Bo60, Bo71, Bo73, Bo78, Bo86, Bo101, B0105, Bo118, Bo132) were mapped completely to intergenic regions. Four sRNAs in M. smegmatis (Sm38, Sm41, Sm90, Sm93) were mapped to the sense strand of annotated protein-coding genes, and four were mapped to the antisense strand (Sm42, Sm67, Sm68, Sm74). One sRNA in M. smegmatis (Sm11) and five in M. bovis BCG (Bo32, Bo47, Bo81, Bo96, Bo130) overlap partially with adjacent genes in the antisense orientation, and four sRNAs in M. bovis BCG (Bo27, Bo46, Bo82, Bo87) overlap partially with adjacent genes in the sense orientation. One sRNA in M. smegmatis (Sm47) and two in M. bovis BCG (Bo13, Bo29) were not classified.
The location of sRNAs relative to protein-coding genes also gives clues as to their function. Regulatory sRNAs that are completely intergenic typically function by base-pairing with distally-encoded mRNAs; however, some of the sRNAs are close to the 5′ end or 3′ end of adjacent genes, suggesting possible alternative regulatory roles. sRNAs antisense to ORFs or UTRs can regulate expression of the overlapping gene [29]. sRNAs located within UTRs or ORFs in the sense orientation may be degradation products or mRNAs or could be important cis-acting regulatory elements such as riboswitches.
sRNAs can be transcribed independently or generated by processing of mRNA UTRs. Several features of the sRNAs identified in this work are consistent with the sRNAs being independently transcribed from their own promoters. First, the Northern blots showed no evidence of larger bands that could correspond to pre-processed mRNAs. Second, 13 sRNAs (Sm35, Sm42, Sm67, Sm68, Sm74, Bo13, Bo32, Bo60, Bo71, Bo73, Bo81, Bo118, Bo130) are orientated away from the surrounding genes. Third, 5 sRNAs (Sm64, Sm82, Bo47, Bo105, Bo132) are located >200 bp from the nearest gene start/stop. Nineteen sRNAs are close to (<200 bp) upstream or downstream coding regions (Sm11, Sm19, Sm32/33, Sm46, Sm47, Sm49, Sm76, Bo27, Bo29, Bo35, Bo46, Bo48, Bo53, Bo78, Bo82, Bo86, Bo87, Bo96, Bo101) and four (Sm38, Sm41, Sm90, Sm93) overlap coding regions in the sense orientation. It is formally possible that these sRNAs are generated by mRNA processing or premature termination, although the Northern blot analysis argues against this. Regardless, sRNAs processed from mRNAs could still have important regulatory functions [3], [30], [31]. Indeed, a recent study identified 3′ UTRs as an abundant source of regulatory sRNAs in Salmonella enterica [32]. Alternatively, sRNAs generated by processing of mRNAs could indicate cis-acting regulatory elements such as riboswitches.
Likely Regulators of sRNAs Identified by Analysis of ChIP-seq Datasets
The regulation of sRNAs can provide important clues as to their biological functions. However, very little is currently known about regulation of mycobacterial sRNAs. The genome-wide binding profiles of many M. tuberculosis transcription factors have recently been determined using ChIP-seq and these data are publicly available [25]. Although we identified sRNAs in M. bovis BCG, it is highly likely that these sRNAs are conserved in M. tuberculosis given the extremely high similarity of the M. bovis BCG and M. tuberculosis genomes [33]. Hence, we searched existing ChIP-seq datasets of M. tuberculosis for transcription factors that bind close to sRNA 5′ ends, including sRNAs identified in earlier studies [12]. We identified 10 ChIP-seq peaks (indicative of a transcription factor binding site) located between 100 bp upstream and 20 bp downstream of sRNA 5′ ends (Table S5). Thus, we have identified likely examples of sRNA regulation. In some cases, the ChIP-seq peak is also close to the start of an annotated protein-coding gene. Hence, the transcription factor may regulate the protein-coding gene rather than the sRNA. Nevertheless, in four cases, the ChIP-seq peak is unambiguously associated with an sRNA 5′ end. The two examples with highest ChIP-seq signal are shown in Figure 5. For each of these examples, the transcription factor is otherwise uncharacterized.
Figure 5. ChIP-seq peaks associated with predicted sRNA homologues in M. tuberculosis.
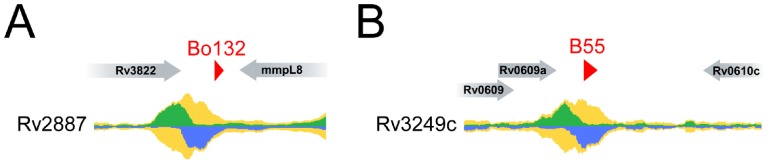
ChIP-seq peaks that are unambiguously associated with sRNA 5′ ends. Raw ChIP-seq data from www.tbdb.org are shown for two transcription factors, (A) Rv2887, and (B) Rv3249c. Data are shown for genomic regions surrounding (A) sRNA Bo132 (this work), and (B) sRNA B55 [12]. The green and blue graphs indicate the relative number of sequence reads mapping to the plus and minus strands, respectively. The yellow graphs indicate the sum of plus and minus strand reads. Annotated genes are shown as gray arrows. sRNAs are shown as red triangles.
Conclusion
In summary, we have identified 17 novel sRNAs in M. smegmatis and 23 novel sRNAs in M. bovis BCG, verified 5′ and 3′ ends, and list these sRNAs according to a recently-proposed annotation nomenclature. Our analysis of sRNA position relative to protein-coding genes suggests various potential roles for these sRNAs in gene regulation. Although the specific biological function of these, and all other known mycobacterial sRNAs, is not understood, we speculate that some of these sRNAs contribute to the biology of pathogenic mycobacterial species. Future studies will focus on the functional characterization of these novel sRNAs.
Supporting Information
Northern blotting analysis for M. smegmatis sRNAs.
(PDF)
Northern blotting analysis of sRNA candidates in M. tuberculosis with M. smegmatis and M. bovis BCG probes.
(PDF)
Northern blotting analysis for M. bovis BCG sRNAs.
(PDF)
Deep-RACE mapped reads of all sRNAs and adjacent gene annotations.
(PDF)
The oligonucleotide sequence of all probes used for Northern Blotting analysis in this study.
(PDF)
The oligonucleotide sequence of all primers used for Deep-RACE PCR.
(PDF)
All 93 sRNA sequences predicted by SIPHT in M. smegmatis.
(PDF)
All 144 sRNA sequences predicted by SIPHT in M. bovis BCG. This list excludes predicted tRNAs.
(PDF)
All transcription factor ChIP-seq peaks located within 100 bp upstream and 20 bp downstream of sRNA 5′ ends.
(PDF)
Acknowledgments
We would like to thank Marquis Martin for his assistance with data analysis, Marlene Belfort for supporting our initial studies (please see funding statement), Todd Gray for helpful discussions and for generously providing materials for this study, Matt Stanger for his assistance with figures, Jeanne DiChiara and Damen Schaak for sample preparation. We thank the Wadsworth Center Applied Genomic Technologies Core Facility for Ion Torrent sequencing.
Funding Statement
This work was supported by the Welch foundation, the DTRA Young Investigator program, National Institutes of Health (NIH) grant 1DP2OD007188 (JTW), and by an appointment (CB) to the Emerging Infectious Diseases (EID) Fellowship Program administered by the Association of Public Health Laboratories (APHL) and funded by the Centers for Disease Control and Prevention (CDC). Initial funding for preliminary studies was provided by (NIH) grants GM39422 and GM44844 to Dr. Marlene Belfort. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.WHO (2012) Global tuberculosis report 2012. WHO. p1.
- 2. Gottesman S, Storz G (2011) Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol 3: a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vogel J, Bartels V, Tang T, Churakov G, Slagter-Jäger J (2003) RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res 31: 6435–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gottesman S (2004) The small RNA regulators of Escherichia coli: roles and mechanisms. Annu Rev Microbiol 58: 303–328. [DOI] [PubMed] [Google Scholar]
- 5. Papenfort K, Vogel J (2010) Regulatory RNA in bacterial pathogens. Cell host & Microbe 8: 116–127. [DOI] [PubMed] [Google Scholar]
- 6. Shinhara A, Matsui M, Hiraoka K, Nomura W, Hirano R, et al. (2011) Deep sequencing reveals as-yet-undiscovered small RNAs in Escherichia coli . BMC Genomics 12: 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raghavan R, Groisman EA, Ochman H (2011) Genome-wide detection of novel regulatory RNAs in E. coli. Genome Res 21: 1487–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T (2010) Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 38: 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Irnov I, Sharma CM, Vogel J, Winkler WC (2010) Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res 38: 6637–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mitschke J, Georg J, Scholz I, Sharma CM, Dienst D, et al. (2011) An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci U S A 108: 2124–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arnvig KB, Comas I, Thomson NR, Houghton J, Boshoff HI, et al. (2011) Sequence-Based Analysis Uncovers an Abundance of Non-Coding RNA in the Total Transcriptome of Mycobacterium tuberculosis . PLoS Pathogens 7: e1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arnvig KB, Young DB (2009) Identification of small RNAs in Mycobacterium tuberculosis . Mol Microbiol 73: 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DiChiara JM, Contreras-Martinez LM, Livny J, Smith D, McDonough K a, et al. (2010) Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis . Nucleic Acids Res 38: 4067–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miotto P, Forti F, Ambrosi A, Pellin D, Veiga DF, et al. (2012) Genome-wide discovery of small RNAs in Mycobacterium tuberculosis . PLoS one 7: e51950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pellin D, Miotto P, Ambrosi A, Cirillo DM, Di Serio C (2012) A Genome-Wide Identification Analysis of Small Regulatory RNAs in Mycobacterium tuberculosis by RNA-Seq and Conservation Analysis. PLoS ONE 7: e32723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pelly S, Bishai WR, Lamichhane G (2012) A screen for non-coding RNA in Mycobacterium tuberculosis reveals a cAMP-responsive RNA that is expressed during infection. Gene 500: 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Livny J, Teonadi H, Livny M, Waldor MK (2008) High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS ONE 3: e3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beauregard A, Smith E, Petrone B, Singh N, Karch C, et al. (2013) Identification and characterization of small RNAs in Yersinia pestis . RNA Biol 10: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lamichhane G, Arnvig KB, McDonough KA (2013) Definition and annotation of (myco)bacterial non-coding RNA. Tuberculosis (Edinburgh, Scotland) 93: 26–29. [DOI] [PubMed] [Google Scholar]
- 20. Livny J (2012) Bioinformatic Discovery of Bacterial Regulatory RNAs Using SIPHT. In: Keiler KC, editor. Bacterial Regulatory RNA: Methods and Protocols. Totowa, NJ: Humana Press, Vol. 905: 3–14. [DOI] [PubMed] [Google Scholar]
- 21. Postic G, Frapy E, Dupuis M, Dubail I, Livny J, et al. (2010) Identification of small RNAs in Francisella tularensis . BMC Genomics 11: 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khoo JS, Chai SF, Mohamed R, Nathan S, Firdaus-Raih M (2012) Computational discovery and RT-PCR validation of novel Burkholderia conserved and Burkholderia pseudomallei unique sRNAs. BMC Genomics 13 Suppl 7S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xia L, Xia W, Li S, Li W, Liu J, et al. (2012) Identification and expression of small non-coding RNA, L10-Leader, in different growth phases of Streptococcus mutans . Nucleic Acid Ther 22: 177–186. [DOI] [PubMed] [Google Scholar]
- 24. Lu X, Goodrich-blair H, Tjaden B (2011) Assessing computational tools for the discovery of small RNA genes in bacteria. RNA 17: 1635–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Galagan J, Lyubetskaya A, Gomes A (2013) ChIP-Seq and the complexity of bacterial transcriptional regulation. Curr Top Microbiol Immunol 363: 43–68. [DOI] [PubMed] [Google Scholar]
- 26. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li SK, Ng PK, Qin H, Lau JK, Lau JP, et al. (2012) Identification of small RNAs in Mycobacterium smegmatis using heterologous Hfq. RNA 19 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olivarius S, Plessy C, Carninci P (2009) High-throughput verification of transcriptional starting sites by Deep-RACE. Biotechniques 46: 130–132. [DOI] [PubMed] [Google Scholar]
- 29. Georg J, Hess WR (2011) cis-antisense RNA, another level of gene regulation in bacteria. Microbiology and molecular biology reviews?: MMBR 75: 286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawano M, Reynolds AA, Miranda-Rios J, Storz G (2005) Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res. 33: 1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, et al. (2009) A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139: 770–779. [DOI] [PubMed] [Google Scholar]
- 32. Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J (2012) An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. The EMBO journal 31: 4005–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garnier T, Eiglmeier K, Camus JC, Medina N, Mansoor H, et al. (2003) The complete genome sequence of Mycobacterium bovis . PNAS 100: 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Northern blotting analysis for M. smegmatis sRNAs.
(PDF)
Northern blotting analysis of sRNA candidates in M. tuberculosis with M. smegmatis and M. bovis BCG probes.
(PDF)
Northern blotting analysis for M. bovis BCG sRNAs.
(PDF)
Deep-RACE mapped reads of all sRNAs and adjacent gene annotations.
(PDF)
The oligonucleotide sequence of all probes used for Northern Blotting analysis in this study.
(PDF)
The oligonucleotide sequence of all primers used for Deep-RACE PCR.
(PDF)
All 93 sRNA sequences predicted by SIPHT in M. smegmatis.
(PDF)
All 144 sRNA sequences predicted by SIPHT in M. bovis BCG. This list excludes predicted tRNAs.
(PDF)
All transcription factor ChIP-seq peaks located within 100 bp upstream and 20 bp downstream of sRNA 5′ ends.
(PDF)