

Abstract
The myelin sheath surrounding axons ensures that nerve impulses travel quickly and efficiently, allowing for the proper function of the vertebrate nervous system. We previously showed that the adhesion G-protein-coupled receptor (aGPCR) Gpr126 is essential for peripheral nervous system myelination, although the molecular mechanisms by which Gpr126 functions were incompletely understood. aGPCRs are a significantly understudied protein class, and it was unknown whether Gpr126 couples to G-proteins. Here, we analyze DhhCre;Gpr126fl/fl conditional mutants, and show that Gpr126 functions in Schwann cells (SCs) for radial sorting of axons and myelination. Furthermore, we demonstrate that elevation of cAMP levels or protein kinase A activation suppresses myelin defects in Gpr126 mouse mutants and that cAMP levels are reduced in conditional Gpr126 mutant peripheral nerve. Finally, we show that GPR126 directly increases cAMP by coupling to heterotrimeric G-proteins. Together, these data support a model in which Gpr126 functions in SCs for proper development and myelination and provide evidence that these functions are mediated via G-protein-signaling pathways.
Introduction
Myelin is essential for the proper function of the vertebrate nervous system. During peripheral nervous system (PNS) development, immature Schwann cells (SCs) are associated with many axons; in a process called radial sorting, immature SCs insert cytoplasmic projections into axon bundles to separate individual axons. Some immature SCs develop into nonmyelinating Remak SCs that ensheath multiple small caliber axons. Other immature SCs become promyelinating SCs that are associated in a 1:1 relationship with a single axonal segment. Myelinating SCs iteratively wrap their membrane around their associated axonal segment to generate the myelin sheath (Jessen and Mirsky, 2005).
We previously showed that the adhesion G-protein-coupled receptor (aGPCR) Gpr126 is essential for PNS myelination. aGPCRs represent a unique protein class defined structurally by a seven-transmembrane helix region (7TM) and an extremely large extracellular N terminus, separated from the 7TM by a GPCR autoproteolysis-inducing (GAIN) domain and a GPCR proteolytic site (GPS) domain. These domains split the receptor during the maturation process into N-terminal and C-terminal fragments that are thought to remain noncovalently attached at the cell surface (Araç et al., 2012). The extracellular regions of many aGPCRs contain domains that are involved in cell–cell or cell–matrix adhesion in other proteins (Yona et al., 2008). The 7TM is classically defined by its ability to bind heterotrimeric G-proteins within a cell, through which signal transduction cascades are activated upon agonist binding. For example, the G-protein subunit Gαs activates adenylyl cyclases to elevate cAMP levels, whereas Gαi inhibits adenylyl cyclases to decrease cAMP levels (Gilman, 1984). Via release of Gβγ dimers, Gi-proteins mediate diverse additional functions, including modulation of phosphatidylinositol 3-kinase (PI3K; Lin and Smrcka, 2011). The majority of aGPCRs remain undercharacterized, and for most family members, the biological functions, activating ligands, and downstream effectors are unknown.
Our previous work defined the first known function of Gpr126 as an essential regulator of PNS myelination. In germline zebrafish and mouse Gpr126 mutants, SCs fail to myelinate axons (Monk et al., 2009, 2011). Analysis of genetic chimeras in zebrafish suggested that Gpr126 is required in SCs for myelin protein expression (Monk et al., 2009). However, mouse Gpr126−/− mutant peripheral nerves have many defects not observed in zebrafish mutants, including radial sorting delays, limb contracture defects, and axon degeneration (Monk et al., 2011), raising questions regarding the cellular autonomy of Gpr126 in mammals. Furthermore, germline deletion of Gpr126 is lethal in mouse, precluding analysis beyond early postnatal stages. Here, we analyze Gpr126 conditional mutant mice, and our data suggest that Gpr126 functions in SCs for timely radial sorting and myelination. We also show that elevating cAMP levels and activating protein kinase A (PKA) restores myelination in Gpr126−/− myelinating cultures, that cAMP is downregulated in Gpr126 conditional mutant nerves, and that GPR126 couples to the Gs-protein and Gi-protein families. These data demonstrate that Gpr126 is required autonomously in SCs for proper development and strongly support a model in which Gpr126, via interactions with G-proteins, modulates cAMP levels in SCs to control differentiation and myelination.
Materials and Methods
Mice.
All animal experiments were performed in compliance with Washington University's institutional animal protocols. Gpr126−/− constitutive knock-out mouse generation and genotyping have been described previously (Monk et al., 2011). Conditional Gpr126 mutant mice (Gpr126fl/fl mice) were generated by Lexicon Pharmaceuticals, and Gpr126fl/+ mice were obtained from Taconic (catalog number TF0269) on a mixed background (129SvEvBrd). We used the following primers to detect the presence of the floxed allele: 5′–CATTGAGCTCTCCCTGTATGG–3′; 5′–CCTGCAGGTACCCACACATG–3′. Gpr126fl/fl mice were crossed to βActinCre mice on a pure C57BL/6 background (Lewandoski et al., 1997), and βActinCre;Gpr126fl/+ mice were crossed to Gpr126fl/fl mice to generate βActinCre;Gpr126fl/fl mice and their sibling controls. βActinCre mice were genotyped as described previously (Abdulkadir et al., 2002). Gpr126fl/fl mice were also crossed to DhhCre mice on a pure C57BL/6 background (Jaegle et al., 2003), and DhhCre;Gpr126fl/+ mice were crossed to Gpr126fl/fl mice to generate DhhCre;Gpr126fl/fl mice and their sibling controls. DhhCre mice were genotyped as described previously (Jaegle et al., 2003). To assess recombination of the floxed allele, we used the ROSA26 LacZ reporter strain (Soriano, 1999). For all mouse experiments, mice of either sex were analyzed, and mutants were always compared with littermate sibling controls.
RT-PCR.
Standard RT-PCR was performed as described previously (Monk et al., 2011) on cDNA derived from βActinCre;Gpr126fl/fl and sibling control postnatal day (P) 1 lung tissue or on cDNA derived from DhhCre;Gpr126fl/fl and sibling control P14 lung or sciatic nerve tissue. For sciatic nerve, we pooled four nerves from two Gpr126fl/+ mice and six nerves from three DhhCre;Gpr126fl/fl mice (all mice were siblings).
In situ hybridization.
Mice were intracardially perfused with 4% PFA. Tissues were dissected and postfixed in the same fixative at 4°C overnight. After protecting in 30% sucrose and embedding in optimal cutting temperature compound, frozen sections were prepared at 7 μm on a cryostat (Leica). In situ hybridization was performed as previously described (VanDunk et al., 2011) on sections from DhhCre;Gpr126fl/fl (n = 2) and sibling control (n = 2 DhhCre;Gpr126fl/+ siblings) P21 sciatic nerve and dorsal root ganglia (DRGs). Antisense and sense control digoxigenen-labeled riboprobes were transcribed as described previously (Patra et al., 2013).
Immunohistochemistry.
For tissue sections, mice were processed as described for in situ hybridization. Antibody staining was performed as described previously (Jeong et al., 2012). For myelinating DRG cultures, cells were fixed at room temperature with 4% PFA in PBS for 30 min. After washing with PBS, cells were permeabilized with ice-cold methanol and plates were kept at −20°C for 20 min. Cells were washed again with PBS and blocked with PBS containing 2% BSA plus 10% goat serum plus 0.1% Triton X-100 for 30 min at room temperature. Primary antibodies were suspended in the blocking buffer and cells were incubated for 1 h at room temperature. Cells were washed with PBS and incubated with appropriate fluorescently labeled secondary antibodies (1:1000; Invitrogen) in blocking buffer for 1 h at room temperature. After washing with PBS, coverslips were mounted on glass slides using Vectashield with DAPI (Vector Labs). We used the following primary antibodies: rat anti-MBP (1:10; AbD Serotec), rabbit anti-s100 (1:400; Dako), rabbit-anti-TUJ1 (1:1000; Covance), chicken anti-β-galactosidase (1:200; Abcam).
Transmission electron microscopy.
Mice were intracardially perfused with modified Karnovsky's fix (2% glutaraldehyde plus 4% PFA in 0.1 m sodium cacodylate, pH 7.4), and sciatic nerves were removed and postfixed in the same fixative at 4°C overnight. Nerves were postfixed and embedded as described previously (Monk et al., 2011). Semithin sections (200 nm) were stained with toluidine blue, viewed on a light microscope (Zeiss AxioImager M2), and images were recorded with an AxioCam MRm. Thin sections (70 nm) were stained with uranyl acetate and Sato's lead stain, and then viewed on a Jeol (JEM-1400) electron microscope. Images were recorded with an Advanced Microscopy Techniques V601 digital camera. We examined three siblings (one Gpr126fl/+, one Gpr126fl/fl, and one DhhCre;Gpr126fl/+) and four DhhCre;Gpr126fl/fl mutants at P1; four siblings (two Gpr126fl/fl and two DhhCre;Gpr126fl/+) and three DhhCre;Gpr126fl/fl mutants at P10; four siblings (four DhhCre;Gpr126fl/+) and three DhhCre;Gpr126fl/fl mutants at P21; three siblings (two Gpr126fl/fl and one DhhCre;Gpr126fl/+) and three DhhCre;Gpr126fl/fl mutants at P42. For quantifications, we quantified all visible axons and their relationships with SCs at 3000× magnification from four to five randomly selected areas of two DhhCre;Gpr126fl/fl mutant and three sibling (DhhCre;Gpr126fl/+) controls.
Proliferation assay and nuclei quantification.
To quantify proliferation, sciatic nerve cryosections were obtained and processed as described for immunocytochemistry. Rabbit anti-Ki67 (1:200; Abcam) and appropriate secondary antibody (1:1000; Invitrogen) were used to identify proliferating cells. The total number of nuclei (DAPI+) and the total number of proliferating cells (Ki67+) cells per sciatic nerve were counted manually for four control (DhhCre;Gpr126fl/+) and four DhhCre;Gpr126fl/fl mutants at P4. To quantify total nuclei, we counted the total number of nuclei from semithin sections obtained from the same P21 animals described for transmission electron microscopy (TEM). SC nuclei were identified based on morphology (see Fig. 3D) and were confirmed by electron microscopy to be associated with axons and in possession of a basal lamina (see Fig. 5F).
Figure 3.
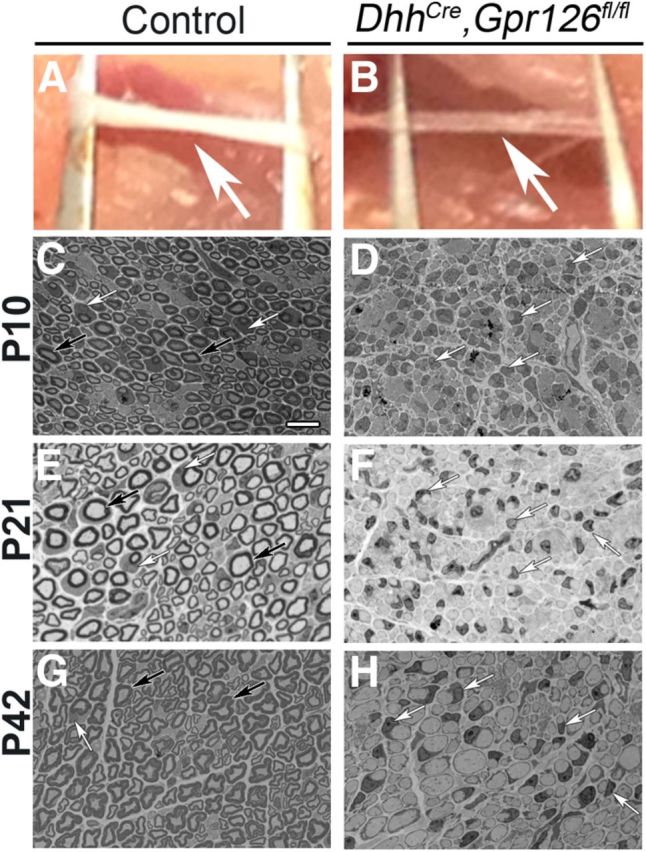
DhhCre;Gpr126fl/fl SCs fail to generate myelin. A, B, Sciatic nerves (arrows) from P21 littermates. A, Control (Gpr126fl/fl shown) nerves are white and opaque. B, DhhCre;Gpr126fl/fl nerves are translucent. C–H, Representative toluidine blue-stained semithin sections of sciatic nerves from sibling control and DhhCre;Gpr126fl/fl mice. C, E, G, Many myelinated axons are observed in control sciatic nerves at P10 (C, Gpr126fl/fl shown), P21 (E, DhhCre;Gpr126fl/+ shown), and P42 (G, DhhCre;Gpr126fl/+ shown). We did not observe differences between any control genotype at any stage examined. Myelinated axons are not observed in DhhCre;Gpr126fl/fl nerves at P10 (D), P21 (F), or P42 (H). Scale bar: (in C) C–H, 10 μm. C–H, Black arrows denote myelin sheaths, and white arrows denote SC nuclei.
Figure 5.

DhhCre;Gpr126fl/fl SCs are hyperproliferative. A, B, Sciatic nerves stained with Ki67 (red) and DAPI (blue) in P4 control (A; DhhCre;Gpr126fl/+ shown) and DhhCre;Gpr126fl/fl mutants (B). C, Quantification of the percentage of Ki67(+) cells shows significantly more proliferating cells in DhhCre;Gpr126fl/fl mutant nerve compared with control nerve at P4. D, Quantification of SC number shows significantly more SCs DhhCre;Gpr126fl/fl in mutant nerve compared with control nerve at P21. Data are given as means ± SD. *p < 0.05; ***p < 0.001.
Myelinating DRG cultures.
Myelinating DRG cultures were performed according to standard methods (Eldridge et al., 1987) with minor modifications. Briefly, DRGs were removed from embryonic day (E) 12.5 mouse embryos and plated on Matrigel (BD Biosciences)-coated coverslips. Embryos were genotyped as described previously (Monk et al., 2011). DRGs were cultured in neurobasal media containing 2% B27 (Invitrogen) and 50 ng/ml NGF (Harlan Labs). Media was changed every other day for the duration of culture. After 7–8 d in vitro, SCs were confluent along axons and 50 μg/ml ascorbic acid (Sigma-Aldrich) was added to induce the formation of basal lamina. We added 5 μm forskolin (Sigma-Aldrich) or 250 μm 8-CTP-cAMP (BioLog) to experimental wells at the same time as ascorbic acid. These conditions were maintained for 3 weeks, at which point the cultures were fixed and subjected to immunohistochemistry. Three independent experiments were performed in triplicate.
In vivo cAMP measurement.
cAMP concentration in the sciatic nerves was measured using cAMP measurement kit (Enzo Life Sciences) according to the manufacturer's instructions. Briefly, sciatic nerves from DhhCre;Gpr126fl/fl mice (n = 3) and control animals (n = 5 Gpr126fl/fl mice) were homogenized in ice-cold trichloroacetic acid. After centrifugation, the supernatant was clarified of cell debris by water-saturated ether, the clarified supernatant was lyophilized overnight, and the dried pellet was resuspended in manufacturer-provided buffer. cAMP levels were measured in this suspension, and cAMP concentrations were quantified using Assay Blaster software (Enzo Life Sciences). Control values were set to 100%, and mutant cAMP concentrations were calculated as a percentage of control.
In vitro functional assays.
Full-length human (NM001032395.2) GPR126 sequence was directly cloned from human monocyte cDNA library (primers: forward, ATGATGTTTCGCTCAGATCGAATG, and reverse, TTAAAACTTTGTGCTGTGGCTG) and inserted into the mammalian expression vector pcDps. The human GPR126 cDNA was N-terminally tagged after the initial signal peptide at amino acid position 38 with a hemagglutinin (HA) epitope and C-terminally tagged with a FLAG epitope by a PCR-based site-directed mutagenesis and fragment replacement strategy.
COS-7 cells were grown in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C and 7% CO2 in a humidified atmosphere. Cells were split into 12-well plates [1 × 105 cells/well, for inositol phosphate (IP3) assay] and 48-well plates (3 × 104 cells/well for cAMP assay) and transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. To measure IP3 formation, transfected COS-7 cells were incubated with 2 μCi/ml myo-[3H]inositol (18.6 Ci/mmol; PerkinElmer Life Sciences) for 16 h. Thereafter, cells were washed once with serum-free DMEM containing 10 mm LiCl followed by incubation with serum-free DMEM containing 10 mm LiCl for 1 h at 37°C. Intracellular IP3 levels were determined by anion-exchange chromatography as described previously (Berridge, 1983). IP3 accumulation data were analyzed using GraphPad Prism version 5.0 for Windows (GraphPad Software). For cAMP measurements, 48 h after transfection, cells were incubated with 3-isobutyl-methyl-xanthine (1 mm)-containing medium for 1 h. Incubation was stopped by washing with ice-cold PBS. Cells were lysed in LI buffer (PerkinElmer Life Sciences) and frozen at −20°C until measurement. To measure cAMP concentration, the AlphaScreen cAMP assay kit (PerkinElmer Life Sciences) was used according to the manufacturer's protocol. The accumulated cAMP was measured in 384-well white OptiPlate microplates (PerkinElmer Life Sciences) with the Fusion AlphaScreen multilabel reader (PerkinElmer Life Sciences). To estimate cell surface expression of receptors carrying an N-terminal HA tag, we used an indirect cellular ELISA as described previously (Schöneberg et al., 1998).
Statistical analysis.
For TEM quantification, we performed a two-tailed Student's t test. For in vitro cAMP assays, we performed a Mann–Whitney U test. For IP3 assays, we performed one-way ANOVA followed by a Dunn's multiple post hoc test. For in vivo cAMP assays, proliferation, and nuclei quantification, we performed one-way ANOVA followed by a Bonferroni post hoc test. p values < 0.05 were considered to be significant; specifically, *p < 0.05; **p < 0.01; ***p < 0.001.
Results
DhhCre;Gpr126fl/fl mutant mice are viable
In an attempt to circumvent the lethality of constituitive Gpr126 mutant mice, we used conditional mutant mice (Gpr126fl/fl). In the mutant allele, exons 2 and 3 are replaced by a selection cassette flanked by loxP sites (Fig. 1A,B). To verify the utility of this line, we globally ablated Gpr126 by mating Gpr126fl/fl mice with mice that express Cre recombinase under control of the β-Actin (β-Act) promoter (Lewandoski et al., 1997). We observed the same limb contracture defects (Fig. 1C) and early lethality in βActCre;Gpr126fl/fl mutants as previously reported in constitutive Gpr126−/− mutants (Monk et al., 2011). Additionally, we did not detect Gpr126 mRNA in lung tissue from βActCre;Gpr126fl/fl mutants (Fig. 1D), though it is normally highly expressed in this tissue (Moriguchi et al., 2004; Haitina et al., 2008).
Figure 1.
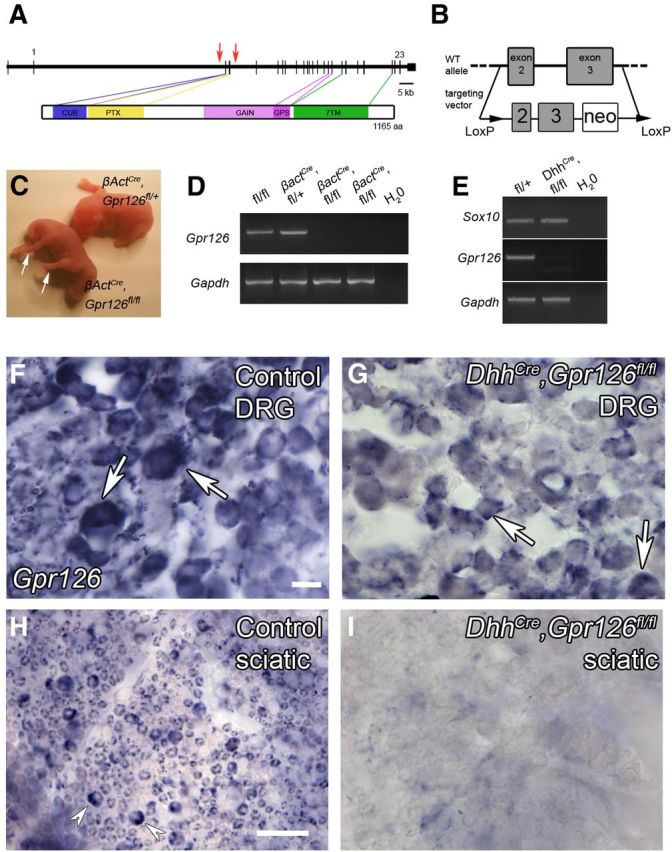
DhhCre;Gpr126fl/fl mutant mice have nerve abnormalities. A, Schematic representations of mouse Gpr126 gene (top) and protein (bottom). In the gene diagram, coding exons 1 and 23 and the targeted locus in Gpr126fl/fl mice (red arrows) are denoted. The protein diagram depicts functional domains in Gpr126 protein: a Complement, Uegf, Bmp1 (CUB) domain, a Pentraxin (PTX) domain, a GPCR autoproteolysis-inducing domain that encompasses a GPCR proteolytic site (GPS) motif, and a type II 7TM domain. B, Targeting strategy used to generate Gpr126fl/fl mice. C, βActCre;Gpr126fl/fl (bottom left) and βActCre;Gpr126fl/+ sibling control (top right) mice at P1. Arrows indicate joint contracture defects. D, RT-PCR analysis of Gpr126 expression in cDNA derived from P1 lung from animals of the indicated genotypes. Gpr126 is not detected in βActCre;Gpr126fl/fl mutant lung (n = 2). E, RT-PCR analysis of Gpr126 expression in cDNA derived from P14 sciatic nerves from animals of the indicated genotypes. Sox10, but not Gpr126, is detected in sciatic nerve from DhhCre;Gpr126fl/fl mice (n = 3 animals, pooled). F–I, In situ hybridization showing Gpr126 expression in DRG (F, G) and sciatic nerve (H, I). Gpr126 is expressed in DRG neurons (arrows) in control DhhCre;Gpr126fl/+ mice (F) and DhhCre;Gpr126fl/fl mutant mice (G). Gpr126 is expressed in sciatic nerve SCs (arrowheads) in control DhhCre;Gpr126fl/+ mice (H) but not in DhhCre;Gpr126fl/fl mutant mice (I) at P21. For F–I, n = 2 animals each genotype; scale bars, 20 μm.
To begin to interrogate the function of Gpr126 in SCs, we crossed Gpr126fl/fl mice with mice that express Cre recombinase under control of the Desert hedgehog (Dhh) promoter, which drives recombination in SC precursors at ∼E12.5 (Jaegle et al., 2003). Unlike constitutive Gpr126−/− mutants, which showed significant embryonic lethality (Monk et al., 2011), DhhCre;Gpr126fl/fl animals were born in near-Mendelian ratios (53:315, 16.8%) and survived for >1 year. Constitutive Gpr126−/− mutants were generally immotile (Monk et al., 2011); in contrast, DhhCre;Gpr126fl/fl mutants were ambulatory, although they exhibited trembling by ∼P10. DhhCre;Gpr126fl/fl mutants were also sometimes smaller than littermate controls, but this phenotype was variable and not fully penetrant. We did not detect Gpr126 mRNA in sciatic nerve of DhhCre;Gpr126fl/fl mutants (Fig. 1E), although Gpr126 was robustly expressed in the lungs of DhhCre;Gpr126fl/fl mutants (data not shown). Because most mRNAs in the sciatic nerve are transcribed by SCs, this result suggests that Gpr126 was efficiently recombined in DhhCre;Gpr126fl/fl mutant SCs. To more thoroughly assess cell type-specific recombination efficiency of Gpr126 in DhhCre;Gpr126fl/fl mutants, we performed in situ hybridization on tissue sections from DRGs and sciatic nerve from DhhCre;Gpr126fl/fl and control DhhCre;Gpr126fl/+ animals. We observed Gpr126 expression in a pattern consistent with DRG neurons in both control and DhhCre;Gpr126fl/fl mutants (Fig. 1F,G). We observed robust Gpr126 expression in control nerve, in a pattern consistent with SCs (Fig. 1H). In contrast, DhhCre;Gpr126fl/fl mutant sciatic nerve did not express Gpr126 (Fig. 1I) and staining was indistinguishable from sense controls (data not shown).
To further ensure that Gpr126 was efficiently recombined in SCs but not in neurons, we examined LacZ expression in DRG, spinal cord, and sciatic nerve of DhhCre;Gpr126fl/+;Rosa26R mice. We detected robust LacZ expression in SCs of the sciatic nerve at P15, but not in DRG neurons or in the CNS (Fig. 2), which is consistent with previous reports (Jaegle et al., 2003; Wu et al., 2008). Importantly, Gpr126fl/fl mice are healthy, fertile, and show no obvious defects in SC development or myelination (Fig. 3A,C; Fig. 4A; data not shown), indicating that the neomycin selection cassette and LoxP sites introduced at the Gpr126 locus (Fig. 1B) did not affect Gpr126 function. Further, we observed no difference in expression of genes flanking Gpr126 (Pex3, Aig1, and Hivep2) in cDNA derived from P4 sciatic nerve of Gpr126fl/fl or DhhCre;Gpr126fl/fl animals compared with Gpr126fl/+ by quantitative PCR (qPCR; data not shown). Similarly, Gpr126 expression was not altered in cDNA derived from P4 lung of Gpr126fl/fl or DhhCre;Gpr126fl/fl animals compared with Gpr126fl/+ by qPCR (data not shown). Therefore, Gpr126fl/fl mice represent a new tool with which to conditionally ablate Gpr126 with cell-type specificity, and DhhCre;Gpr126fl/fl mutants represent a useful model to examine the function of Gpr126 in SCs.
Figure 2.

DhhCre drives recombination in SCs but not neurons. A–F, Cross-sections of DRGs (A, B), ventral horn of spinal cord (C, D), and sciatic nerve (E, F) from P15 DhhCre;Gpr126fl/+;Rosa26R mice. β-Galactosidase/LacZ stain (green) indicates a cell in which Cre-mediated recombination has occurred. A, B, DRGs stained with LacZ (green), DAPI, and TUJ1 (A, red) or MBP (B, red) show no expression of LacZ in sensory neuronal cell bodies. A' and B' show higher magnification of boxed region in A and B, respectively. C, D, Ventral horn of the spinal cord stained with LacZ, DAPI, and TUJ1 (C) or MBP (D) show no expression of LacZ in motor neuron cell bodies or any other cell type in the spinal cord. C', D', Higher magnification of boxed region in C and D, respectively. E, F, Sciatic nerve stained with LacZ, DAPI, and TUJ1 (E) or MBP (F) show robust expression of LacZ in SCs (arrows). Scale bars: (in A, E) A, B, E, F, 50 μm; (in C) C, D, 20 μm.
Figure 4.

DhhCre;Gpr126fl/fl SCs show multiple ultrastructural defects. A–C, TEMs from P1 animals. A, In control animals (Gpr126fl/fl shown), thin myelin is present around some axons (m), and many axons are ensheathed by promyelinating SCs (p). B, C, Radial sorting is significantly delayed in DhhCre;Gpr126fl/fl mutants. B, Low magnification shows SC nuclei (s) localized to the axon bundle edges, and very few axons are segregated by SCs. C, Higher magnification shows a SC nucleus on the periphery of many naked axons. D–F, TEMs from P21 animals. D, In control animals (DhhCre;Gpr126fl/+ shown), axons are either well myelinated (m) or ensheathed by Remak SC (R) cytoplasm. E, F, Myelin is absent and radial sorting delays persist in DhhCre;Gpr126fl/fl mutants. E, Low magnification shows axons that have been sorted 1:1 by SCs (black asterisks), but remain unmyelinated. Bundles of unsorted axons persist (white asterisks), and several of these axons are large caliber (>1 μm diameter). F, Higher magnification shows aberrant cytoplasmic protrusions (arrowheads) in DhhCre;Gpr126fl/fl mutant SCs. G–I, TEMs from P42 animals. G, In control animals (DhhCre;Gpr126fl/+ shown), similar to P21, axons are well myelinated or ensheathed by Remak SCs. H, I, Nerve phenotypes observed at P21 persist in DhhCre;Gpr126fl/fl mutants at P42. H, Low magnification shows that although more axons have been sorted 1:1 by SCs (black asterisks), some large-caliber axons remain unsorted (white asterisks). In bundles of unsorted axons, some axons remain in direct contact with each other and have no SC cytoplasmic interdigitations. I, Higher magnification shows persistence of aberrant cytoplasmic protrusions (arrowheads). Scale bars: (in A) A, B, D, E, G, H, 500 nm; C, F, 1 μm; I, 2 μm. J–M, Quantification of axons and their relationships with SCs in the sciatic nerves of P21 animals. Data are given as means ± SD. ***p < 0.001.
DhhCre;Gpr126fl/fl mutant mice show defects in radial sorting and myelination
Gross examination revealed that DhhCre;Gpr126fl/fl mutant sciatic nerves were less opaque than control nerves at P21 (Fig. 3A,B), indicative of reduced myelination. To test this, we examined sciatic nerve ultrastructure at multiple developmental stages between P1 and P42 by transmission electron microscopy (TEM). Analysis of semithin sections stained by toluidine blue revealed a lack of myelination in DhhCre;Gpr126fl/fl mutants at P10, P21, and P42 (Fig. 3D,F,H), and ultrastructural analyses revealed multiple defects in the mutants (Fig. 4). At P1, axons in control nerves were well sorted; many axons were in a 1:1 relationship with SCs, and occasional axons were surrounded by a thin myelin sheath (Fig. 4A). In contrast, radial sorting was dramatically impaired in sciatic nerves of DhhCre;Gpr126fl/fl mutants at P1. Mutant SC nuclei were observed at the edges of axon bundles (Fig. 4B) with very little cytoplasmic interdigitation into the bundles (Fig. 4C). Interestingly, we did not observe perinatal axon degeneration in DhhCre;Gpr126fl/fl mutant sciatic nerve, although this phenotype was evident in constitutive Gpr126−/− mutants (Monk et al., 2011). Future analyses will determine whether Gpr126 has a neuronal function in survival/axonal maintenance or whether neurons require a Gpr126-mediated signal from SCs before E12.5. In sum, this analysis suggests that Gpr126 is required in SCs for timely radial sorting.
At P21 and P42, large-caliber axons in control nerves were well myelinated while small-caliber axons were organized into Remak bundles (Fig. 4D,G). In DhhCre;Gpr126fl/fl mutant sciatic nerves at these stages, many axons were sorted into a 1:1 relationship by SCs (Fig. 4E,H,J), although no myelin was observed and radial sorting defects persisted (Fig. 4E,F,H–M). Furthermore, we observed aberrant abaxonal cytoplasmic protrusions in mutant SCs (Fig. 4F,I), similar to defects previously reported in Rac1 mutants (Benninger et al., 2007; Nodari et al., 2007; Guo et al., 2012). These results suggest that Gpr126 is required in SCs for myelination, which is consistent with chimeric analysis in zebrafish mutants (Monk et al., 2009). These results also suggest that Gpr126, like Rac1, is required in SCs for membrane stabilization.
DhhCre;Gpr126fl/fl mutant SCs are hyperproliferative
Examination of semithin sections stained by toluidine blue suggested that more SC nuclei were present in DhhCre;Gpr126fl/fl mutant nerves compared with control (Fig. 3). We assessed proliferation in P4 sciatic nerve by Ki67 immunostaining. Proliferation was significantly increased in DhhCre;Gpr126fl/fl mutant nerves compared with control nerves (Fig. 5A–C), and this increase in SC number persisted at P21 (Fig. 5D). These data indicate that loss of Gpr126 causes SCs to overproliferate and show that defects in radial sorting and myelination are not due to a reduction in SC number.
cAMP elevation suppresses myelin defects in Gpr126 mutants
For the majority of aGPCRs, downstream signaling effectors are unknown or not well defined. Moreover, although previous studies have supported a link between aGPCRs and G-protein signaling (Liebscher et al., 2013), direct G-protein coupling has only been recently demonstrated for two family members, GPR133 and GPR114 (Bohnekamp and Schöneberg, 2011; Gupte et al., 2012). We previously reported that forskolin treatment to elevate cAMP suppresses myelin defects in gpr126 mutant zebrafish (Monk et al., 2009), suggesting that Gpr126 might couple to Gs-protein and elevate cAMP. To begin to dissect this signaling pathway, we first tested the hypothesis that cAMP elevation would suppress myelin defects in Gpr126 mouse mutants. To this end, we performed myelinating assays in vitro using DRGs derived from constitutive Gpr126+/+, Gpr126+/−, or Gpr126−/− animals with or without stimulation by compounds that elevate cAMP. Myelin internodes, as assessed by myelin basic protein [MBP(+)] segments, were observed in Gpr126+/+ and Gpr126+/− (control) cultures after stimulation with ascorbic acid to promote SC basal lamina formation and myelination (Eldridge et al., 1987; Fig. 6A,G). No differences were observed between Gpr126+/+ and Gpr126+/− cultures (data not shown). MBP(+) internodes were never observed in Gpr126−/− cultures treated with ascorbic acid alone (Fig. 6B,G). Myelination was potentiated in control cultures upon addition of 5 μm forskolin (Fig. 6C,G) to activate adenylyl cyclase and elevate cAMP or upon addition of 250 μm 8-(4-chlorophenylthio)-adenosine-3′,5′-cAMP (8-CPT-cAMP; Fig. 6E,G), a cell-permeable cAMP analog that activates PKA (Dostmann et al., 1990). In contrast to ascorbic acid alone, both forskolin and 8-CPT-cAMP stimulated MBP(+) segment formation in Gpr126−/− cultures (Fig. 6D,F,G). These data show that cAMP elevation suppresses Gpr126−/− myelin defects in mammals. Moreover, since 8-CPT-cAMP is a site-selective activator of PKA (Dostmann et al., 1990), these results also support a model in which Gpr126 elevates cAMP in SCs to activate PKA before myelination, which is consistent with a recent report in zebrafish (Glenn and Talbot, 2013).
Figure 6.

cAMP elevation rescues amyelination in Gpr126−/− mutants. A–F, In vitro DRG myelinating cultures from Gpr126+/+ (control; A, C, E) and Gpr126−/− embryos (B, D, F) stained with MBP (red), s100 (green), and DAPI (blue). A, Gpr126+/+ SCs myelinate axons after 2 weeks in culture following ascorbic acid addition (AA). This is potentiated by forskolin (FSK; C) or by 8-CPT-cAMP (E) addition. B, Gpr126−/− SCs do not myelinate axons following AA addition. MBP(+) segments are observed after addition of FSK (D) or 8-CPT-cAMP (F). Scale Bar: (in A) A–F, 100 μm. G, Quantification shows the number of MBP(+) segments observed in 1 mm2 in Gpr126+/+ (control) cocultures (gray bars) and in Gpr126−/− cocultures (black bars). Data are given as means ± SD of three independent experiments, each performed in triplicate. ***p < 0.001.
GPR126 elevates cAMP and couples to heterotrimeric G-proteins
The ability of cAMP elevation to suppress mutant phenotypes in Gpr126 zebrafish and mouse mutants suggested that Gpr126 elevates cAMP. Since an agonist is not known for Gpr126, we took advantage of the fact that GPCRs without bound agonists exist in equilibrium between inactive and active conformations (Lefkowitz et al., 1993). Receptor overexpression does not change this equilibrium but rather increases the number of receptors in both conformations. At some point, a threshold is reached and the active conformation can be detected due to constitutive activation of signaling pathways, which are normally activated after agonist stimulation. The coupling ability of several receptors, including “orphan” receptors, has been characterized by this overexpression in the absence of an agonist (Eggerickx et al., 1995; Schulz and Schöneberg, 2003; Preuss et al., 2007). We transfected cDNA-encoding full-length human GPR126 into COS-7 cells and measured cAMP accumulation using a second messenger assay. GPR126-transfected COS-7 cells showed a concentration-dependent increase of cAMP levels (Fig. 7A). As a control, we transfected human P2Y12, a known Gαi-coupling receptor (Yang et al., 2002). As expected, P2Y12-transfected COS-7 cells did not show a concentration-dependent increase in cAMP, and cAMP accumulation was significantly lower than in GPR126-transfected cells (Fig. 7A). Proper cell-surface expression of GPR126 was detected by ELISA (Fig. 7B). These results show that full-length GPR126 can localize to the plasma membrane and elevate cAMP via the Gs/adenylyl cyclase pathway in a concentration-dependent manner.
Figure 7.
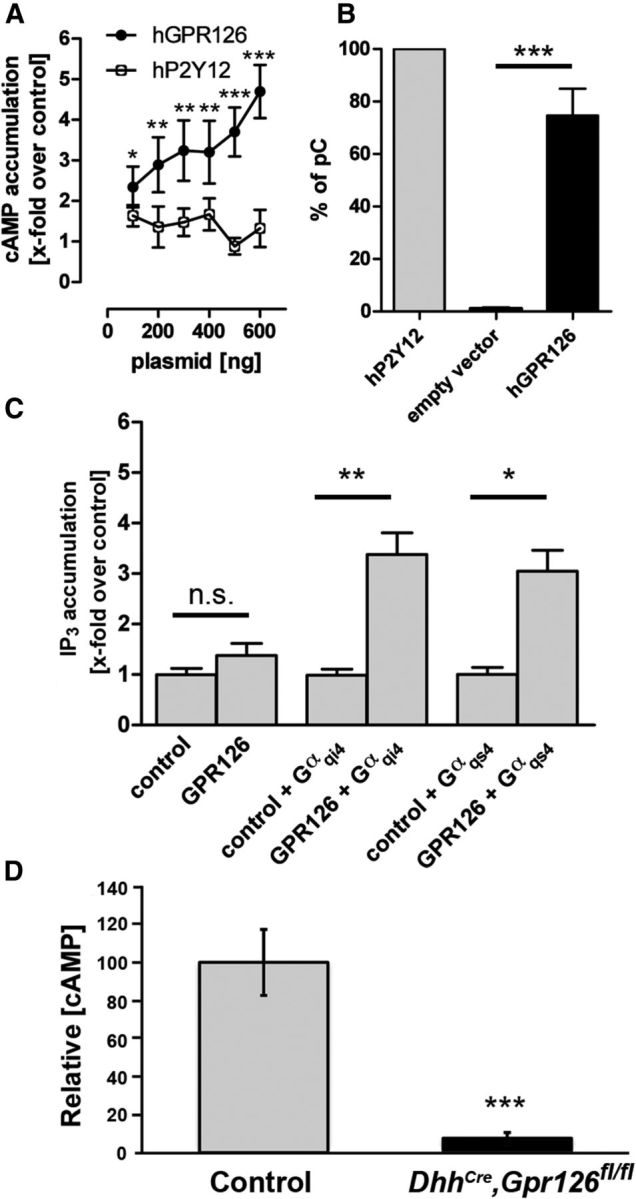
GPR126 directly elevates cAMP, couples to Gas and Gai, and cAMP is downregulated in DhhCre;Gpr126fl/fl mutant nerves. A, COS-7 cells transfected with increasing amounts (100–600 ng/well) of plasmids encoding either human GPR126 (black circles) or human P2Y12 (white circles). After 2 d, intracellular cAMP levels were measured. cAMP concentration is shown as fold change over empty vector (pcDps) control. GPR126, but not P2Y12, caused a dose-dependent increase of cAMP. Data are given as means ± SD of four independent experiments, each performed in triplicate. B, COS-7 cells were transfected with 300 ng of plasmid encoding human GPR126 (black bar), human P2Y12 (gray bar), or empty vector (pcDps). After 2 d, cell surface expression levels were determined. OD values are given as the percentage of human P2Y12, which served as a positive control (percentage of positive control). Data are given as means ± SD of three independent experiments, each performed in triplicate. C, To specify G-protein-coupling abilities of GPR126, we performed IP3 accumulation assays, which detect Gαq-mediated activation of PLC and subsequent increase in IP3. Through the use of chimeric Gαqi4-proteins or Gαqs4-proteins, Gαi-coupling and Gαs-coupling GPCRs can be recognized and directed via the Gαq-mediated pathway. For each assay, 1500 ng of GPR126 plasmid was cotransfected with 100 ng of either chimeric G-protein or empty vector. IP3 levels increased only with cotransfection of GPR126 with Gαqi4 or Gαqs4, showing that GPR126 couples to both Gαs-proteins and Gαi-proteins, but not to Gαq-proteins. Data are given as means ± SD of three independent experiments, each performed in triplicate. D, cAMP is significantly downregulated in P4 DhhCre;Gpr126fl/fl mutant nerve (black bar, n = 3) compared with Gpr126fl/fl nerve (control, gray bar, n = 5). Data are given as means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
To further define the G-protein-coupling abilities of GPR126 to Gi-proteins and Gq-proteins, we performed IP3 accumulation assays. Transfection of GPR126 into COS-7 cells alone did not lead to an increase in IP3 levels, indicating that this receptor does not couple to Gq (Fig. 7C). Chimeric G-proteins, which have the C-terminal 4 aa of a Gαq-protein exchanged for the corresponding amino acids of Gαs or Gαi, can redirect the intracellular signaling cascades of these respective GPCRs toward a classic Gαq pathway, resulting in the production of IP3 as a downstream effector of PLC (Conklin et al., 1993). Using this well established system, we cotransfected GPR126 and chimeric G-proteins Gαqi4 (to assess Gαi coupling) or Gαqs4 (to assess Gαs coupling). In both experiments, we observed a robust increase in IP3 levels (Fig. 7C). Therefore, we conclude that GPR126 couples to both Gi-proteins and Gs-proteins, but not Gq-proteins. Finally, we measured cAMP concentration in DhhCre;Gpr126fl/fl mutant sciatic nerve at P4 and found that cAMP levels in mutant nerves were significantly reduced compared with control (Fig. 7D). Together, these data show that GPR126 elevates cAMP and, like many Gs-coupled GPCRs (Gudermann et al., 1997, Liebscher et al., 2013), couples to Gi-proteins as well as Gs-proteins.
Discussion
Gpr126 couples to G-proteins and elevates cAMP
A role for cAMP in SC development and myelination has long been hypothesized. Elegant studies spanning decades of work have firmly established that axonal signals are required for SC differentiation and myelination in vivo and in vitro (Wood and Bunge, 1975; Weinberg and Spencer, 1978; Salzer and Bunge, 1980; Salzer et al., 1980a). In vitro, embryonic SCs grown in the presence of neurons display a similar developmental progression as SCs in vivo–they migrate along neurites, proliferate, sort axons, and myelinate axons (Wood, 1976). These behaviors rely on the presence of axons because in their absence, SCs fail to differentiate. The requirement for neurons can be overcome in vitro by the addition of axonal membrane fragments (Salzer et al., 1980a, 1980b) or by elevating levels of cAMP in the SCs. Upon exogenous axon membrane application or cAMP elevation, SCs upregulate the expression of myelin-related molecules and downregulate the expression of immature SC markers (Sobue et al., 1984; Mokuno et al., 1988; Monuki et al., 1989; Mirsky et al., 1990; Morgan et al., 1991; Scherer et al., 1994). Thus, an in vivo role for cAMP in SC myelination was hypothesized; however, the identity of a receptor that might alter its levels was only recently discovered.
Our previous work in zebrafish offered the first piece of evidence that cAMP elevation is essential for SC myelination in vivo. We showed that treatment of gpr126 mutant zebrafish larvae with forskolin, an adenylyl cyclase activator that elevates cAMP, could suppress myelin defects in vivo (Monk et al., 2009). This suggested, albeit indirectly, that Gpr126 elevates cAMP. Here, we extend these studies to mammals and show that addition of forskolin or 8-CPT-cAMP, a cell-permeable cAMP analog that activates PKA, can suppress Gpr126 mutant phenotypes in myelinating DRG cultures (Fig. 6D,F,G). These results are consistent with our previous studies in zebrafish and support the notion that Gpr126 elevates cAMP. To more definitively test this hypothesis, we show that GPR126 directly elevates cAMP in COS-7 cells (Fig. 7A), and that GPR126 couples to Gi-proteins and Gs-proteins (Fig. 7C). Together, our data support a model in which Gpr126, via interactions with Gαs, functions in SCs to elevate cAMP and drive myelination. GPR126 also couples to Gi-proteins, and although it is not uncommon for GPCRs to couple to more than one G-protein class (Gudermann et al., 1997; Liebscher et al., 2013), this was not predicted from our rescue experiments. In mouse SCs in vitro, low levels of cAMP promote neuregulin-dependent proliferation, while high levels of cAMP promote differentiation (Arthur-Farraj et al., 2011). Consistent with this model, we observe overproliferation of SCs in cAMP-depleted DhhCre;Gpr126fl/fl mutant sciatic nerve (Fig. 5). Via interactions with both Gi-proteins and Gs-proteins and via integration of signals from other essential pathways, Gpr126 could precisely regulate the concentration of cAMP required for a given stage of SC development.
Cellular autonomy of Gpr126
In zebrafish, we previously generated genetic chimeras in an attempt to define the autonomy of Gpr126 in myelination. In these transplantation experiments, wild-type (WT) SCs in mutant hosts were capable of expressing MBP, even though they were associated with gpr126 mutant axons. This suggested that Gpr126 functions in SCs for myelination. However, MBP expression was also rescued when WT SCs and neurons were present in mutant hosts, and importantly, we did not perform TEM analyses to determine whether WT SCs and neurons together rescued myelination more efficiently than SCs alone (Monk et al., 2009). Furthermore, Gpr126 constitutive mutant mice showed many defects that were not observed in gpr126 mutant zebrafish, including perineurial-like cell invasion throughout the endoneurium, radial sorting delays, severe axon loss, limb contracture defects, and lethality (Monk et al., 2011). These observations raised the question of whether Gpr126 has essential functions in multiple cell types during PNS development.
Our analysis of DhhCre;Gpr126fl/fl mutants suggests that Gpr126 is required in SCs for timely radial sorting and for myelination (Figs. 3, 4). We observed a lack of myelination in DhhCre;Gpr126fl/fl mutants at all ages examined up to P42 (Figs. 3, 4). We also observed severe delays in radial sorting in DhhCre;Gpr126fl/fl mutants at all stages examined (Fig. 4). Interestingly, we did not observe limb contracture defects, perineurial-like cell invasion, axon degeneration, or significant lethality in DhhCre;Gpr126fl/fl mutants, though these phenotypes were observed in constitutive Gpr126 mutants. The lethality of constitutive Gpr126 mutants is likely due to an essential function for Gpr126 in heart development (Waller-Evans et al., 2010; Patra et al., 2013). However, it is unclear whether the other defects observed in constitutive Gpr126 mutant PNS are due to a Gpr126-dependent signal from SCs before E12.5 or whether Gpr126 might function in other cells types in the developing nerve. Previous RT-PCR experiments were inconclusive regarding DRG expression of Gpr126 (Monk et al., 2011), but our in situ hybridization results demonstrate that Gpr126 is expressed in a pattern consistent with DRG neurons (Fig. 1F,G). Importantly, this expression was maintained in DhhCre;Gpr126fl/fl mutants, although the signal was less strong than in control nerve. It is also important to note that DhhCre drives recombination in endoneurial fibroblasts (Joseph et al., 2004), so we cannot formally exclude the possibility that Gpr126 is also required in this cell type. Future work is needed to define the function(s) of Gpr126 in other cell types during peripheral nerve development.
Phenotypic similarities between Gpr126 and Rac1 mutants
In addition to radial sorting delays and amyelination, we also observed cytoplasmic protrusion defects in DhhCre;Gpr126fl/fl mutants at P21 and P42 (Fig. 4F,I) strikingly similar to defects previously reported in SC-conditional Rac1 mutants (Benninger et al., 2007; Nodari et al., 2007; Guo et al., 2012). The Rac1-like defects were likely not observed in constitutive Gpr126 mutants because these animals could not be examined past P12 due to lethality. The phenotypic similarities between Gpr126 and Rac1 mutants suggest that these pathways interact. β1 Integrin is known to activate Rac1 in SCs (Nodari et al., 2007), and deletion of Itgβ1 or Rac1 in SCs leads to radial sorting delays similar to those observed in Gpr126−/− and DhhCre;Gpr126fl/fl mutants (Feltri et al., 2002; Benninger et al., 2007; Nodari et al., 2007; Guo et al., 2012). Gpr126 might indirectly activate Rac1, perhaps by modulating β1 integrin, which would in turn activate Rac1. Other aGPCRs have been shown to bind extracellular matrix molecules via their N termini (Xu et al., 2006; Luo et al., 2011). It is conceivable that the N terminus of Gpr126 binds components of the basal lamina in SCs and that this could modulate β1 integrin/laminin signaling. This is an attractive hypothesis to pursue in future work, as Gpr126 mutant SCs also phenocopy radial sorting defects observed in Laminin mutants (Chen and Strickland, 2003; Yu et al., 2005). Gpr126 may thus indirectly modulate Rac1, and Gpr126 may also directly activate Rac1. The βγ-dimers of Gi-coupled GPCRs are known to activate Rac1 via PI3K (Vogt et al., 2007), and PKA also promotes Rac1 activation via stimulation of Sif-like and Tiam1-like exchange factor (Goto et al., 2011).
In summary, our data support a model in which Gpr126 functions in SCs for timely radial sorting and myelination, and we show that this aGPCR couples to Gi-proteins and Gs-proteins. Via these G-protein interactions, Gpr126 can modulate precise levels of cAMP required for different stages of SC development. We hypothesize that both signals from Gpr126, cAMP/PKA activity via Gs-proteins and βγ-subunits release from Gi-proteins, are essential for SC differentiation and myelination.
Footnotes
This work was supported by a grant to K.R.M. from the National Institutes of Health (R01 NS079445). A.E.B. was supported by a W.M. Keck Foundation postdoctoral fellowship, F.B.E. was supported by the Alexander von Humboldt Foundation Sofja Kovalevskaja Award, T.S. was supported by grants from the German Research Foundation, and I.L. was supported by Formel-1 funding (University of Leipzig). We thank Josh Rubin and members of the Monk laboratory for helpful discussions and suggestions. We thank Andreu Viader for myelinating DRG culture training and Jeffrey Milbrandt for the use of laboratory space and reagents. We thank Dies Meijer for DhhCre mice, Paul Gray for assistance with in situ hybridization, Marilyn Levy for assistance with the TEM, and Nicole Warrington for assistance with the in vivo cAMP assays. We thank Michael Bruchas, Dave Lyons, and Xianhua Piao for helpful comments on the manuscript.
The authors declare no competing financial interests.
References
- Abdulkadir SA, Magee JA, Peters TJ, Kaleem Z, Naughton CK, Humphrey PA, Milbrandt J. Conditional loss of Nkx3.1 in adult mice induces prostatic intraepithelial neoplasia. Mol Cell Biol. 2002;22:1495–1503. doi: 10.1128/MCB.22.5.1495-1503.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araç D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Südhof TC, Brunger AT. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 2012;31:1364–1378. doi: 10.1038/emboj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur-Farraj P, Wanek K, Hantke J, Davis CM, Jayakar A, Parkinson DB, Mirsky R, Jessen KR. Mouse Schwann cells need both NRG1 and cAMP to myelinate. Glia. 2011;59:720–733. doi: 10.1002/glia.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger Y, Thurnherr T, Pereira JA, Krause S, Wu X, Chrostek-Grashoff A, Herzog D, Nave KA, Franklin RJ, Meijer D, Brakebusch C, Suter U, Relvas JB. Essential and distinct roles for cdc42 and rac1 in the regulation of Schwann cell biology during peripheral nervous system development. J Cell Biol. 2007;177:1051–1061. doi: 10.1083/jcb.200610108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Rapid accumulation of inositol triphosphate reveals that agonists hydrolyse polyphosphoinositides instead of phosphatidylinositol. Biochem J. 1983;212:849–858. doi: 10.1042/bj2120849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnekamp J, Schöneberg T. Cell adhesion receptor GPR133 couples to Gs protein. J Biol Chem. 2011;286:41912–41916. doi: 10.1074/jbc.C111.265934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZL, Strickland S. Laminin gamma1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J Cell Biol. 2003;163:889–899. doi: 10.1083/jcb.200307068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Substitution of three amino acids switches receptor specificity of Gq alpha to that of Gi alpha. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- Dostmann WR, Taylor SS, Genieser HG, Jastorff B, Døskeland SO, Ogreid D. Probing the cyclic nucleotide binding sites of cAMP-dependent protein kinases I and II with analogs of adenosine 3′,5′-cyclic phosphorothioates. J Biol Chem. 1990;265:10484–10491. [PubMed] [Google Scholar]
- Eggerickx D, Denef JF, Labbe O, Hayashi Y, Refetoff S, Vassart G, Parmentier M, Libert F. Molecular cloning of an orphan G-protein-coupled receptor that constitutively activates adenylate cyclase. Biochem J. 1995;309:837–843. doi: 10.1042/bj3090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge CF, Bunge MB, Bunge RP, Wood PM. Differentiation of axon-related Schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J Cell Biol. 1987;105:1023–1034. doi: 10.1083/jcb.105.2.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, Graus Porta D, Previtali SC, Nodari A, Migliavacca B, Cassetti A, Littlewood-Evans A, Reichardt LF, Messing A, Quattrini A, Mueller U, Wrabetz L. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J Cell Biol. 2002;156:199–209. doi: 10.1083/jcb.200109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG. G proteins and dual control of adenylate cyclase. Cell. 1984;36:577–579. doi: 10.1016/0092-8674(84)90336-2. [DOI] [PubMed] [Google Scholar]
- Glenn TD, Talbot WS. Analysis of Gpr126 function defines distinct mechanisms controlling the initiation and maturation of myelin. Development. 2013;140:3167–3175. doi: 10.1242/dev.093401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto A, Hoshino M, Matsuda M, Nakamura T. Phosphorylation of STEF/Tiam2 by protein kinase A is critical for Rac1 activation and neurite outgrowth in dibutyryl cAMP-treated PC12D cells. Mol Biol Cell. 2011;22:1780–1790. doi: 10.1091/mbc.E10-09-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudermann T, Schöneberg T, Schultz G. Functional and structural complexity of signal transduction via G-protein-coupled receptors. Annu Rev Neurosci. 1997;20:399–427. doi: 10.1146/annurev.neuro.20.1.399. [DOI] [PubMed] [Google Scholar]
- Guo L, Moon C, Niehaus K, Zheng Y, Ratner N. Rac1 controls Schwann cell myelination through cAMP and NF2/merlin. J Neurosci. 2012;32:17251–17261. doi: 10.1523/JNEUROSCI.2461-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte J, Swaminath G, Danao J, Tian H, Li Y, Wu X. Signaling property study of adhesion G-protein-coupled receptors. FEBS Lett. 2012;586:1214–1219. doi: 10.1016/j.febslet.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Haitina T, Olsson F, Stephansson O, Alsiö J, Roman E, Ebendal T, Schiöth HB, Fredriksson R. Expression profile of the entire family of adhesion G protein-coupled receptors in mouse and rat. BMC Neurosci. 2008;9:43. doi: 10.1186/1471-2202-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaegle M, Ghazvini M, Mandemakers W, Piirsoo M, Driegen S, Levavasseur F, Raghoenath S, Grosveld F, Meijer D. The POU proteins Brn-2 and Oct-6 share important functions in Schwann cell development. Genes Dev. 2003;17:1380–1391. doi: 10.1101/gad.258203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SJ, Luo R, Li S, Strokes N, Piao X. Characterization of G protein-coupled receptor 56 protein expression in the mouse developing neocortex. J Comp Neurol. 2012;520:2930–2940. doi: 10.1002/cne.23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Joseph NM, Mukouyama YS, Mosher JT, Jaegle M, Crone SA, Dormand EL, Lee KF, Meijer D, Anderson DJ, Morrison SJ. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development. 2004;131:5599–5612. doi: 10.1242/dev.01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Cotecchia S, Samama P, Costa T. Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol Sci. 1993;14:303–307. doi: 10.1016/0165-6147(93)90048-O. [DOI] [PubMed] [Google Scholar]
- Lewandoski M, Wassarman KM, Martin GR. Zp3-cre, a transgenic mouse line for the activation or inactivation of loxP-flanked target genes specifically in the female germline. Curr Biol. 1997;7:148–151. doi: 10.1016/S0960-9822(06)00059-5. [DOI] [PubMed] [Google Scholar]
- Liebscher I, Schöneberg T, Prömel S. Progress in demystification of adhesion GPCR. Biol Chem. 2013;394:937–950. doi: 10.1515/hsz-2013-0109. [DOI] [PubMed] [Google Scholar]
- Lin Y, Smrcka AV. Understanding molecular recognition by G protein βγ subunits on the path to pharmacological targeting. Mol Pharmacol. 2011;80:551–557. doi: 10.1124/mol.111.073072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Jeong SJ, Jin Z, Strokes N, Li S, Piao X. G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination. Proc Natl Acad Sci U S A. 2011;108:12925–12930. doi: 10.1073/pnas.1104821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirsky R, Dubois C, Morgan L, Jessen KR. O4 and A007-sulfatide antibodies bind to embryonic Schwann cells prior to the appearance of galactocerebroside; regulation of the antigen by axon-Schwann cell signals and cyclic AMP. Development. 1990;109:105–116. doi: 10.1242/dev.109.1.105. [DOI] [PubMed] [Google Scholar]
- Mokuno K, Sobue G, Reddy UR, Wurzer J, Kreider B, Hotta H, Baron P, Ross AH, Pleasure D. Regulation of Schwann cell nerve growth factor receptor by cyclic adenosine 3′,5′-monophosphate. J Neurosci Res. 1988;21:465–472. doi: 10.1002/jnr.490210237. [DOI] [PubMed] [Google Scholar]
- Monk KR, Naylor SG, Glenn TD, Mercurio S, Perlin JR, Dominguez C, Moens CB, Talbot WS. A G protein-coupled receptor is essential for Schwann cells to initiate myelination. Science. 2009;325:1402–1405. doi: 10.1126/science.1173474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk KR, Oshima K, Jörs S, Heller S, Talbot WS. Gpr126 is essential for peripheral nerve development and myelination in mammals. Development. 2011;138:2673–2680. doi: 10.1242/dev.062224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monuki ES, Weinmaster G, Kuhn R, Lemke G. SCIP: a glial POU domain gene regulated by cAMP. Neuron. 1989;3:783–793. doi: 10.1016/0896-6273(89)90247-X. [DOI] [PubMed] [Google Scholar]
- Morgan L, Jessen KR, Mirsky R. The effects of cAMP on differentiation of cultured Schwann cells: progression from an early phenotype (04+) to a myelin phenotype (P0+, GFAP-, N-CAM-, NGF-receptor-) depends on growth inhibition. J Cell Biol. 1991;112:457–467. doi: 10.1083/jcb.112.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi T, Haraguchi K, Ueda N, Okada M, Furuya T, Akiyama T. DREG, a developmentally regulated G protein-coupled receptor containing two conserved proteolytic cleavage sites. Genes Cells. 2004;9:549–560. doi: 10.1111/j.1356-9597.2004.00743.x. [DOI] [PubMed] [Google Scholar]
- Nodari A, Zambroni D, Quattrini A, Court FA, D'Urso A, Recchia A, Tybulewicz VL, Wrabetz L, Feltri ML. Beta1 integrin activates Rac1 in Schwann cells to generate radial lamellae during axonal sorting and myelination. J Cell Biol. 2007;177:1063–1075. doi: 10.1083/jcb.200610014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra C, van Amerongen M, Ghosh S, Ricciardi F, Sajjad A, Novoyatleva T, Mogha A, Monk KR, Mühlfeld C, Engel FB. Organ-specific function of adhesion G protein-coupled receptor GPR126 is domain dependent. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1304837110. doi: 10.1073/pnas.1304837110. Advance online publication. Retrieved Oct. 11, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss H, Ghorai P, Kraus A, Dove S, Buschauer A, Seifert R. Constitutive activity and ligand selectivity of human, guinea pig, rat, and canine histamine H2 receptors. J Pharmacol Exp Ther. 2007;321:983–995. doi: 10.1124/jpet.107.120014. [DOI] [PubMed] [Google Scholar]
- Salzer JL, Bunge RP. Studies of Schwann cell proliferation. I. An analysis in tissue culture of proliferation during development, Wallerian degeneration, and direct injury. J Cell Biol. 1980;84:739–752. doi: 10.1083/jcb.84.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer JL, Williams AK, Glaser L, Bunge RP. Studies of Schwann cell proliferation. II. Characterization of the stimulation and specificity of the response to a neurite membrane fraction. J Cell Biol. 1980a;84:753–766. doi: 10.1083/jcb.84.3.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer JL, Bunge RP, Glaser L. Studies of Schwann cell proliferation. III. Evidence for the surface localization of the neurite mitogen. J Cell Biol. 1980b;84:767–778. doi: 10.1083/jcb.84.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Wang DY, Kuhn R, Lemke G, Wrabetz L, Kamholz J. Axons regulate Schwann cell expression of the POU transcription factor SCIP. J Neurosci. 1994;14:1930–1942. doi: 10.1523/JNEUROSCI.14-04-01930.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöneberg T, Schulz A, Biebermann H, Grüters A, Grimm T, Hübschmann K, Filler G, Gudermann T, Schultz G. V2 vasopressin receptor dysfunction in nephrogenic diabetes insipidus caused by different molecular mechanisms. Hum Mutat. 1998;12:196–205. doi: 10.1002/(SICI)1098-1004(1998)12:3<196::AID-HUMU7>3.0.CO%3B2-F. [DOI] [PubMed] [Google Scholar]
- Schulz A, Schöneberg T. The structural evolution of a P2Y-like G-protein-coupled receptor. J Biol Chem. 2003;278:35531–35541. doi: 10.1074/jbc.M303346200. [DOI] [PubMed] [Google Scholar]
- Sobue G, Brown MJ, Kim SU, Pleasure D. Axolemma is a mitogen for human Schwann cells. Ann Neurol. 1984;15:449–452. doi: 10.1002/ana.410150508. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- VanDunk C, Hunter LA, Gray PA. Development, maturation, and necessity of transcription factors in the mouse suprachiasmatic nucleus. J Neurosci. 2011;31:6457–6467. doi: 10.1523/JNEUROSCI.5385-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt A, Lutz S, Rümenapp U, Han L, Jakobs KH, Schmidt M, Wieland T. Regulator of G-protein signaling 3 redirects prototypical Gi-coupled receptors from Rac1 to RhoA activation. Cell Signal. 2007;19:1229–1237. doi: 10.1016/j.cellsig.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Waller-Evans H, Prömel S, Langenhan T, Dixon J, Zahn D, Colledge WH, Doran J, Carlton MB, Davies B, Aparicio SA, Grosse J, Russ AP. The orphan adhesion-GPCR GPR126 is required for embryonic development in the mouse. PLoS One. 2010;5:e14047. doi: 10.1371/journal.pone.0014047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg HJ, Spencer PS. The fate of Schwann cells isolated from axonal contact. J Neurocytol. 1978;7:555–569. doi: 10.1007/BF01260889. [DOI] [PubMed] [Google Scholar]
- Wood PM. Separation of functional Schwann cells and neurons from peripheral nerve tissue. Brain Res. 1976;115:361–375. doi: 10.1016/0006-8993(76)90355-3. [DOI] [PubMed] [Google Scholar]
- Wood PM, Bunge RP. Evidence that sensory axons are mitogenic for SCs. Nature. 1975;256:662–664. doi: 10.1038/256662a0. [DOI] [PubMed] [Google Scholar]
- Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, Stemmer-Rachamimov AO, Cancelas JA, Ratner N. Plexiform and dermal neurofibromas and pigmentation care caused by Nf1 loss in desert hedgehog expressing cells. Cancer Cell. 2008;13:105–116. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma growth and metastasis. Proc Natl Acad Sci U S A. 2006;103:9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wu J, Jiang H, Mortensen R, Austin S, Manning DR, Woulfe D, Brass LF. Signaling through Gi family members in platelets. Redundancy and specificity in the regulation of adenylyl cyclase and other effectors. J Biol Chem. 2002;277:46035–46042. doi: 10.1074/jbc.M208519200. [DOI] [PubMed] [Google Scholar]
- Yona S, Lin HH, Siu WO, Gordon S, Stacey M. Adhesion-GPCRs: emerging roles for novel receptors. Trends Biochem Sci. 2008;33:491–500. doi: 10.1016/j.tibs.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Yu WM, Feltri ML, Wrabetz L, Strickland S, Chen ZL. Schwann cell-specific ablation of laminin γ1 causes apoptosis and prevents proliferation. J Neurosci. 2005;25:4463–4472. doi: 10.1523/JNEUROSCI.5032-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]