

Abstract
Intracellular tau aggregates are the neuropathological hallmark of several neurodegenerative diseases, including Alzheimer's disease, progressive supranuclear palsy, and cases of frontotemporal dementia, but the link between these aggregates and neurodegeneration remains unclear. Neuronal models recapitulating the main features of tau pathology are necessary to investigate the molecular mechanisms of tau malfunction, but current models show little and inconsistent spontaneous tau aggregation. We show that dorsal root ganglion (DRG) neurons in transgenic mice expressing human P301S tau (P301S-htau) develop tau pathology similar to that found in brain and spinal cord and a significant reduction in mechanosensation occurs before detectable fibrillar tau formation. DRG neuronal cultures established from adult P301S-htau mice at different ages retained the pattern of aberrant tau found in vivo. Moreover, htau became progressively hyperphosphorylated over 2 months in vitro beginning with nonsymptomatic neurons, while hyperphosphorylated P301S-htau-positive neurons from 5-month-old mice cultured for 2 months died preferentially. P301S-htau-positive neurons grew aberrant axons, including spheroids, typically found in human tauopathies. Neurons cultured at advanced stages of tau pathology showed a 60% decrease in the fraction of moving mitochondria. SEG28019, a novel O-GlcNAcase inhibitor, reduced steady-state pSer396/pSer404 phosphorylation over 7 weeks in a significant proportion of DRG neurons showing for the first time the possible beneficial effect of prolonged dosing of O-GlcNAcase inhibitor in vitro. Our system is unique in that fibrillar tau forms without external manipulation and provides an important new tool for understanding the mechanisms of tau dysfunction and for screening of compounds for treatment of tauopathies.
Introduction
Insoluble aggregates made of microtubule-associated protein tau represent the pathological hallmark of several neurodegenerative diseases, including Alzheimer's disease, Pick's disease, progressive supranuclear palsy, corticobasal degeneration, and frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17T; Goedert et al., 2012; Spillantini and Goedert, 2013). Tau is a soluble phospho-protein that is expressed in neurons of the CNS and peripheral nervous system (PNS), where it is found mainly in axons (Cleveland et al., 1977; Binder et al., 1985). Tau is implicated in the assembly and stabilization of microtubules (MTs), as well as in neuritogenesis, axonal maintenance, and transport (Morris et al., 2011; Mandelkow and Mandelkow, 2012). The identification of mutations in the tau gene MAPT in cases of FTDP-17T demonstrated the relevance of tau for neurodegeneration (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998) and led to the development of cell and animal models of tau pathology.
While several transgenic models have been created that reproduce features of tau pathology and tau-related cell death, cellular models with consistent and reproducible filamentous tau aggregates are not available. Cells that reproduce the key hallmarks of tau pathology would provide a means of studying the as yet unclear basic mechanisms associated with tau pathophysiology and toxicity, and would be an important tool for screening potential therapeutic compounds.
To establish cultures of primary neurons that would recapitulate the main features of tau dysfunction in a cell-autonomous manner, we focused on neurons from dorsal root ganglia (DRG) of transgenic mice expressing human P301S tau under the control of the Thy1.2 promoter (Allen et al., 2002). In a homozygous state these mice develop a stereotypic tauopathy that culminates in severe neurodegeneration at ∼5–6 months of age. DRG neurons constitute an ideal cellular model, since they can be cultured from adult mice for months, potentially enabling the progression of pathological features to develop in a time-dependent manner. Furthermore, DRG neurons are relevant as a model of tau pathology, because filamentous tau has been reported in lumbar DRG neurons from progressive supranuclear palsy (PSP) patients (Nishimura et al., 1993), as well as in other peripheral ganglia in Alzheimer's disease (AD; Kawasaki et al., 1987).
Here we show that tau pathology progresses spontaneously in cultures of DRG neurons from adult P301S-htau mice. We demonstrate that the presence of pathological tau in P301S-htau DRG neurons is associated in vivo with a reduction in mechanosensation. Moreover, DRG neuronal cultures from P301S-htau mice develop age-dependent tau pathology similar to CNS neurons in vivo, morphological alterations in axons similar to those described in human tauopathies, and time-dependent alterations in mitochondrial axonal transport. Furthermore, tau hyperphosphorylation develops progressively in DRG neurons kept in culture for 8 weeks. We demonstrate the model's utility for drug screening using a novel potent and selective iminosugar O-GlcNAcase inhibitor (Hart et al., 2011; Horne et al., 2011) and show a reduction in phosphorylation of tau at pSer396/pSer404, the PHF-1 epitope. Our data show that P301S-htau+ve DRG neurons are a valuable model to investigate tau pathology and potential treatments.
Materials and Methods
Antibodies.
Anti-phospho-tau AT8 (epitope pS202/pT205), AT100 (epitope pT212/pS214), AT180 (epitope pT231), and anti-human tau HT7 (epitope 159–163) mouse monoclonal antibodies (Biernat et al., 1992; Goedert et al., 1995; Yoshida and Goedert, 2006) were from Innogenetics NV or Thermo Scientific. PHF-1 (epitope pS396/pS404) and MC1 (conformational antibody; Jicha et al., 1997; Weaver et al., 2000; Jeganathan et al., 2008) (mouse monoclonals) were a kind gift from Dr. P Davies, Albert Einstein College of Medicine, New York. Other antibodies include rabbit polyclonal anti-Tau (A0024, Dako), rabbit polyclonal βIII tubulin (Covance; PRB-435P-100), Anti-O-GlcNAc antibody RL2 (ab2739; Abcam), anti-Tau C terminus BR134 (Spillantini et al., 1996; Allen et al., 2002), and isolectin IB4 conjugated to FITC was from Vector Laboratories. All the primary antibodies used for DRG immunolabeling were used at 1:500–1000 with the exceptions of RL2, which was used at 1:300, and IB4-FITC used at 1:100. All secondary antibodies were used at 1:1000 for immunocytochemistry and 1:5000–10,000 for immunoblotting.
Animals.
Homozygous P301S-htau mice (Allen et al., 2002) and C57BL/6S (C57BL/6 OlaHsd; Harlan) control mice were maintained on a 12 h light/dark cycle in a temperature-controlled room (22°C) in cages with free access to food and water. No difference was observed in DRG cultures from male and female P301S-htau mice, therefore, both male and female mice were used in the study. All procedures were performed in accordance with the UK Animal (Scientific Procedures) Act 1986 for the welfare of laboratory animals and were approved by the Cambridge University local ethical committee.
Dissociated adult mouse DRG cultures.
Protocols for culturing dissociated DRG neurons from adult mice were adapted from Malin et al., 2007 and Gumy et al., 2008. Briefly, mice were killed by CO2 inhalation followed by cervical dislocation and the total pool of DRGs per mouse was collected in L15 medium (Life Technologies). Meninges and roots were removed and ganglia were digested with 2 mg/ml collagenase (Sigma-Aldrich) at 37°C for 1 h followed by addition of 0.1% trypsin (Sigma-Aldrich) for 10 min. DRGs were collected into DMEM (Life Technologies) containing 9% fetal bovine serum (FBS; Life Technologies), triturated into a homogenous cell suspension, and centrifuged at 1000 × g for 2 min. The cell pellet was resuspended in culture medium (DMEM supplemented with 1% FBS, penicillin-streptomycin-fungizone PSF; Life Technologies) and 20 ng/ml NGF (Sigma-Aldrich). For long-term cultures, Neurobasal A medium (Life Technologies) containing B27 (Life Technologies), Glutamax (Life Technologies), PSF, NGF, and a mixture of 20 μm 5-fluoro-2′-deoxyuridine/uridine and 0.5 μg/ml mitomycin C (Sigma-Aldrich) was used. DRG cells were plated onto poly-d-lysine and laminin-coated glass coverslips or glass-bottomed dishes (MatTek). Cells were grown at 37°C in 7% CO2 and the medium was changed twice a week. Each culture was obtained using the whole DRG set from a single mouse. For SEG28019 testing, DRG neurons were cultured from 3-month-old P301S-htau mice. SEG28019 (Summit plc; 20 mg) was dissolved in 100 μl of dimethylsulfoxide, and water was added to make a 30 mm stock solution. Aliquots were kept at −20°C. SEG28019 was freshly diluted into long-term culture medium to a final concentration of 10 μm just before addition to the neurons. Medium containing SEG28019 was first added after 2 days in vitro (DIV), and half the volume of media of both untreated and treated cultures was changed twice a week.
Immunocytochemistry.
DRG cultures were fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature, washed in PBS, permeabilized, and blocked in PBS/0.01% Triton X-100 (PBST) containing 5% serum of the appropriate species, or in PBS/0.3% Triton X-100 without blocking, and incubated with primary antibodies in PBST overnight at 4°C. Secondary antibodies conjugated with Alexa fluorochromes (Life Technologies) were incubated for 1 h at room temperature and nuclei stained with 1 μg/ml Hoechst 33258 (Sigma-Aldrich). Cultures were mounted in FluorSave (MerckMillipoe). For dual labeling of dissociated DRG cultures with the monoclonal antibodies PHF-1/AT8/AT100 and HT7, incubations were performed sequentially in two rounds: cells were incubated with the anti-phospho-tau antibody together with the anti-βIII tubulin antibody followed by the appropriate secondary antibodies, then with HT7 followed by a secondary antibody that showed no spectral overlap. For DRG sections, mice were anesthetized by intraperitoneal injection of sodium pentobarbital (Euthatal; 2 ml/kg; Rhône-Mérieux), and perfused transcardially with PBS followed by 4% PFA (Sigma-Aldrich) in 0.1 m phosphate buffer, pH 7.4. DRGs were postfixed in 4% PFA for 30 min at 4°C, washed, and cryopreserved in PBS/15% sucrose containing 0.02% sodium azide at 4°C. Tissue was embedded in OCT mounting medium (Thermo Scientific), frozen in cold 2-methylbutane for 5 min, and sectioned using a Leica CM 3050S cryostat. Cryostat sections (12 μm) were mounted onto poly-l-lysine-coated glass slides (SuperFrost Plus; VWR), stained, and then coverslipped in FluorSave. Labeling was visualized using a Leica TMDM 6000B fluorescence microscope and images were acquired with Leica LAS AF software.
Quantification of P301S-htau expression and phosphorylation.
The total number of neurons in nine fields (20× objective; 200–400 neurons; Leica DM6000 microscope) was counted per coverslip per culture, and the percentage of P301S-htau+ve out of βIII tubulin+ve neurons, or phosphorylated P301S-htau+ve out of total P301S-htau+ve neurons was calculated. Significance was determined by one-way ANOVA adjusted with Bonferroni post hoc adjustment. For analysis of PHF-1 expression in SEG28019-treated neurons, the intensity of labeling of PHF-1 was measured and normalized to that of HT7, after confirmation that intensities were in the linear range. All the positive neurons on the coverslip were scored. Because the data did not follow a normal distribution, the nonparametric Mann–Whitney test was applied to determine significance and p values.
Axon length and growth cone area quantification.
Low-density dissociated DRG cultures from P301S-htau and C57BL/6S mice of various ages were grown for 2 DIV and costained with anti-βIII tubulin and anti-tau HT7 antibodies. Images of dissociated DRG neurons were acquired using a Leica DM6000 microscope. The length of the longest axonal process per neuron and the area of the associated growth cone (70–90 neurons each of HT7−ve, HT7+ve in P301S-htau and C57BL/6S wild-type mice per independent culture) were determined with ImageJ software (National Institutes of Health; http://rsbweb.nih.gov/ij/index.html). The drawing tool was used to trace and measure the length of the longest neurite per cell from the axon hillock to the growth cone. The same method was used to trace around the growth cone to obtain the area and perimeter in micrometers. Three and four independent DRG cultures from, respectively, control and transgenic animals at each age were analyzed. Significance was tested by two-way ANOVA followed by Bonferroni post hoc adjustment.
Protein extraction.
DRGs, spinal cord, and brain (cortex or brainstem) were dissected from a 5-month-old mouse and immediately frozen on dry ice. For soluble tau extraction, DRGs from four P301S-htau mice were pooled. For sarkosyl-insoluble preparations, DRGs were pooled from five P301S-htau and five control mice. For sarkosyl-insoluble preparations from 2 DIV cultures, DRGs were from a single 5-month-old P301S-htau or control mouse. Soluble tau was extracted essentially as previously described (Goedert and Jakes, 1990). Briefly, tissue was homogenized in 2.5% (v/v) perchloric acid (0.5 ml/200 mg; Sigma-Aldrich), centrifuged at 12,000 × g for 10 min at 4°C, and the supernatant dialyzed against 50 mm Tris-HCl, pH 7.4, 0.1 mm EDTA and 1 mm phenylmethylsulfonyl fluoride (Sigma-Aldrich) overnight at 4°C. Half of the sample was dephosphorylated with 18 U/ml Escherichia coli alkaline phosphatase (AP) (Promega) in 50 mm Tris-HCl, pH 7.4, containing 5 mm MgCl2 for 3–4 h at 65°C. Sarkosyl-insoluble tau extraction was performed as previously reported (Allen et al., 2002), samples were homogenized in A68 buffer (10 mm Tris-HCl, pH 7.4, 0.8 m NaCl, 1 mm EGTA, and 10% sucrose). The homogenate was centrifuged at 15,000 × g for 20 min at 4°C and the supernatant was collected, while the pellet was resuspended in A68 buffer and recentrifuged. The supernatants were combined and incubated in 1% sarkosyl (Sigma-Aldrich) containing cOmplete protease inhibitor cocktail (Roche) for 1 h at room temperature and centrifuged at 100,000 × g for 1 h at 4°C (Optima MAX Ultracentrifuge; Beckman Coulter). The pellet was reconstituted in 50 mm Tris-HCl, pH 7.4, and used for immunoblotting. The same protocol was used for extraction of insoluble tau from tissue and DRG cultures except that the cells were collected and homogenized directly in A68 containing 1% sarkosyl and protease/phosphatase (Sigma-Aldrich) inhibitors. After spinning at 100,000 × g for 1 h at 4°C, the pellet was rinsed once in the same buffer, reconstituted in sample buffer, and half of the aliquot loaded on the gel.
Immunoblotting.
Proteins were solubilized in gel-loading buffer, resolved on 10% SDS-PAGE, and transferred onto PVDF membranes (Merck). Nonspecific background was blocked in 5% w/v Marvel milk (Premier International Foods) in TBS-T and incubated with the primary antibody overnight at 4°C, followed by 1 h at room temperature in the appropriate peroxidase-labeled secondary antibody. Blots were developed with ECL Plus (GE Healthcare). Each experiment was repeated three times using independent sets of samples.
Immunogold electron microscopy.
Sarkosyl-insoluble proteins were extracted from DRG tissue or cell pellets as described above. Immunogold electron microscopy (EM) was performed as previously reported (Crowther, 1991). Briefly, sarkosyl-insoluble samples were placed on carbon-coated 400 mesh grid and labeled with anti-tau BR134 or AT8 antibodies and secondary antibodies linked to gold particles and negatively stained with 1% potassium phosphotungstate. Micrographs were recorded at a nominal magnification of 40,000× on a Philips EM208S microscope.
Behavioral tests.
Sensory function in 1- and 3-month-old P301S-htau mice and age-matched C57BL/6S controls was evaluated in a blinded fashion according to protocols commonly used to assess sensory dysfunction due to peripheral nerve injury (Wang et al., 2011; Gladman et al., 2012). Mice were placed in a Plexiglas box with a wire mesh floor (mechanical test) or a Plexiglas floor (thermal test), and habituated to the chamber until they became calm. In the thermal sensitivity test, a stimulus from an infrared light source (55°C; Ugo Basile) was directed perpendicularly against the mouse left hindpaw. The period from application of the stimulus to the withdrawal of the paw was recorded. If the mouse was not responding to the stimulus within 15 s, the trial was ended to avoid tissue damage. Mechanical threshold for paw withdrawal was assessed using a dynamic plantar aesthesiometer (model 1601C; Life Science Instruments). Probe was gently applied to the right hindpaw of the animal and an increasing vertical force was applied until withdrawal of the paw. The force at which the mouse withdrew its hindpaw was measured in grams by a force transducer. If the mouse did not respond to a maximum force of 100 g, the trial was aborted. For each animal, five trials were performed with a minimum of 10 min rest interval between each recording. Significance was tested by one-way ANOVA followed by Bonferroni post hoc adjustment.
Live imaging and mitochondrial transport.
Dissociated DRG neurons from P301S-htau and C57BL/6S mice at 3 and 5 months were grown on glass-bottomed dishes for 40 h before incubation with 10 nm MitoTracker Orange (Life Technologies) for 5 min at 37°C. Mitochondrial dynamics along the axon were recorded with a 63× oil-immersion objective using a Leica AF7000 microscope with a Hamamatsu EM CCD C9100 camera connected to Leica LAS AF software. The environment was controlled with a heated stage set at 37°C (Heating Insert P; PeCon) and a CO2 chamber set to 5%. Time lapse images of mitochondrial movements were acquired every 5 s for 5 min (61 frames in total). Neuronal cell bodies were distinguished from non-neuronal cells by phase contrast. To determine the directionality of mitochondrial movements, part of the neuronal soma was included in the imaged field. Neurons from P301S-htau mice were imaged blindly. To identify whether the recordings were made from HT7−ve and HT7+ve neurons in the cultures from P301S-htau mice, the x,y coordinates for each imaged cell were saved with LAS AF software. After recording, cells were fixed with 4% PFA, stained with the HT7 antibody, and the x,y coordinates of each previously imaged neuron were recalled under the microscope using the Leica software. For dissociated DRG cultures from 3-month-old mice, 11 axons from four independent culture preparations were imaged, yielding a total of 251, 283, and 212 mitochondria for C57BL/6S, P301S-htau HT7−ve, and HT7+ve neurons, respectively. For animals at 5 months, 20 axons from six independent culture preparations were imaged, yielding a total of 456, 443, and 418 mitochondria for C57BL/6S, HT7−ve or HT7+ve neurons, respectively. QuickTime movies and kymographs of time-lapse image series were obtained with MetaMorph software (version 7; Molecular Devices) and directionality of movement, distance, time traveled, and average speed were determined. Mitochondria were classified as stationary if they did not move >2 μm during the entire recording period (Calkins and Reddy, 2011). For each age, the proportion of stationary and moving mitochondria and the average speed were calculated by pooling the mitochondria from all the imaged axons according to their directionality. Normality of the data was estimated by both Kolmogorov–Smirnoff normality test and D'Agostino–Pearson omnibus normality test. For each imaged axon, the number of stationary, anterograde, and retrograde moving mitochondria was calculated. Datasets for transport were normally distributed so the significance of the differences in the mitochondrial movements in C57BL/6S, P301S-htau HT7−ve, and P301S-htau HT7+ve neurons from 3- and 5 month-old mice was tested by two-way ANOVA followed by Bonferroni post hoc adjustment. Since average velocity datasets were not normally distributed, data are presented as cumulative frequency distribution and the significance of the differences was determined using the Kruskal–Wallis test (for three groups) or Mann–Whitney test (for two groups; Falzone et al., 2009).
Statistics.
Data were analyzed using GraphPad Prism version 5, IBM SPSS version 21, or MetaMorph version 7 software. Where data followed a normal distribution, one- or two-way ANOVA where appropriate was followed by Bonferroni post hoc adjustment. For data that did not follow a normal distribution, the Mann–Whitney or Kruskal–Wallis nonparametric tests were applied where data from two or three groups, respectively, were compared. Details of statistical tests and exact p values are given in the figure legends. The symbols * or #, ** or ##, *** or ### denote p values < 0.05, 0.01, and 0.001 respectively.
Results
DRG neurons from P301S-htau mice contain insoluble tau aggregates
Transgenic P301S-htau mice expressing human mutant tau under the Thy1.2 promoter show extensive development of hyperphosphorylated tau and filamentous tau aggregates (neurofibrillary tangles, NFT) in brain and spinal cord at 5 months of age (Allen et al., 2002). To establish whether DRG neurons from these mice express transgenic P301S-htau, sections from the fourth lumbar ganglion (L4) of 5-month-old mice were immunolabeled for the presence of human tau (HT7 antibody) and hyperphosphorylated tau (antibody AT8). Immunofluorescent imaging showed that several DRG neurons stained positively for human tau (Fig. 1A,a), including hyperphosphorylated tau (Fig. 1A,c). Some AT8+ve neurons showed cytoplasmic ring or crescent-like structures typical of NFTs (Fig. 1A,e). The occurrence of hyperphosphorylated tau in lumbar as well as cervical and thoracic DRGs from 5-month-old transgenic mice was further confirmed by immunolabeling with the phospho-tau antibodies AT8 and AT180 (data not shown). There was no specific labeling with any of the anti-phospho-tau antibodies in non-neuronal cells, or in DRG neurons from age-matched C57BL/6S controls (Fig. 1A,b,d).
Figure 1.

DRG neurons in P301S-htau mice contain hyperphosphorylated tau, NFTs, and an associated mechanosensory deficit. A, Sections (12 μm) from DRGs of 5-month-old P301S-htau and C57BL/6S control mice were stained with anti-htau (HT7; a and b) or anti-phospho-tau (AT8; c–e) and visualized by fluorescence microscopy. The detail (e) shows typical ring and crescent-like shapes formed by pathological htau. Scale bars: A, a–d, 200 μm; A, e, 50 μm. B, Perchloric acid-soluble protein extracts from P301S-htau DRG and spinal cord (SC) separated by SDS-PAGE alongside a SC extract from a control animal. Half of each sample was treated with alkaline phosphatase (AP+). Blots were probed with anti-total tau antibody (Dako); white arrows: dephosphorylated P301S-htau aligned with recombinant 0N4R htau band; *, murine tau isoforms. C, Hyperphosphorylation at pS202/pT205 detected with AT8 in perchloric acid-soluble tau extracts. D, Sarkosyl-insoluble tau prepared from DRGs from 5-month-old P301S-htau or control mice and a P301S-htau mouse brain probed with AT8. No insoluble tau is present in the control sample while a 64 kDa band (arrow) is present in DRG and brain of P301S htau mice. E, Immunogold EM of the sarkosyl-insoluble tau from DRGs probed with AT8 antibody shows the presence of tau filaments. Bkg, background immunogold labeling. No filaments were observed in extracts from control mice. Scale bars: 100 nm. F, Sensory tests in P301S-htau and C57BL/6S mice at 1 and 3 months of age show no alterations in thermal sensitivity (left) but a significant change in mechanosensitivity, as shown by the pressure test (right), which is altered in 3-month-old P301S-htau mice [(mean ± SEM; n = 7–10; one-way ANOVA (p = 0.012) followed by Bonferroni post hoc adjustment p = 0.021 (P301S-1m/P301S-3m); p = 0.032 (P301S-3m/C57BL6-3m); p = 0.018 (P301S-3m/C57BL6-1m)].
The expression of transgenic P301S-htau was also investigated by immunoblotting using an anti-tau antibody that recognizes both human and murine tau isoforms. To resolve the individual isoforms of tau, the samples were dephosphorylated with alkaline phosphatase (AP+). A prominent band of ∼50 kDa (Fig. 1B) corresponding to the transgenic human P301S tau aligned with a band from spinal cord (SC) of P301S-htau mice, and with the 0N4R recombinant isoform of human tau. This band was absent in spinal cord extracts from C57BL/6S controls, where only three bands corresponding to the three endogenous mouse tau isoforms were clearly visible in the AP+-treated samples. The presence of hyperphosphorylated tau was confirmed by immunoblotting with the AT8 antibody (Fig. 1C). Two closely spaced bands of ∼64 kDa, possibly representing different levels of tau phosphorylation, were present and aligned with transgenic P301S-htau from spinal cord.
To determine whether DRGs from P301S-htau mice contain filamentous insoluble tau, Sarkosyl-insoluble extracts were analyzed by immunoblotting with AT8 and by immunogold EM. Immunoblotting showed a 64 kDa band in DRG and brain extracts from transgenic P301S-htau mice (Allen et al., 2002) but not in DRGs from age-matched C57BL/6S mice (Fig. 1D). Furthermore, immunogold EM of the Sarkosyl-insoluble extract revealed the presence of AT8+ve tau filaments resembling the straight and narrow twisted filaments found in human tauopathies and in the CNS of P301S-htau mice (Allen et al., 2002) (Fig. 1E). These data demonstrate that DRGs from P301S-htau mice develop pathology that mimics the features of tauopathy found in CNS of P301S-htau mice and human disease.
Reduced mechanosensory response in P301S-htau mice
Because DRG neurons showed tau pathology, we asked whether P301S-htau mice develop sensory deficits. Therefore, thermal and mechanical sensitivities were studied in transgenic and control mice at 1 and 3 months of age to avoid the interference of possible sensory deficits due to loss of motor functions. No deficits were observed in the thermal acuity test in P301S-htau mice compared with control mice (Fig. 1F, left), indicating that pain and thermal sensations were intact. However, the pressure that was required to elicit withdrawal of the hindpaw in 3-month-old P301S-htau mice was significantly increased compared with age-matched C57BL/6S and 1-month-old P301S-htau mice (Fig. 1F, right; ANOVA p = 0.012, Bonferroni post hoc p = 0.018–0.032), indicating a reduction in mechanosensory function. The likely reason why the increase was relatively modest compared with effects observed after sciatic nerve crush (Wang et al., 2011; Gladman et al., 2012) is that only a small proportion of the neurons that respond to this stimulus express pathological forms of P301S-htau, as shown next.
Dissociated adult DRG cultures from P301S-htau mice retain tau pathology
To examine whether tau pathology found in the intact ganglia is preserved in culture, DRG neurons were isolated from adult P301S-htau mice at different ages and cultured for 2 DIV. In neurons from 2-week-old mice, P301S-htau was mainly localized in the soma and only a few neurons showed human tau (HT7) labeling in their processes (Fig. 2A). However, in neurons from 1- to 5 month-old mice, HT7 labeling was found in the processes as well as in the soma. In some neurons from 5-month-old P301S-htau mice, a patchy pattern of labeling was observed in the axons. HT7 labeling always colocalized with βIII tubulin+ve labeling, confirming the neuronal expression of the transgene, as noted previously by Thy1.2 GFP mice (Bradman et al., 2011; Fig. 2A). The percentage of neurons showing P301S-htau in culture increased according to the age of the mouse from which the DRG were taken, reaching a plateau in cultures from 3-month-old mice (HT7/βIII tubulin+ve neurons: 6% at 2 weeks, 12% at 1 month, 18% at 3 months, and 17.8% at 5 months; Fig. 2B). No labeling for human tau (HT7) was detected in neurons from C57BL/6S mice, in keeping with the results shown in Figure 1.
Figure 2.
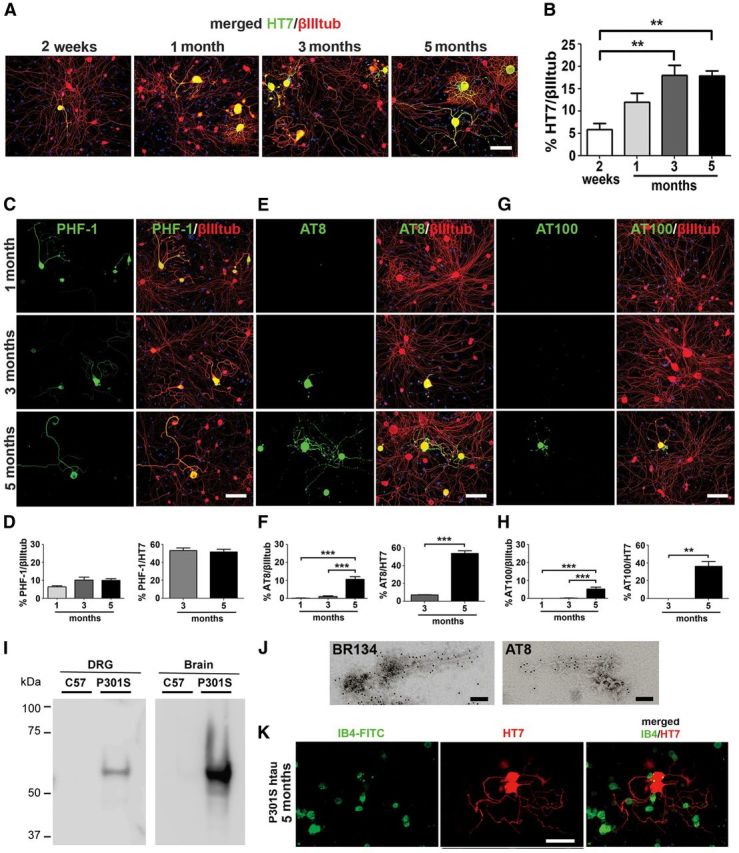
Dissociated DRG neurons from P301S-htau mice at different ages develop progressive tau pathology. Dissociated DRG neurons were prepared from P301S-htau mice at 2 weeks and 1, 3, and 5 months of age, cultured for 2 DIV, and immunolabeled with the indicated anti-human tau (HT7) or anti-phospho-tau antibodies and anti-βIII tubulin antibody. A, Developmental profile of HT7+ve immunolabeling at 2 weeks and 1, 3, and 5 months. Note weak labeling of axons in sample cultured at 2 weeks and complex pattern of outgrowth in neurons cultured at 1, 3, and 5 months. B, The proportion of cells expressing P301S-htau (HT7+ve) out of the total βIII tubulin+ve neurons is shown as a function of mouse age (mean ± SEM, n = 6; one-way ANOVA (p = 0.003) followed by Bonferroni post hoc adjustment; p = 0.0086 (2w/3m); p = 0.0093 (2w/5m)). C, E, G, Dissociated DRG neurons cultured for 2 DIV from P301S-htau mice aged 1, 3, and 5 months, immunolabeled with the phosphorylation-dependent anti-tau antibodies PHF-1, AT8, and AT100. Scale bar, 100 μm. D, F, H, Quantification of the proportion of PHF-1/AT8/AT100+ve neurons out of the total βIII tubulin+ve neurons (n = 6), or HT7+ve neurons (n = 3–4). Mean ± SEM, one-way ANOVA followed by Bonferroni post hoc adjustment. F, AT8/βIII tubulin ANOVA p = 5.1 × 10−7; p = 0.0011 (1m/5m); p = 0.0017 (3m/5m); Student's t test; AT8/HT7 p = 0.0002. H, AT100/βIII tubulin ANOVA p = 1.2 × 10−5, p = 0.016 (1m/5m), p = 0.017 (3m/5m); Student's t test; AT100/HT7 p = 0.0012. I, Sarkosyl-insoluble tau was extracted from dissociated DRG neuron cultures (2 DIV) from 5-month-old P301S-htau or C57BL/6S mice and analyzed by immunoblotting using the HT7 antibody. Brain extracts from the same mice were used as positive controls. Note that the samples were run on the same blot but to visualize both sets of bands, the image on the left was exposed for 10 min whereas the image on the right was exposed for 1 min. J, Sarkosyl-insoluble tau was extracted from dissociated DRG neuron cultures (2 DIV) from 5-month-old mice and analyzed by immunogold EM with the phosphorylation-independent and phosphorylation-dependent anti-tau antibodies BR134 and AT8, respectively. Tau filaments from the cultured cells appear as straight or narrow twisted filaments and are similar to those found in DRG tissue from 5-month-old mice shown in Figure 1. Scale bars: 100 nm. K, DRG neurons from 3-month-old P301S-htau mice were cultured for 2 d, fixed, and stained with IB4-FITC (green) and HT7 (red), and imaged by fluorescence microscopy. Note lack of overlap between the two probes.
To evaluate the presence of hyperphosphorylated tau, neurons were immunolabeled with different phosphorylation-dependent anti-tau antibodies. PHF-1 labeling was present in ∼5% of HT7+ve neurons cultured from 1-month-old mice, increasing to 10% in neurons cultured from 3- and 5-month-old P301S-htau mice (Fig. 2C,D). PHF-1 labeling was homogeneously distributed throughout the neurons though in some neurons PHF-1 labeling was found only in the soma. DRG neurons from 5-month-old mice expressing PHF-1 comprised ∼55% of the HT7+ve neurons (Fig. 2D), demonstrating that this was not a rare event.
AT8+ve labeling (Fig. 2E) was rarely observed in P301S-htau cultures from 1-month-old mice (Fig. 2E,F, 0.15% of βIII tubulin+ve), increasing slightly in cultures from 3-month-old mice (1.1%). However, in cultures from 5-month-old P301S-htau mice, 10.6% of the neurons stained AT8+ve, with both the cell body and the processes showing strong labeling. In some axons this labeling was patchy, supporting the data obtained with HT7 and suggesting the formation of aggregates. Similar to PHF-1 labeling, in cultures from 5-month-old mice, AT8+ve neurons comprised ∼55% of HT7+ve neurons (Fig. 2F).
Labeling by AT100 has been associated with the presence of tau filaments in our P301S-htau mouse line (Allen et al., 2002; Yoshida and Goedert, 2006; Delobel et al., 2008). No AT100 immunoreactivity was present in cultures from 1-month-old P301S-htau mice and was rarely detected in cultures from 3-month-old P301S-htau mice (Fig. 2G,H; 0–0.3% AT100+ve/HT7+ve neurons). However, 5.3% of the neurons from 5-month-old mice were AT100+ve, comprising 32% of HT7+ve neurons. Patchy labeling patterns were often observed in the axons, indicating tau aggregation.
To directly establish the presence of filaments, Sarkosyl-insoluble tau was extracted from neurons prepared from 5-month-old P301S-htau mice after 2 DIV and analyzed by immunoblotting and immunogold EM. Figure 2J shows that cultured DRG neurons from 5-month-old P301S-htau mice, but not aged-matched C57BL/6S control mice, contained a Sarkosyl-insoluble tau band similar to that present in brains of 5-month-old P301S-htau mice. AT8+ve tau filaments that were also decorated by the phosphorylation-independent anti-tau antibody BR134 were detected by immunogold EM in Sarkosyl-insoluble cell extracts (Fig. 2J). Thus, cultured neurons from the DRG of transgenic P301S-htau mice preserve the attributes of pathological tau found in the intact DRG and the CNS.
To investigate the subtype of neurons that express human tau (HT7+ve), we stained the cultures with the Griffonia simplicifolia isolectin IB4, which detects small-diameter nonpeptidergic heat-sensitive nociceptors (Silverman and Kruger, 1988; Snider and McMahon, 1998; Stucky and Lewin, 1999). HT7+ve neurons were uniformly negative for IB4 labeling (Fig. 2K), consistent with the presence of a mechanosensory deficit and the lack of a temperature-sensing deficit observed in vivo, as shown in Figure 1.
P301S-htau-expressing DRG neurons are stained by the pathological conformation-specific MC1 antibody
To further demonstrate that P301S-htau+ve DRG neurons express a disease-related conformation of tau, we stained cultures with the antibody MC1, which detects a disease-specific tau conformation in human AD (Jicha et al., 1997; Weaver et al., 2000; Jeganathan et al., 2008; Fig. 3). Cultured P301S-htau+ve neurons from 4- to 5-month-old mice, but not those from 3-month-old or younger mice, showed the pathological conformational change detected by MC1 (Fig. 3A). MC1 reactivity occurred when PHF-1, AT8, and AT100 antibodies already detected tau hyperphosphorylation. No MC1 reactivity was detected in C57BL/6S control neurons. Thus, although MC1 is not a phospho-epitope-specific antibody, our data agree with biochemical studies that indicate that MC1 recognizes a conformation that depends on there being prior phosphorylation at all three (PHF-1/AT8/AT100) epitopes (Jeganathan et al., 2008). In neurons from 4-month-old P301S-htau mice cultured for 2.5 weeks, MC1 showed a patchy labeling suggestive of tau aggregation (Fig. 3B).
Figure 3.
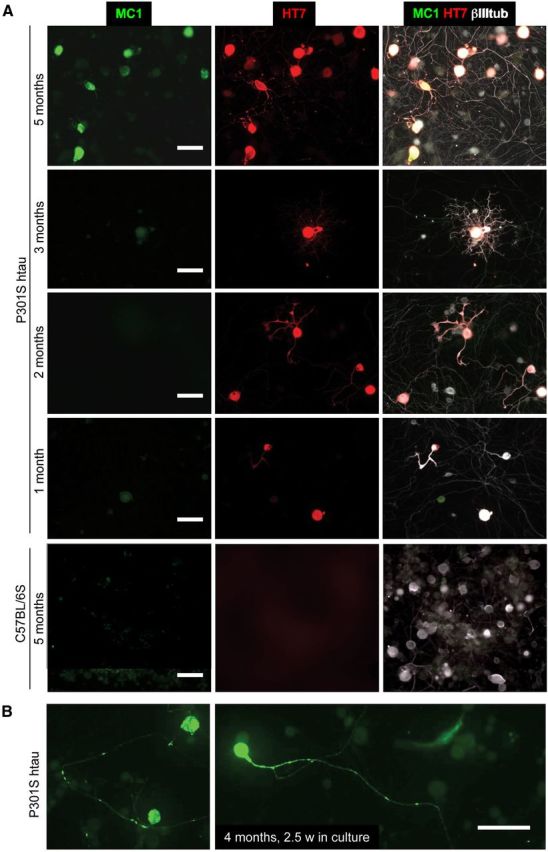
MC1 reactivity develops in culture at a late stage of pathology. A, DRG neurons cultured for 2 DIV from 1-, 2-, 3-, and 5-month-old P301S-htau or 5-month-old C57BL/6S mice were stained with antibody MC1 (green), HT7 (red); merged images also show labeling for βIII tubulin (white). B, DRG neurons from 4-month-old mice stained with MC1 (green). Note MC1-positive aggregates in the axons and cell bodies, Scale bars: 50 μm.
P301S-htau-expressing DRG neurons display aberrant morphologies
We next examined axon regeneration of P301S-htau+ve DRG neurons. Abnormal morphologies were observed in P301S-htau-expressing (HT7+ve) DRG neurons grown for 2 DIV from 1-, 3-, and 5-month-old P301S-htau transgenic mice (Fig. 4), while the growth of HT7−ve neurons present in the same cultures was similar to that of the nerve cells from age-matched C57BL/6S controls. The abnormal patterns consisted of short neurites with enlarged and amorphous growth cones, and thickened axonal processes with increased branching and accumulation of tau at the branch point (Fig. 4 A,B) as has been observed in neurons in tauopathies. These changes were already evident in neurons from 1-month-old mice. Moreover, P301S-htau HT7+ve neurons, in particular those from 3- and 5-month-old P301S-htau mice, showed disorganized MTs. For example, at the leading edge of the growth cone, MTs were not dynamically protruding towards the finger-like filopodia, but were bent and looped (Fig. 4C). Such structures are similar to those previously reported in resting growth cones that have lost their direction of growth due to structural changes of MTs (Conde and Cáceres, 2009; Lowery and Van Vactor, 2009), or in growth cones whose MTs have been overly stabilized due to lack of tip proteins or treatment with taxol (Gumy et al., 2013). Most interestingly, large axonal swellings similar to the spheroids found in human tauopathies (Murrell et al., 1999) were found along the axons, where MTs stained with an anti-βIII tubulin antibody seemed to have lost an ordered structure (Fig. 4D). A peculiar feature of dissociated DRG cultures from 5-month-old P301S-htau mice was the presence of structures that were highly immunoreactive for phospho-tau (PHF-1, AT8 or AT100) but which had reduced or absent βIII tubulin immunoreactivity and nuclear DNA staining (Fig. 4E). In some cases these structures were reminiscent of “ghost-like” tangles found in AD, where tau labeling shows the shape of a neuron with very little or no βIII tubulin labeling and no nuclear staining (Duyckaerts et al., 2009). To assess the impact of P301S-htau expression on axon growth, we quantified the length of the longest axon per neuron, and the areas of the associated growth cones. Axons from P301S-htau HT7+ve neurons were significantly shorter and displayed significantly larger growth cones compared with HT7−ve neurons from the same cultures or C57BL/6S nerve cells (Fig. 4F). This effect was already evident in cultures from 1-month-old mice, indicating that problems with reorganization of the MTs necessary for regeneration after injury (Gumy et al., 2013) may be an early response to the presence of P301S-htau.
Figure 4.
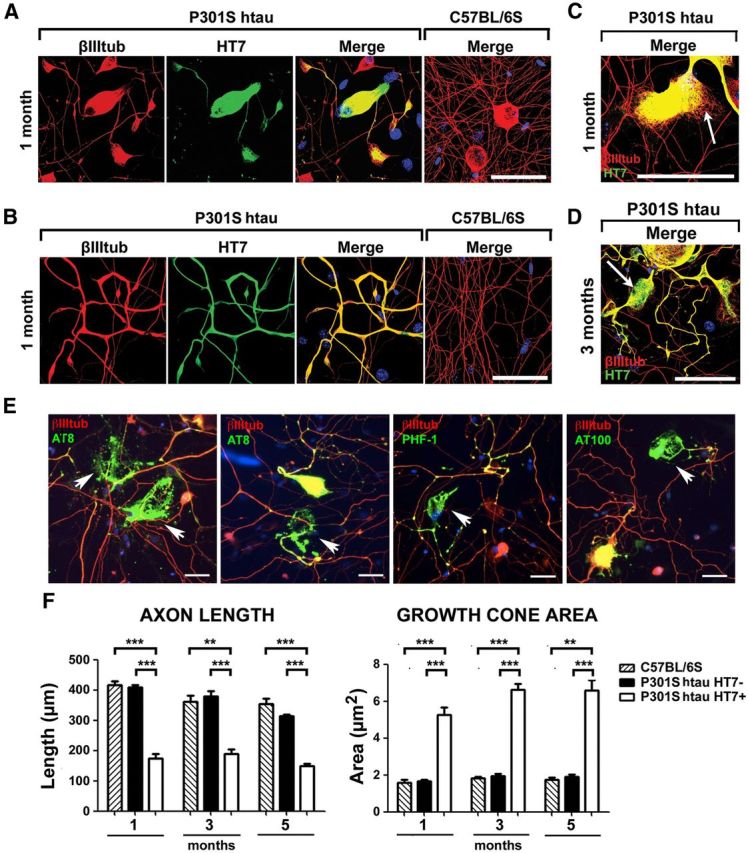
Cultured P301S-htau-expressing DRG neurons display early morphological abnormalities. Low-density dissociated DRG cultures were grown for 2 DIV, fixed, and immunolabeled with HT7 and anti-βIII tubulin antibodies. A–C, Enlarged and stunted growth cones with splayed MTs and thickened, looped, and bent axons are present in neurons from 1-month-old P301S-htau mouse but not C57BL/6S control. D, A spheroid bulge in an axon from a 3-month-old mouse. Spheroids are often found in human tauopathy brains. E, Residual tangles of aggregated tau in degenerating cells with little or no βIII tubulin (red) or nuclear stain (blue) in cultures from 5-month-old mice. Arrows point to typical tau deposits that stain for PHF-1, AT8, or AT100 (green). Merged images were captured with a fluorescence microscope using a 20× objective. Scale bars: A–D, 50 μm; E, 20 μm. F, Quantification of axon length and growth cone area in cultures from 1-, 3-, and 5-month-old mice show reduction in axon length and an increase in growth cone area in DRGs with P301S-htau compared with DRG without htau (P301S-HT7−ve) or from C57BL/6S control mice (mean ± SEM, n = 3 for C57BL/6S cultures and n = 4 for P301S-htau cultures, two-way ANOVA followed by Bonferroni post hoc adjustment). (Axon length: ANOVA C57BL/6S/P301S-HT7−ve/P301S-HT7+ve p = 4.5 × 10−5, 1m/3m/5m p = 1 × 10−5, Interactions p = 0.0368; p = 0.0002 (1m C57BL/6S/ P301S-HT7+ve), p = 1.3 × 10−5 (1m P301S-HT7−ve/P301S-HT7+ve), p = 0.0025 (3m C57BL/6S/ P301S-HT7+ve), p = 0.0005 (3m P301S-HT7−ve/P301S-HT7+ve); p = 0.0003 (5m C57BL/6S/ P301S-HT7+ve), p = 1.2 × 10−5 (5m P301S-HT7−ve/P301S-HT7+ve); Growth cone area: ANOVA C57BL/6S/P301S-HT7−ve/P301S-HT7+ve p = 0.0256, 1m/3m/5m p = 5 × 10−4, p = 0.0009 (1m C57BL/6S/P301S-HT7+ve), p = 0.0008 (1m P301S-HT7−ve/P301S-HT7+ve), p = 0.0002 (3m C57BL/6S/P301S-HT7+ve), p = 2.5 × 10−5 (3m P301S-HT7−ve/P301S-HT7+ve); p = 0.0018 (5m C57BL/6S/ P301S-HT7+ve), p = 0.0004 (5m P301S-HT7−ve/P301S-HT7+ve).
Mitochondrial trafficking is altered differently in P301S-htau-expressing neurons at different ages
To test whether there were any changes in mitochondrial transport in relation to the temporal development of tau pathology, mitochondria were imaged in the axons of live DRG neurons (2 DIV) after labeling with MitoTracker Orange (Fig. 5A, right). Recordings were blinded as to whether the neurons were HT7+ve or HT7−ve. After recording, the HT7 status of the imaged neurons was determined by immunofluorescence. Time-lapse recordings were analyzed by kymography, typical profiles of which are shown in Figure 5A). Quantification of stationary and anterogradely and retrogradely moving mitochondria showed that the percentage of stationary and motile mitochondria in P301S-htau HT7+ve neurons did not differ from that of C57BL/6S and P301S-htau HT7-negative axons in DRG neurons from 3-month-old mice (Fig. 5B). The distance traveled by mitochondria was also not altered in the three neuronal groups in cultures from 3-month-old or 5 month-old mice, nor did we note any systematic change in mitochondrial length between the three groups such as that recently reported (Duboff et al., 2012). However, in neurons from 5-month-old animals the percentage of stationary mitochondria was significantly increased by ∼40% in P301S-htau HT7+ve neurons compared with HT7−ve neurons, while movement was significantly reduced by ∼50% in the anterograde direction and by ∼30% in the retrograde direction compared with P301S-htau HT7−ve and C57BL/6S groups, respectively (Fig. 5B). Conversely, cumulative distribution analysis of average velocities among the three groups showed that in 3-month-old P301S-htau HT7+ve axons, mitochondria were moving significantly faster in both directions compared with C57BL/6S and P301S-htau HT7−ve neurons, whereas similar average velocities were found in the three experimental groups from 5-month-old mice (Fig. 5C), indicating that the remaining motile mitochondria were pacing along the axon with a speed comparable to controls. These data demonstrate that there are two independent outcomes of mitochondrial transport in tau-expressing DRG neurons: at early time points, when there are no detectable tau aggregates, mitochondria move slightly faster than in wild-type controls, whereas in neurons associated with filamentous tau aggregates, there is a significant global loss of mitochondrial movement.
Figure 5.

Mitochondrial movement is differentially altered in DRG neurons from P301S-htau mice. DRG neurons from 3-month-old or 5 month-old P301S-htau or C57BL/6S mice were cultured for 40 h, then incubated in medium containing 10 nm MitoTracker Orange for 5 min at 37°C just before imaging. P301S-htau+ve neurons were relocated after the experiment by immunolabeling with HT7 antibody. A, Representative time-lapse image series (1 frame every 5 s for 5 min) of C57BL/6S, P301S-htau HT7−ve, and P301S-htau HT7+ve neurons from 3- and 5-month-old mice converted into kymographs. Vertical tracks represent stationary mitochondria while oblique tracks depict the moving organelles. A typical snapshot of mitochondria in a P301S-htau HT7−ve and HT7+ve axon is shown to the right of the kymographs. Note that the density and shapes of polarized mitochondria in the axons are similar between the HT7−ve and HT7+ve neurons. B, Quantification of the proportion of stationary and moving mitochondria in the anterograde and retrograde directions (mean ± SEM, n = 11 axons from 3-month-old mice, n = 20 axons from 5-month-old mice; two-way ANOVA with Bonferroni post hoc adjustment; Stationary: ANOVA p = 5 × 10−7; p = 0.0006 (5m P301S-HT7+ve/5m P301S-HT7−ve), p = 5 × 10−7; (5m P301S-HT7+ve/5m C57BL/6S), p = 4.7 × 10−5 (5m P301S-HT7+ve/3m C57BL/6S), p = 0.1 × 10−7 ( (5m P301S-HT7+ve/3m P301S-HT7−ve), p = 5 × 10−7 (5m P301S-HT7+ve/3m P301S-HT7+ve), p = 0.0099 (5m P301S-HT7−ve/3m P301S-HT7−ve); Anterograde: ANOVA p = 2 × 10−6; p = 0.0003 (5m P301S-HT7+ve/5m P301S-HT7−ve), p = 2.1 × 10−5 (5m P301S-HT7+ve/5m C57BL/6S), p = 8 × 10−6 (5m P301S-HT7+ve/3m C57BL/6S), p = 0.0333 (5m P301S-HT7+ve/3m P301S-HT7+ve); Retrograde: ANOVA p = 0.0041, p = 0.0325 (5m P301S-HT7+ve/5m C57BL/6S), p = 0.0159 (5m P301S-HT7+ve/3m C57BL/6S), p = 0.0457. C, Analysis of cumulative anterograde and retrograde average velocity distributions. Cumulative distribution statistics: Anterograde: p = 0.0592 (3m C57BL/6S/3m P301S-HT7−ve), p = 2.5 × 10−5 (3m C57BL/6S/3m P301S-HT7+ve), p = 0.0221 (3m P301S-HT7−ve/3m P301S-HT7+ve); Retrograde: p = 0.7128 (3m C57BL/6S/3m P301S-HT7−ve), p = 1.12 × 10−5 (3m C57BL/6S/3m P301S-HT7+ve), p = 0.0008 (3m P301S-HT7−ve/3m P301S-HT7+ve); Average speed in both directions was increased in HT7+ve neurons from 3-month-old mice compared with HT7−ve neurons or C57BL/6S controls, but not in cultures from 5-month-old animals. The average speed of mitochondria in P301S-htau HT7+ve neurons from 3-month-old mice was also significantly greater than that from 5-month-old mice; p = 0.0199 (3m/5m P301S-HT7+ve Anterograde), p = 0.0418 (3m/5m P301S-HT7+ve Retrograde).
DRG cultures established from nonsymptomatic age develop tau pathology in vitro
Cultured neurons from DRGs would be most valuable for studying the development of tau pathology if its progression could be replicated in vitro. To examine whether neurons cultured from P301S-htau DRGs at early stages, before detection of tau modification, develop tau pathology, dissociated DRG cultures were established from 3-month-old P301S-htau mice and grown for ∼8 weeks. Coverslips were removed every few days and stained for the presence of AT8, HT7, and βIII tubulin reactivities (Fig. 6A shows an example of AT8+ve neurons from 3-month-old mice after 52 DIV). Despite starting off as AT8−ve cultures, within 2 weeks ∼10% of the HT7+ve neurons developed AT8+ve labeling (Fig. 6B), which was initiated in the soma and eventually spread through the axon. The percentage of AT8+ve neurons out of the HT7+ve cohort increased with time in culture, reaching ∼50% of HT7+ve neurons after 8 weeks (Fig. 6B), similar to the 50% value found when neurons were directly cultured from the DRGs of 5-month-old P301S-htau mice (Fig. 2). The pattern of AT8+ve labeling in the cell body ranged from being uniform to being patchy, aggregated, or forming ring-like structures (Fig. 6C), similar to profiles observed in histological sections of DRG from 5-month-old P301S-htau mice (Fig. 1A). These data indicate that the development of pathological forms of tau is recapitulated in vitro in DRG neurons from P301S-htau mice, and thus progression of tau pathology can be followed in vitro from early stages.
Figure 6.

DRG neurons from P301S-htau mice develop tau pathology during long-term culture in vitro. A, Profile of a typical long-lived AT8+ve/HT7+ve DRG neuron cultured from 3-month-old mice after 8 weeks in vitro. B, DRG neurons cultured from 3-month-old P301S-htau mice were fixed at the days indicated, and the proportion of HT7+ve neurons out of the total βIII tubulin+ve neuron population (●), or the proportion of AT8+ve neurons out of the total HT7+ve population (○) was quantified. Each point is the mean value of replicates from one culture of three mice. The lines depict a linear regression and the dashed lines show the 95% confidence intervals. For ratio of HT7+ve/βIII tub+ve: R2 = 0.08, F test for slope being different from zero, p = 0.32 (not significant); for AT8/HT7+ve: R2 = 0.71, F test for slope being different from zero, p = 0.0002. C, Detail of three adjacent neurons stained with AT8 (red) and βIII tubulin (white) after 8 weeks, showing typical profiles of pathological tau: *, AT8+ve large cytoplasmic aggregates; **, AT8+ve small cytoplasmic aggregates; ***, AT8+ve cytoplasmic perinuclear crescent. D, E, DRG neurons from 5-month-old P301S-htau mice cultured for 73 d. Left, Quantification of the proportion of AT8+ve (D) and AT100+ve (E) neurons out of the total HT7+ve population (○), and the proportion of HT7+ve neurons out of the total βIII tubulin+ve neuron population (●). Each point shows the mean value ± SEM independent cultures from three to four mice except for the result at 73 d, which shows the average ± range from two mice. Points without error bars are because the errors were smaller than the size of the point. The lines depict a linear regression through the total cohort of raw values for each condition (17 data points). For ratio of AT8/HT7+ve: R2 = 0.36, F test for slope being different from zero, p = 0.0173; For overall ratio of AT100/HT7+ve: R2 = 0.19, F test for slope being different from zero, p = 0.0993; HT7+ve/βIII tub+ve in samples stained for AT8, p = 0.052; AT100, p = 0.062 (not significant).
We also examined whether we could maintain long-term cultures of neurons from 5-month-old mice, when DRG neurons show tau abnormalities, thereby extending their lifetime to an age equivalent to that of 7-month-old mice, which is beyond the age the animals are permitted to survive. Over 2 months in culture the number of AT8+ve neurons significantly decreased relative to the total number HT7+ve neurons (p = 0.0173), indicating that neurons with htau hyperphosphorylated at this site die preferentially. For AT100+ve neurons, the results were more variable; in two experiments there was a similar extent of loss of neurons as that found for AT8+ve neurons (p = 0.0413), but in two other experiments the decrease over time did not reach significance, maybe because some neurons were still developing the AT100 epitope while others were dying. The reduction in the percentage of total HT7+ve neurons relative to that of βIII tub+ve population was also not significant (p = 0.052–0.062), most likely because of the relatively small contribution made by AT8/AT100+ve neurons to the total neuron population present. These data indicate that long-term cultures from 5-month-old animals may be useful to study the mechanisms of tau-induced cell death mainly related to the decrease in the population of neurons that are stained by AT8.
Long-term treatment of DRG cultures with the O-GlcNAcase inhibitor SEG28019 reduces PHF-1+ve labeling
To investigate the feasibility of using P301S-htau+ve DRG neurons as a platform for testing drugs that may ameliorate tau pathology, we treated the cultures with SEG28019, a novel inhibitor of O-GlcNAcase, the enzyme that removes O-GlcNAcylated groups from serines and threonines (Hart et al., 2011). Inhibition of this enzyme has been shown to increase protein O-GlcNAcylation in lieu of phosphorylation (Hart et al., 2011). Tau is normally modified by O-GlcNAc at some of the same sites that are hyperphosphorylated in AD and other tauopathies, and the amount of tau O-GlcNAcylation is consistently reduced in AD brains, leading to the idea that O-GlcNAc inhibitors might prevent tau phosphorylation at specific sites and ameliorate tau pathology (Lefebvre et al., 2003; Liu et al., 2004). One epitope that is differentially O-GlcNAcylated in tau is Ser396, the motif recognized by the antibody PHF-1 (Yuzwa et al., 2008, 2012; Yu et al., 2012). We added 10 μm SEG28019 to the growth medium of DRG neurons cultured from 3-month-old P301S-htau mice over 7 weeks and tested whether there were any changes in steady-state tau phosphorylation at the PHF-1 epitope. Figure 7A shows that total O-GlcNAcylation was significantly higher in SEG28019-treated neurons compared with untreated cohorts. When the intensity of PHF-1 immunoreactivity was measured in individual HT7+ve neurons, a significant reduction in PHF-1 labeling relative to HT7 labeling intensity was evident in the SEG28019-treated cohort compared with the untreated control (Fig. 7B). This change was subtle in that there was no significant difference between the two treatment groups when total PHF-1 immunofluorescence was averaged; the difference was only revealed when the intensity of PHF-1 labeling was normalized to that of anti-human tau HT7 labeling intensity, indicating that some, but not all, of the neurons are affected by the drug at any one time. Thus, using DRG neurons from P301S-htau mice, we have teased out small differential effects of the OGA inhibitor SEG28019 after long-term treatment, similar to the period of treatment others have applied in vivo. Although a subtle yet significant change was observed for the SEG28019 in DRG neurons, this may be sufficient to elicit a change in disease pathology and hence explain the observed benefits of O-GlcNAcase inhibition in tauopathy models (Yuzwa et al., 2012).
Figure 7.

The O-GlcNAcase inhibitor SEG28019 reduces PHF-1 immunoreactivity in DRG neurons. A, DRG neurons were cultured from 5-month-old P301S-htau mice for 6 d after which SEG28019 (SEG; 10 μm) was added for 22 h. Cells were fixed and stained for the presence of O-GlcNAc modification using the RL2 antibody (green) and anti-βIII tubulin (white) and visualized by fluorescence microscopy. Bar graph shows average cytoplasmic intensity of RL2 labeling from two independent cultures. Mean ± SEM, NoSEG28019, n = 91 neurons; +SEG28019, n = 151 neurons. ANOVA p = 1.48 × 10−15; t test, p = 2.16 × 10−7. Scale bar, 20 μm. B, DRG neurons were cultured from two different litters of 3-month-old P301S-htau mice. SEG28019 (10 μm) was added at day two and freshly added two times a week over 7 weeks. Cultures were fixed and stained with anti-phospho tau antibody PHF-1 and for human tau (HT7) and visualized by fluorescence microscopy. The intensity of PH-1 labeling was normalized to the intensity of HT7 labeling to offset fluctuations due to different amounts of htau expression. The graphs show the ratios plotted as fractional cumulative distribution plots for three independent cultures from litter a (left and middle) and litter b (right). Note that there is a higher fraction of neurons with lower PHF-1/HT7 intensity in the SEG28019-treated cultures compared with the nontreated cultures. Data were analyzed using a nonparametric Mann–Whitney test. Left plot, No SEG28019 n = 89, +SEG28019 n = 59, p = 0.0029. Middle plot, No SEG28019 n = 91, +SEG28019 n = 91, p = 0.0013. Right plot, No SEG28019 n = 150, + SEG28019 n = 146, p = 0.0012.
Discussion
This study shows that in transgenic P301S-htau mice, htau+ve DRG neurons develop tau pathology with similar characteristics to those found in human tauopathies. This pathology was associated with some evidence of reduced sensory function. Moreover, htau+ve DRG neurons cultured from adult P301S-htau mice of different ages displayed the same developmental pattern of tau pathology as DRG and CNS neurons in vivo. Furthermore, functionally, axonal transport of mitochondria was differentially altered in the htau+ve neurons depending on whether tau was aggregated or just hyperphosphorylated. Remarkably, long-term cultures of DRG neurons from presymptomatic P301S-htau transgenic mice recapitulated aspects of progressive tau pathology as they matured, as indicated by increasing AT8 labeling over 2 months starting with neurons from 3-month-old mice. Furthermore, there was preferential neuronal death of AT8+ve neurons and some AT100+ve neurons over 2 months when starting with neurons from advanced stage disease (5-month-old mice). Thus, these cultures provide a unique system where tau filament formation is self-generating without a requirement for special manipulations or seeding of exogenous tau, differently from cultures recently described (Vogelsberg-Ragaglia et al., 2000; Sato et al., 2002; Ferrari et al., 2003; Pérez et al., 2003; Alonso Adel et al., 2006; Iliev et al., 2006; Wang et al., 2007; Guo and Lee, 2011, 2013), and neuronal death associated with hyperphosphorylated/conformationally altered htau occurs spontaneously as we showed in the cortex of the same transgenic line in vivo (Hampton et al., 2010). Our results also differ from those described in a wild-type htau transgenic mouse, where no increase in hyperphosphorylated or aggregated tau in DRG neurons was observed (Nuydens et al., 2002).
Although sensory deficits are not hallmarks of human tauopathies, there may be prevalent tau pathology in the PNS, as indicated by studies where tau pathology was found in lumbar DRG neurons of several PSP patients (Nishimura et al., 1993) and in upper cervical ganglia of AD patients (Kawasaki et al., 1987). To investigate the consequences of tau pathology in the DRGs, we tested whether expression of P301S-htau was associated with sensory deficit, studying 1- and 3-month-old mice to avoid complications due to the motor neuron death and motor phenotype that occurs at 5 months of age. At 1 month, when there was little AT8+ve labeling, no sensory deficits were detected. However, by 3 months of age, when 50% of the HT7+ve neurons were AT8+ve, thermal nociceptive thresholds were normal, but there was a significant loss of mechanosensation. Since there was little detectable AT100 labeling at 3 months, which reflects tau filament formation in the P301S-htau mice (Allen et al., 2002), these results indicate that more advanced pathology was not necessary for revealing sensory deficits, as shown previously in this mouse model (Scattoni et al., 2010). Moreover, the late appearance of MC1+ve-positive neurons suggests that tau hyperphosphorylation at AT8 and PHF-1 epitopes occurs in DRG neurons before this disease-linked conformational alteration is detectable. In keeping with the observation of a sensory-specific impairment, we show that P301S-htau+ve neurons are IB4−ve, whereas IB4+ve neurons are mainly small-diameter nonpeptidergic heat-sensitive nociceptors (Silverman and Kruger, 1988; Snider and McMahon, 1998; Stucky and Lewin, 1999). Whether the P301S-htau+ve neurons belong to one class or modality will require closer scrutiny.
Neurons expressing P301S-htau showed several aberrant patterns of axonal outgrowth reminiscent of pathological forms found in human tauopathies. Most notably, spheroids formed within axons, and the MTs were splayed and bent in the growth cones, indicating the predominance of static over dynamic MTs, similar to the patterns we (data not shown) and others (Letourneau and Ressler, 1984) observed when low (10–100 nm) concentrations of taxol were added to enhance MT stability. Where axon growth occurred, the pattern of growth was interrupted by kinks and bends, and the overall axon length was markedly reduced, as previously observed in retinal explants from P301S-htau mice (Gasparini et al., 2011). Also similar to human tauopathies, some degenerating neurons cultured from 5-month-old mice lost most of their MT labeling and in some cases nuclear staining was also missing, leaving AT8/AT00+ve deposits possibly reminiscent of ghost-like tangles. To our knowledge, this has not been previously reported in cultured neurons. Hence, reorganization of the cytoskeleton to provide dynamic MTs necessary for proper regenerative outgrowth appears to be deficient in the transgenic neurons. Some of the aberrant patterns could have been due to expression of excess tau rather than mutant tau (Nuydens et al., 2002), but these changes were not because of overexpression per se as we did not observe them in DRG neurons cultured from 5-month-old Thy1.2-YFP transgenic mice, where YFP is overexpressed (data not shown). In keeping with this notion, reduced outgrowth length was already pronounced in DRG neurons from 1-month-old P301S-htau mice, a stage that is before consistent tau phosphorylation as detected by AT8 or AT100. Whether this deficit is linked to phosphorylation at the PHF-1 epitope, which is already present in ∼50% of the HT7+ve neurons at 1 month of age, will require further study.
To test for functional tau-related deficits at the cellular level, we studied the movement of polarized mitochondria in the axons of DRG neurons grown for 2 DIV. Unlike reports of axonal transport in other htau transgenic mice (Spittaels et al., 1999; Leroy et al., 2007; Ittner et al., 2008), the number or shape of mitochondria in DRG axons did not differ between P301S-htau+ve, P301S-htau−ve, and C57BL/6S neurons. Hence, delivery of polarized mitochondria to axons that are >100 μm long was not limited, nor was there any evidence for the accumulation of mitochondria in cell bodies, as found in early differentiated N2A cells or in primary neurons (Ebneth et al., 1998; Stamer et al., 2002). Instead, mitochondrial movement displayed two distinct patterns: in HT7+ve neurons from 5-month-old mice, there was an increase of ∼50% in the proportion of stationary mitochondria compared with HT7−ve neurons from the same culture or to control neurons. This occurred with a concomitant reduction in both anterograde and retrograde moving fractions. There were no changes in these parameters in neurons from 3-month-old mice. Interestingly, in vivo studies in the optic nerve found alterations in anterograde and retrograde transport of cholera toxin B at both 3 and 5 months (Bull et al., 2012). It is likely that cholera toxin B and mitochondria use different mechanisms of transport (e.g., endosomal vs Milton/Miro; Vershinin et al., 2007; Saxton and Hollenbeck, 2012), different supply of ATP (Zala et al., 2013), and/or that the timescale of the experiments and travel distance reveal different aspects of axonal transport pathology. Only in P301S-htau HT7+ve DRG axons from 3-month-old, but not 5-month-old mice, a proportion of mitochondria moved significantly faster compared with C57BL/6S and P301S-tau HT7−ve neurons. There were no differences in the run length between the three groups at any age, so the increased velocity was an intrinsic feature. The reasons underlying mitochondria increased velocity in 3-month-old DRG axons remain unclear. A related phenomenon was recently reported for mitochondria in the tibial nerve of P301L tau knockin mice with no tau aggregation (Gilley et al., 2011), where an increased fraction of anterogradely moving mitochondria was found in young mice, switching to a decreased fraction in old mice. Thus, opposing switches in the rate or fraction of moving mitochondria, are not necessarily related to differences between intact nerves and cultured axons. Another reason for the slightly higher motility of mitochondria in axons from our 3-month-old mice may relate to altered axonal metabolic demands (Miller and Sheetz, 2004). Our preliminary evidence suggests that most mitochondria are polarized in the axons of HT7+ve neurons regardless of age, but it is possible that the metabolic demands of the axon change depending on the phosphorylation status of P301S-htau, such that initially the cell increases anterograde mitochondrial movement to provide higher energetic supply, but with the progression of tau pathology, this regulatory step fails.
To demonstrate the usefulness of DRG neurons as a platform for drug testing, we used SEG28019, a novel inhibitor of O-GlcNAcase (Hart et al., 2011; Horne et al., 2011). Inhibition of O-GlcNAcase is predicted to increase tau O-GlcNAcylation inhibiting its detrimental phosphorylation in specific sites such as the epitope of PHF-1 antibody Ser 396. However, recent studies (Yuzwa et al., 2008, 2012; Yu et al., 2012) could not detect a significant reduction in tau phosphorylation following O-GlcNAcase inhibition with Thiamet G leading to the conclusion that its effect was possibly not mediated via tau phosphorylation. Here we show that SEG28019 reduced tau phosphorylation at the PHF-1 epitope, while preliminary results showed no change in AT8 phosphorylation or labeling of the MC1 epitope. These observations show how specific changes can be dissected out in our model with developing tau pathology.
In conclusion, we show a highly reproducible and versatile neuronal model where tau hyperphosphorylation and aggregation develop without additional manipulation. This model will be useful for investigating mechanisms of tau-related neuronal dysfunction, and to test new therapies for tauopathies.
Footnotes
This work was supported by a UK Medical Research Council Industrial Case PhD studentship (M.G.S., M.M.) with a contribution from GlaxoSmithKline, Cambridge Home and EU Scholarship Scheme PhD studentship (D.K.), the UK Medical Research Council (R.A.C., M.G.), Summit (Technology Strategy Board Biomedical Catalyst Fund, Project 130996), and the Alzheimer's Research UK (M.G.S., M.M.). We Are grateful to Dr. Laura F. Gumy (University of Cambridge, UK) for advice on culturing dissociated adult DRG neurons and Dr. Richard Eva (University of Cambridge, UK) for both technical advice on live imaging and for helpful discussions, Dr. Peter Davies (Albert Einstein College of Medicine, New York) for the gift of the PHF-1 antibody, Isabelle Lavenir for help with transgenic mice, and Drs. Stephen D. Skaper (University of Padua, Italy) and Jill C. Richardson (GlaxoSmithKline, UK) for their comments on the project.
J.T. and G.H. are employed by Summit plc, and provided the OGA inhibitor (SEG28019) and financial support for the OGAi experiment. M.G.S. received financial grant support from Summit plc to carry out the OGAi experiment.
References
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso Adel C, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A. 2006;103:8864–8869. doi: 10.1073/pnas.0603214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernat J, Mandelkow EM, Schröter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MJ, Morris R, McArdle A, Jackson MJ, Thippeswamy T. The effects of L-NAME on neuronal NOS and SOD1 expression in the DRG-spinal cord network of axotomised Thy 1.2 eGFP mice. Neuron Glia Biol. 2011;7:129–141. doi: 10.1017/S1740925X12000051. [DOI] [PubMed] [Google Scholar]
- Bull ND, Guidi A, Goedert M, Martin KR, Spillantini MG. Reduced axonal transport and increased excitotoxic retinal ganglion cell degeneration in mice transgenic for human mutant P301S tau. PLoS One. 2012;7:e34724. doi: 10.1371/journal.pone.0034724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins MJ, Reddy PH. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim Biophys Acta. 2011;1812:507–513. doi: 10.1016/j.bbadis.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116:207–225. doi: 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- Conde C, Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- Crowther RA. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci U S A. 1991;88:2288–2292. doi: 10.1073/pnas.88.6.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delobel P, Lavenir I, Fraser G, Ingram E, Holzer M, Ghetti B, Spillantini MG, Crowther RA, Goedert M. Analysis of tau phosphorylation and truncation in a mouse model of human tauopathy. Am J Pathol. 2008;172:123–131. doi: 10.2353/ajpath.2008.070627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboff B, Götz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75:618–632. doi: 10.1016/j.neuron.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone TL, Stokin GB, Lillo C, Rodrigues EM, Westerman EL, Williams DS, Goldstein LS. Axonal stress kinase activation and tau misbehavior induced by kinesin-1 transport defects. J Neurosci. 2009;29:5758–5767. doi: 10.1523/JNEUROSCI.0780-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari A, Hoerndli F, Baechi T, Nitsch RM, Götz J. beta-Amyloid induces paired helical filament-like tau filaments in tissue culture. J Biol Chem. 2003;278:40162–40168. doi: 10.1074/jbc.M308243200. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Crowther RA, Martin KR, Berg N, Coleman M, Goedert M, Spillantini MG. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiol Aging. 2011;32:419–433. doi: 10.1016/j.neurobiolaging.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Gilley J, Seereeram A, Ando K, Mosely S, Andrews S, Kerschensteiner M, Misgeld T, Brion JP, Anderton B, Hanger DP, Coleman MP. Age-dependent axonal transport and locomotor changes and tau hypophosphorylation in a “P301L” tau knockin mouse. Neurobiol Aging. 2011;33 doi: 10.1016/j.neurobiolaging.2011.02.014. 621 e621–621 e615. [DOI] [PubMed] [Google Scholar]
- Gladman SJ, Huang W, Lim SN, Dyall SC, Buddy S, Kang JX, Knight MM, Priestly JV, Michael-Titus AT. Improved outcome after peripheral nerve injury in mice with measured levels of endogenous omega-3 polyunsaturated fatty acids. J Neurosci. 2012;32:563–571. doi: 10.1523/JNEUROSCI.3371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Crowther RA, Vanmechelen E, Probst A, Götz J, Bürki K, Cohen P. Molecular dissection of the paired helical filament. Neurobiol Aging. 1995;16:325–334. doi: 10.1016/0197-4580(95)00017-9. [DOI] [PubMed] [Google Scholar]
- Goedert M, Ghetti B, Spillantini MG. Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006254. doi: 10.1101/cshperspect.a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumy LF, Bampton ET, Tolkovsky AM. Hyperglycaemia inhibits Schwann cell proliferation and migration and restricts regeneration of axons and Schwann cells from adult murine DRG. Mol Cell Neurosci. 2008;37:298–311. doi: 10.1016/j.mcn.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Gumy LF, Chew DJ, Tortosa E, Katrukha EA, Kapitein LC, Tolkovsky AM, Hoogenraad CC, Fawcett JW. The kinesin-2 family member KIF3C regulates microtubule dynamics and is required for axon growth and regeneration. J Neurosci. 2013;33:11329–11345. doi: 10.1523/JNEUROSCI.5221-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VM. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett. 2013;587:717–723. doi: 10.1016/j.febslet.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton DW, Webber DJ, Bilican B, Goedert M, Spillantini MG, Chandran S. Cell-mediated neuroprotection in a mouse model of human tauopathy. J Neurosci. 2010;30:9973–9983. doi: 10.1523/JNEUROSCI.0834-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne G, Wilson FX, Tinsley J, Williams DH, Storer R. Iminosugars past, present and future: medicines for tomorrow. Drug Discov Today. 2011;16:107–118. doi: 10.1016/j.drudis.2010.08.017. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Iliev AI, Ganesan S, Bunt G, Wouters FS. Removal of pattern-breaking sequences in microtubule binding repeats produces instantaneous tau aggregation and toxicity. J Biol Chem. 2006;281:37195–37204. doi: 10.1074/jbc.M604863200. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Fath T, Ke YD, Bi M, van Eersel J, Li KM, Gunning P, Götz J. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc Natl Acad Sci U S A. 2008;105:15997–16002. doi: 10.1073/pnas.0808084105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(SICI)1097-4547(19970415)48:2<128::AID-JNR5>3.0.CO%3B2-E. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Murayama S, Tomonaga M, Izumiyama N, Shimada H. Neurofibrillary tangles in human upper cervical ganglia. Morphological study with immunohistochemistry and electron microscopy. Acta Neuropathol. 1987;75:156–159. doi: 10.1007/BF00687076. [DOI] [PubMed] [Google Scholar]
- Lefebvre T, Ferreira S, Dupont-Wallois L, Bussière T, Dupire MJ, Delacourte A, Michalski JC, Caillet-Boudin ML. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins–a role in nuclear localization. Biochim Biophys Acta. 2003;1619:167–176. doi: 10.1016/S0304-4165(02)00477-4. [DOI] [PubMed] [Google Scholar]
- Leroy K, Bretteville A, Schindowski K, Gilissen E, Authelet M, De Decker R, Yilmaz Z, Buée L, Brion JP. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am J Pathol. 2007;171:976–992. doi: 10.2353/ajpath.2007.070345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneau PC, Ressler AH. Inhibition of neurite initiation and growth by taxol. J Cell Biol. 1984;98:1355–1362. doi: 10.1083/jcb.98.4.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery LA, Van Vactor D. The trip of the tip: understanding the growth cone machinery. Nat Rev Mol Cell Biol. 2009;10:332–343. doi: 10.1038/nrm2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc. 2007;2:152–160. doi: 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, Goedert M. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol. 1999;58:1207–1226. doi: 10.1097/00005072-199912000-00002. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Namba Y, Ikeda K, Akiguchi I, Oda M. Neurofibrillary tangles in the neurons of spinal dorsal root ganglia of patients with progressive supranuclear palsy. Acta Neuropathol. 1993;85:453–457. doi: 10.1007/BF00230481. [DOI] [PubMed] [Google Scholar]
- Nuydens R, Van Den Kieboom G, Nolten C, Verhulst C, Van Osta P, Spittaels K, Van den Haute C, De Feyter E, Geerts H, Van Leuven F. Coexpression of GSK-3beta corrects phenotypic aberrations of dorsal root ganglion cells, cultured from adult transgenic mice overexpressing human protein tau. Neurobiol Dis. 2002;9:38–48. doi: 10.1006/nbdi.2001.0454. [DOI] [PubMed] [Google Scholar]
- Pérez M, Hernández F, Lim F, Díaz-Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis. 2003;5:301–308. doi: 10.3233/jad-2003-5405. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Sato S, Tatebayashi Y, Akagi T, Chui DH, Murayama M, Miyasaka T, Planel E, Tanemura K, Sun X, Hashikawa T, Yoshioka K, Ishiguro K, Takashima A. Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J Biol Chem. 2002;277:42060–42065. doi: 10.1074/jbc.M202241200. [DOI] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125:2095–2104. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scattoni ML, Gasparini L, Alleva E, Goedert M, Calamandrei G, Spillantini MG. Early behavioural markers of disease in P301S tau transgenic mice. Behav Brain Res. 2010;208:250–257. doi: 10.1016/j.bbr.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Silverman JD, Kruger L. Lectin and neuropeptide labeling of separate populations of dorsal root ganglion neurons and associated “nociceptor” thin axons in rat testis and cornea whole-mount preparations. Somatosens Res. 1988;5:259–267. doi: 10.3109/07367228809144630. [DOI] [PubMed] [Google Scholar]
- Snider WD, McMahon SB. Tackling pain at the source: new ideas about nociceptors. Neuron. 1998;20:629–632. doi: 10.1016/S0896-6273(00)81003-X. [DOI] [PubMed] [Google Scholar]
- Spillantini M, Crowther RA, Goedert M. Comparison of the neurofibrillary patholofy in Alzheimer's disease and familial presenile dementia with tangles. Acta Neuropath. 1996;92:42–48. doi: 10.1007/s004010050487. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–622. doi: 10.1016/S1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucky CL, Lewin GR. Isolectin B(4)-positive and -negative nociceptors are functionally distinct. J Neurosci. 1999;19:6497–6505. doi: 10.1523/JNEUROSCI.19-15-06497.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vershinin M, Carter BC, Razafsky DS, King SJ, Gross SP. Multiple-motor based transport and its regulation by Tau. Proc Natl Acad Sci U S A. 2007;104:87–92. doi: 10.1073/pnas.0607919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Bruce J, Richter-Landsberg C, Zhang B, Hong M, Trojanowski JQ, Lee VM. Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell. 2000;11:4093–4104. doi: 10.1091/mbc.11.12.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Ichiyama RM, Zhao R, Andrews MR, Fawcett JW. Chondroitinase combined with rehabilitation promotes recovery of forelimb function in rats with chronic spinal cord injury. J Neurosci. 2011;31:9332–9344. doi: 10.1523/JNEUROSCI.0983-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YP, Biernat J, Pickhardt M, Mandelkow E, Mandelkow EM. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci U S A. 2007;104:10252–10257. doi: 10.1073/pnas.0703676104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging. 2000;21:719–727. doi: 10.1016/S0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Goedert M. Sequential phosphorylation of tau protein by cAMP-dependent protein kinase and SAPK4/p38delta or JNK2 in the presence of heparin generates the AT100 epitope. J Neurochem. 2006;99:154–164. doi: 10.1111/j.1471-4159.2006.04052.x. [DOI] [PubMed] [Google Scholar]
- Yu Y, Zhang L, Li X, Run X, Liang Z, Li Y, Liu Y, Lee MH, Grundke-Iqbal I, Iqbal K, Vocadlo DJ, Liu F, Gong CX. Differential effects of an O-GlcNAcase inhibitor on tau phosphorylation. PLoS One. 2012;7:e35277. doi: 10.1371/journal.pone.0035277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- Zala D, Hinckelmann MV, Yu H, Lyra da Cunha MM, Liot G, Cordelières FP, Marco S, Saudou F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152:479–491. doi: 10.1016/j.cell.2012.12.029. [DOI] [PubMed] [Google Scholar]