

Abstract
Sialic acid-binding Ig-like lectins (Siglecs) are members of the Ig superfamily that recognize sialic acid residues of glycoproteins. Siglec-E is a mouse CD33-related Siglec that preferentially binds to sialic acid residues of the cellular glycocalyx. Here, we demonstrate gene transcription and protein expression of Siglec-E by cultured mouse microglia. Siglec-E on microglia inhibited phagocytosis of neural debris and prevented the production of superoxide radicals induced by challenge with neural debris. Soluble extracellular Siglec-E receptor protein bound to the neural glycocalyx. Coculture of mouse microglia and neurons demonstrated a neuroprotective effect of microglial Siglec-E that was dependent on neuronal sialic acid residues. Increased neurotoxicity of microglia after knockdown of Siglece mRNA was neutralized by the reactive oxygen species scavenger Trolox. Data suggest that Siglec-E recognizes the intact neuronal glycocalyx and has neuroprotective function by preventing phagocytosis and the associated oxidative burst.
Introduction
Sialic acid-binding Ig-like lectin-E (Siglec-E) is a CD33-related member of the mouse Siglec family (Crocker et al., 2007). Siglec-E is broadly expressed on tissue macrophages, splenic dendritic cells, neutrophils, and a subset of mature natural killer cells (Zhang et al., 2004). Previous studies suggested that Siglec-E mainly bound to α2–8-linked disialic acid residues of the glycocalyx but also recognized α2–3-linked and weakly α2–6-linked sialyllactose residues (Zhang et al., 2004). Recent data demonstrate that Siglec-E binds to a wide range of sialyloligosaccharides with a preference for N-acetyl neuraminic acid (Redelinghuys et al., 2011). Siglec-E consists of three extracellular Ig-like domains, a transmembrane region, and a cytoplasmic tail bearing one immunoreceptor tyrosine-based inhibitory motif (ITIM) and one ITIM-like domain that recruits the inhibitory phosphatases SHP-1/SHP-2 (Crocker et al., 2007). Interestingly, the inhibitory activity of ITIMs counterbalances the immunoreceptor tyrosine-based activation motif (ITAM) signaling of DAP12/TYROBP (Linnartz and Neumann, 2013).
Recent data of gene expression profiles from distinct mouse tissue macrophages suggest that Siglece is also detected in microglia (Gautier et al., 2012), the resident immune cells of the CNS. Microglia execute innate immunity, participate in adaptive immune responses, and facilitate tissue homeostasis by clearance of apoptotic cells, cellular debris, and unwanted synaptic structures (Neumann et al., 2009; Schafer et al., 2012). Phagocytic clearance of apoptotic neurons by microglia is mediated via triggering receptor expressed on myeloid cells-2 and DAP12 in vitro (Wakselman et al., 2008; Linnartz and Neumann, 2013). Furthermore, microglial complement receptor-3 signaling via DAP12 is involved in synaptic pruning and neurons during development (Wakselman et al., 2008; Schafer et al., 2012). The ITAM-containing adaptor DAP12 also leads to the activation of the phagocytic NADPH oxidase NOX2 and the production of reactive oxygen species (ROS) (Graham et al., 2007), a process that is called oxidative burst.
We now detected gene transcription and protein expression of Siglec-E in microglia. Knockdown of Siglece mRNA of microglia prevented phagocytic uptake of neural debris and the oxidative burst. Siglec-E recognized sialic acid residues on the neural glycocalyx and had neuroprotective effects by preventing ROS production in a microglia–neuron coculture.
Materials and Methods
Cultured microglia, astrocytes, and neurons.
Primary microglia, astrocytes, and neurons were prepared from brains of C57BL/6 mice of either sex as described previously (Gorlovoy et al., 2009). Embryonic stem cell-derived microglia were used as the microglial line (Beutner et al., 2010). Microglia were treated with 500 ng/ml lipopolysaccharide (LPS; Sigma-Aldrich), 100 U/ml mouse interferon (IFN)-γ (R&D Systems), 1000 U/ml mouse IFN-α (Hycult Biotech), or 20 ng/ml tumor necrosis factor-α (TNF-α; R&D Systems).
Gene transcript analysis of cells.
Total RNA was isolated from cells by RNeasy Mini (Qiagen), reverse transcribed, and amplified by PCR. Samples without cDNA and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were applied as controls.
Flow cytometry analyses.
Microglial cells were mechanically detached and incubated with a Siglec-E antibody (1:200; MBL International) followed by a phycoerythrin (PE)-conjugated secondary antibody (Dianova). For flow cytometry of ex vivo microglia, cells were isolated from C57BL/6 mice of either sex by density gradient. For double labeling of microglia, cells were first incubated for Fc block (anti-CD16/CD32; BD Biosciences) and then stained with biotinylated anti-Siglec-E (1:200; MBL International), followed by Alexa Fluor 647-conjugated streptavidin (Dianova), a PE-conjugated anti-CD11b (eBioscience), and a V450-conjugated anti-CD45 (BD Horizon). Isotype-matched control antibodies (BD Biosciences) were used as controls. Analysis was done with a FACS Calibur or FACS CantoII flow cytometer and FlowJo Software (BD Biosciences).
Plasmid construction, viral particle production, and transduction.
Plasmids containing the Gfp (Invitrogen) or Siglece gene linked to the GFP-variant gene citrine (gift from Prof. Fleischer, Hamburg, Germany) were cloned into the lentiviral backbone pll3.7 behind a cytomegalovirus promoter together with a cassette of phosphoglycerate-kinase promoter and neomycin resistance gene. Constructs for lentiviral knockdown of Siglece (shRNASigE1: TRCN0000094526, target sequence 5′-CCCAATTCGTAAAGCAGTGAA-3′; shRNASigE2: TRCN0000094527, target sequence 5′-GCCACAAATAACCCAATTCGT-3′) were obtained from a knockdown library in a pLKO.1 backbone. HEK293FT cells were transfected with the targeting and packaging plasmids (Invitrogen). Viral particles were collected and applied to the target cells three times.
Phagocytosis of neural debris.
Primary neural cells were incubated with 40 nm okadaic acid and mechanically disrupted to obtain neural debris. Microglia were incubated with prestained (celltracker cM-DiI; Invitrogen) neural debris for 2 h at 37°C. Cells were fixed and incubated with an anti-iba1 antibody (1:1000; Wako Chemicals), followed by a secondary Alexa Fluor 488-conjugated antibody (Invitrogen). Images were obtained with a confocal laser scanning microscope (Fluoview 1000; Olympus), and 3D reconstruction was performed. To determine the ratio of cells having ingested fluorescently labeled material, all cells of the collected images (n = 21) per experimental group were quantified using NIH ImageJ software.
Microglia–neuron coculture and immunocytochemistry.
Primary cultured neurons were either untreated or treated with neuraminidase (25 mU/ml, EC3.2.1.18; Roche) for 2.5 h to remove sialic acids from the cell surface. For ROS scavenging experiments, 40 nm Trolox (Sigma-Aldrich) was added to the medium before starting the coculture. The coculture and immunocytochemistry was performed as described previously (Wang and Neumann, 2010). The mean length of βIII-tubulin-positive neurites or the density of βIII-tubulin-positive cell bodies were analyzed versus iba1-positive cells in all collected images (n = 15) per experimental group using NIH ImageJ/NeuronJ software.
Detection of superoxide by dihydroethidium and cytokine transcript analysis during phagocytosis of neural debris.
Microglia were treated with 5 μg/μl neural debris for 1 h for superoxide measurement or for 16 h for RNA isolation. For measurement of superoxide, cells were incubated in 30 μm dihydroethidium (DHE; Invitrogen) for 15 min at 37°C, fixed, and analyzed by confocal microscopy. Trolox (40 nm) or superoxide dismutase-1 (SOD1; 20 μg/ml; Serva) were added as indicated. For quantification, DHE intensity of all cells of the collected images (n ≥ 18) per experimental group was determined by NIH ImageJ software. Quantification of gene transcripts was performed using qRT-PCR and the δδ-CT method.
Detection of superoxide by Amplex Red.
Quantitative rates of ROS generation of microglia incubated with 10 μg/μl neural debris were determined using a Shimadzu RF-5001PC spectrofluorimeter with the Amplex Red/peroxidase-coupled method (1 μm Amplex Red plus 20 U/ml horseradish peroxidase) in the additional presence of 20 μg/ml SOD1. Excess SOD1 allowed the quantification of extracellular superoxide production in hydrogen peroxide (H2O2) equivalents (Malinska et al., 2009). All measurements were performed at 35°C in oxygen-saturated PBS as described previously (Malinska et al., 2009).
Binding of extracellular Siglec-E:Fc fusion protein.
Neurons and astrocytes were either untreated or treated with neuraminidase and then incubated with Siglec-E:Fc fusion protein (R&D Systems) for 1 h. Cells were fixed and incubated with rabbit anti-mouse IgG Fcγ (1:200; Dianova) and Alexa Fluor 488-conjugated goat anti-rabbit IgG, followed by mouse anti-βIII-tubulin (1:500; Sigma-Aldrich) or mouse anti-GFAP (1:500; Abcam) and Cy3-conjugated goat anti-mouse IgG antibody (1:500; Fab fragment; Dianova). Images were collected by confocal microscopy.
Statistical analyses.
Data are presented as mean ± SEM of at least three independent experiments. Data were analyzed by ANOVA, followed by Bonferroni's test (SPSS software).
Results
Detection of Siglec-E on microglia
Because the expression of Siglec-E on microglia was only indicated by microarray data (Gautier et al., 2012), we first investigated expression of Siglec-E on microglia. Although no Siglece mRNA was detected in primary neurons, primary microglia and a microglial line showed gene transcripts of Siglece (Fig. 1A). Next, we determined the protein expression of Siglec-E on microglia. Low constitutive expression of Siglec-E was detected by flow cytometry on the microglial line (Fig. 1B). Treatment with interferons (IFN-γ or IFN-α) did not change Siglece transcription as determined by qRT-PCR (data not shown) but slightly increased the cell-surface expression of Siglec-E (Fig. 1B). Furthermore, we observed low expression of Siglec-E on primary microglia isolated from the brain of adult mice (ex vivo microglia) as identified by antibodies directed against CD45 and CD11b (Fig. 1C).
Figure 1.
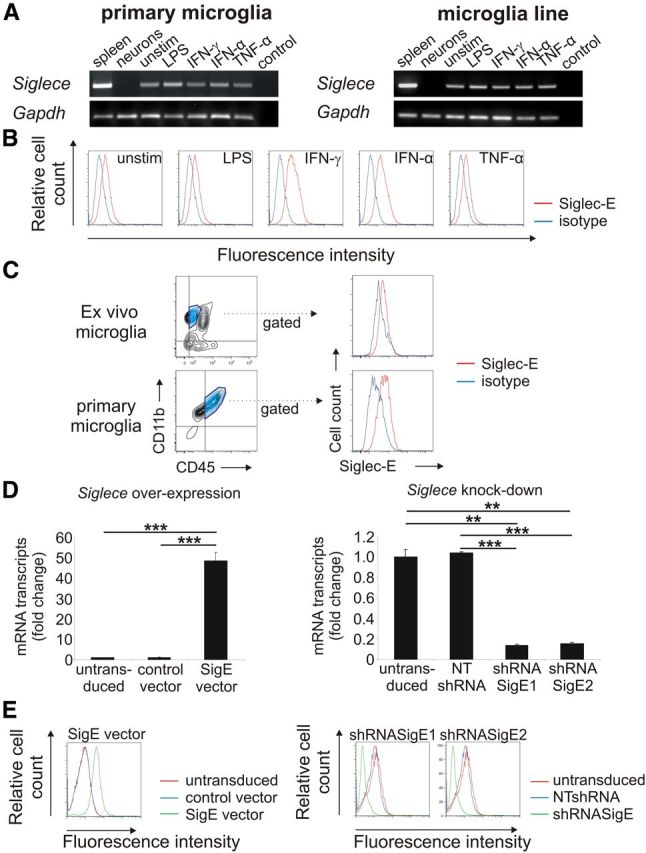
Detection of Siglec-E on microglia. A, Detection of Siglece mRNA in primary microglia and the microglia line. Spleen tissue served as positive control. Siglece transcripts were detected in unstimulated microglia (unstim.) as well as in microglia stimulated with LPS, IFN-γ, IFN-α, and TNF-α. No Siglece transcripts were detected in primary neurons. Gapdh mRNA served as housekeeping standard. Representative data of three independent experiments are shown. Control, Water control. B, Flow cytometry analysis of the microglial line. Siglec-E was detected on unstimulated (unstim.) microglia at low levels. Treatment with interferons (IFN-γ, IFN-α) slightly increased expression of Siglec-E, whereas treatment with LPS or TNF-α had no effect. Representative data of three independent experiments are shown. Isotype, Isotype control antibody. C, Flow cytometry analysis of ex vivo and primary microglia. Low constitutive expression of Siglec-E on CD11b+ and CD45low cells was detected. Representative data of three independent experiments are shown. Isotype, Isotype control antibody. D, Microglia were transduced with lentiviral vectors expressing Siglece (SigE vector) or a control vector. Furthermore, lentiviral knockdown was performed by two lentiviral short-hairpin constructs targeting Siglece (shRNASigE1; shRNASigE2) or a corresponding nontargeting control vector (NTshRNA). qRT-PCR confirmed the successful modification of the microglial line by showing an increased (left graph) or decreased (right graph) Siglece cDNA, respectively. **p ≤ 0.01, ***p ≤ 0.001. E, Flow cytometry analysis confirmed overexpression (left graph) and reduced expression (right graph) of the Siglec-E protein. Representative data of three independent experiments are shown.
Lentiviral overexpression or knockdown of Siglece does not change the microglial phenotype
After lentiviral overexpression, transcription for Siglece was increased, but after knockdown, it was decreased (Fig. 1D). Flow cytometry confirmed overexpression and knockdown of Siglec-E (Fig. 1E). Next, we asked whether the expression level of Siglec-E changes the microglial phenotype. Therefore, we analyzed inflammatory gene transcripts by qRT-PCR and cell-surface markers by flow cytometry. No changes in gene transcription of interleukin-1β (Il-1β), TNF-α (tnsfsf2), and nitric oxide synthase-2 (Nos2) were observed after lentiviral transduction (data not shown). Furthermore, we did not observe any changes in CD11b, CD11c, CD18, CD31, CD34, CD45, CD80, CD86, and F4/80 after lentiviral knockdown or overexpression of Siglece (data not shown). Thus, Siglec-E expression levels do not alter the overall microglial phenotype.
Siglec-E prevents phagocytosis of neural debris
Because Siglec-E signals via an inhibitory ITIM that is known to negatively interfere with activatory ITAM phagocytosis signaling (Linnartz and Neumann, 2013), we analyzed the role of Siglec-E in phagocytosis of neural debris. Engulfment of fluorescently labeled neural debris into the microglial line was determined by confocal microscopy and 3D reconstruction (Fig. 2A). Although overexpression of Siglec-E decreased the uptake of neural debris, reduced expression of Siglec-E led to an increase of uptake (Fig. 2B). In detail, Siglec-E overexpression reduced the percentage of microglia having engulfed neural debris from 34.17 ± 1.30% (control vector) to 17.55 ± 1.56% (SigE vector; p < 0.001; Fig. 2B). In contrast, reduced Siglec-E expression increased the percentage of phagocytozing microglia from 31.0 ± 0.06% [nontargeting short-hairpin RNA (NTshRNA)] to 44.32 ± 0.86% (shRNASigE1; p < 0.001) and 44.03 ± 0.71% (shRNASigE2; p < 0.001), respectively (Fig. 2B). Thus, Siglec-E inhibits the engulfment of neural debris.
Figure 2.
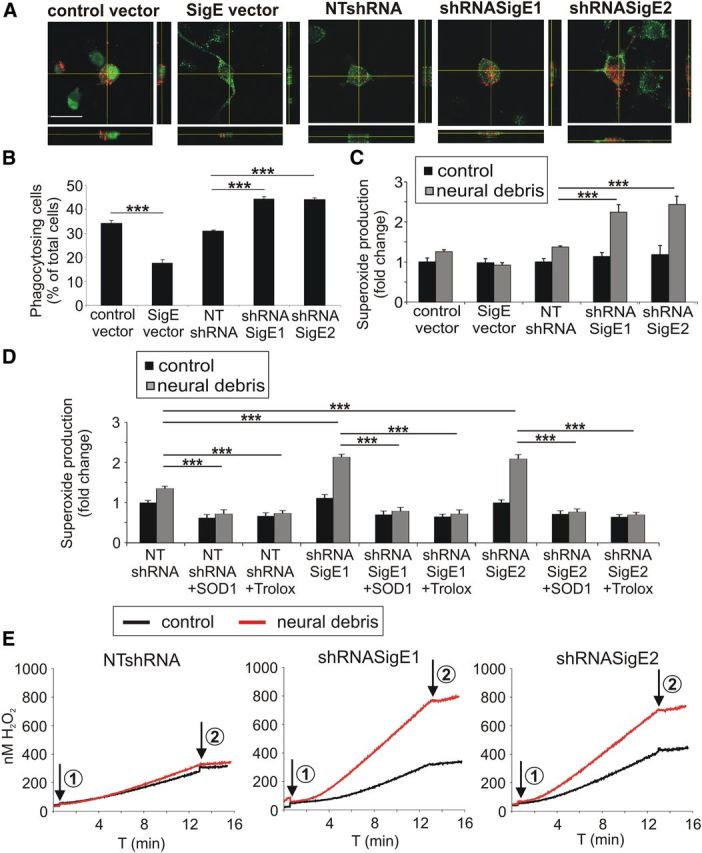
Siglec-E prevents phagocytosis and the associated reactive oxygen burst after challenge with neural debris. A, Uptake of red fluorescent-labeled neural debris into the microglial line was determined by confocal microscopy and 3D reconstruction. Microglial cells were transduced with the control vector, the Siglece overexpressing vector (SigE vector), the Siglece knockdown vectors (shRNASigE1, shRNASigE2) or the nontargeting vector (NTshRNA). Representative images of three independent experiments are shown. Scale bar, 20 μm. B, Phagocytosis of neural debris was quantified. Overexpression of Siglece mRNA reduced the uptake of neural material, whereas knockdown of Siglece increased the uptake of neural debris. ***p ≤ 0.001. C, Level of superoxide production as determined by DHE staining was quantified in the microglial line. After stimulation with neural debris, DHE intensity was increased after Siglece knockdown compared with the NTshRNA. ***p ≤ 0.001. D, Quantification of superoxide production as determined by DHE staining. After stimulation with neural debris in the presence of either 20 μg/ml SOD1 or 40 nm Trolox, increased DHE intensity after Siglece knockdown was antagonized. ***p ≤ 0.001. E, Quantification of superoxide production of microglial cells using the Amplex Red method. Knockdown of microglial Siglece via shRNASigE1 or shRNASigE2 increased the endogenous production of H2O2 equivalents after stimulation with neural debris compared with cells transduced with a control construct (NTshRNA). Arrow 1, Addition of cells; arrow 2, addition of 12,000 U/ml catalase. Representative data of three independent experiments are shown.
Siglec-E prevents superoxide release triggered by neural debris
Next, we analyzed whether Siglec-E also interferes with the phagocytosis-associated oxidative burst. Treatment with neural debris increased microglial superoxide production after knockdown of Siglec-E as determined by the intensity of the superoxide-sensitive fluorescent dye DHE (Fig. 2C). In the untreated situation, the superoxide production was comparable between the different microglial cells, although the levels were significantly increased with neural debris after knockdown of Siglece mRNA (Fig. 2C). In detail, the relative DHE intensity after incubation with neural debris was increased after knockdown with shRNASigE1 or shRNASigE2 from 1.38 ± 0.03 to 2.25 ± 0.19 (p < 0.001) or 2.4 ± 0.21 (p < 0.001), respectively (Fig. 2C). Overexpression of Siglec-E had no significant effect on the superoxide production triggered by phagocytosis of neural debris (Fig. 2C). The increased DHE staining of microglia having phagocytozed neural debris after knockdown of Siglec-E was abrogated by addition of SOD1 or the radical scavenger Trolox, a vitamin E derivate that is known to scavenge radicals (Fig. 2D). Next, we performed measurements using the Amplex Red method, which selectively detects extracellularly produced ROS. Similar to the DHE experiments, addition of neural debris stimulated the production of superoxide of microglial cells ∼1.1-fold in the control vector transduced microglia (Fig. 2E, left), whereas the knockdown of Siglec-E resulted in an approximate twofold stimulation of superoxide production rates (Fig. 2E, middle and right). In detail, endogenous production of 12.10 ± 1.10 pmol H2O2 equivalents/min/mg protein in untreated cells transduced with a control construct (NTshRNA) increased to 13.6 ± 1.34 pmol H2O2 equivalents/min/mg protein after addition of neural debris (Fig. 2E). In Siglece knockdown microglia production of superoxide increased from 15.43 ± 0.47 to 32.39 ± 0.39 pmol H2O2 equivalents/min/mg protein (by shRNASigE1; p < 0.001) or from 16.13 ± 0.65 to 28.15 ± 1.12 pmol H2O2 equivalents/min/mg protein (by shRNASigE2; p < 0.001) after addition of neural debris (Fig. 2E). Thus, Siglec-E acted as negative regulator of extracellular superoxide released by microglia after challenge with neural debris.
Siglec-E prevents production of proinflammatory cytokines triggered by neural debris
Next, we analyzed whether Siglec-E interferes with the production of proinflammatory mediators triggered by stimulation with neural debris. Gene transcription of Il-1β and Tnfsf2 (TNF-α) was significantly increased in microglia challenged with neural debris after knockdown of Siglece mRNA, although no effect on Nos2 mRNA was observed (Fig. 3A). In detail, gene transcription of Il-1β was increased from 10.0 ± 0.13 to 17.42 ± 2.26 after knockdown with shRNASigE1 (p = 0.001) and to 16.56 ± 1.10 after knockdown with shRNASigE2 (p = 0.003). Gene transcription of Tnfsf2 (TNF-α) was increased from 9.07 ± 1.39 to 14.21 ± 0.73 after knockdown with shRNASigE1 (p = 0.011) and to 15.27 ± 1.15 after knockdown with shRNASigE2 (p = 0.001; Fig. 3A). Overexpression of Siglec-E had no significant effect on the gene transcription of proinflammatory mediators after neural debris challenge (Fig. 3A). In summary, Siglec-E of microglia acted anti-inflammatory and prevented the production of the proinflammatory cytokines IL-1β and TNF-α after stimulation with neural debris.
Figure 3.
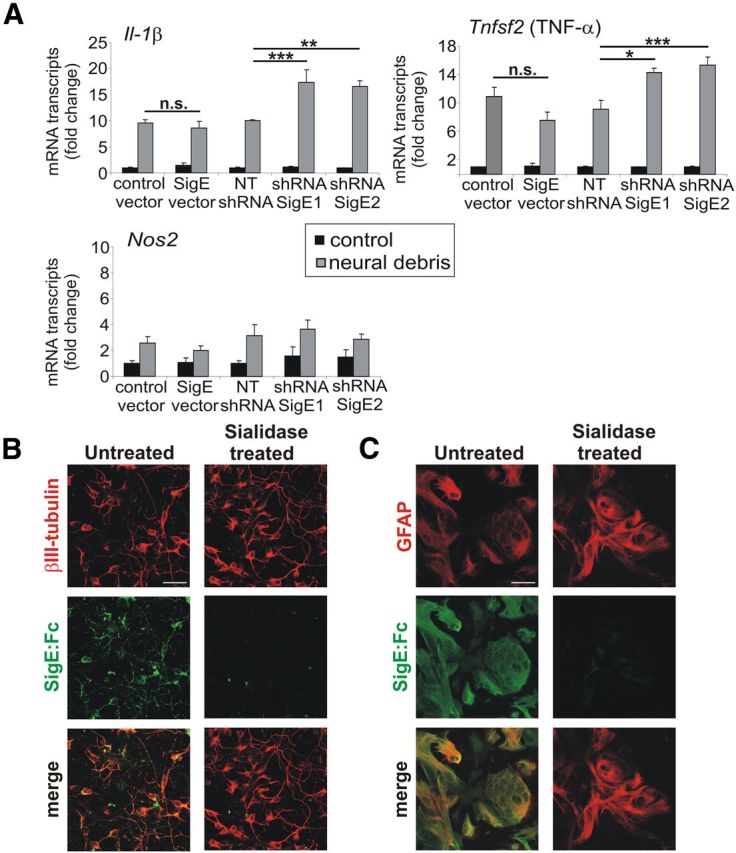
Siglec-E has anti-inflammatory effects and binds to neurons and astrocytes. A, qRT-PCR to detect Il-1β, Tnfsf2 (TNF-α), and Nos2 cDNA after 16 h incubation of microglia with neural debris. Microglia with knockdown of Siglece (shRNASigE1, shRNASigE2) showed a significant increase in gene transcription of Il-1β and Tnfsf2 (TNF-α) in the presence of neural debris compared with the control vector (NTshRNA). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; n.s., not significant. B, C, Binding of Siglec-E to sialic acid residues of neural cells. Neurons/astrocytes were either untreated or treated with sialidase and then incubated with the Siglec-E:Fc fusion protein. Removal of sialic acids led to a decreased binding of Siglec-E:Fc to neurons (B) and astrocytes (C). Representative images of three independent experiments are shown. Scale bar, 30 μm.
Binding of Siglec-E:Fc fusion protein to sialic acid residues of neurons
Siglec-E has a broad binding spectrum for different sialic acid linkages (Zhang et al., 2004), suggesting that the intact glycocalyx could act as natural ligand for Siglec-E. Therefore, we used a fusion protein consisting of the extracellular domains linked to an Ig (Siglec-E:Fc fusion protein) and demonstrated that Siglec-E recognized neurons and astrocytes as demonstrated via immunofluorescence staining (Fig. 3B,C). The staining was abrogated after enzymatic removal of the sialic acid residues on the glycocalyx (Fig. 3B,C). Siglec-E also bound to microglia but to a lower extent (data not shown). Thus, Siglec-E of microglia can sense the glycocalyx of the neighboring cells.
Protective effect of Siglec-E on neurites in neuron–microglia interaction
Next, we analyzed the role of Siglec-E in a microglia–neuron coculture system. Primary neurons were either untreated or treated with sialidase to remove the sialic acids from the neuronal glycocalyx and then cocultured with microglia displaying different expression levels of Siglec-E. In cocultures with an intact neuronal glycocalyx, overexpression of Siglec-E increased, whereas knockdown of Siglec-E decreased the relative neurite length (Fig. 4A). In detail, the neurite length was increased from 100 ± 9.97% (control vector) to 150 ± 2.19% in coculture with microglia overexpressing Siglece (p < 0.001; Fig. 4B). Knockdown of Siglece reduced the relative neurite length from 100 ± 5.31% (NTshRNA) to 70 ± 1.45% (shRNASigE1; p < 0.001) and 69 ± 1.2% (shRNASigE2; p < 0.001). Pretreatment of the neurons with sialidase abolished both effects (Fig. 4B). No change in the neuronal cell body density was observed after overexpression or reduced expression of Siglec-E in microglia (Fig. 4B). Thus, Siglec-E of microglia recognized sialic acid residues on neurons and inhibited the removal of neurites.
Figure 4.

Neurite protective effect of Siglec-E in neuron–microglia cocultures. A, Loss of neurites after knockdown of Siglece in microglia. Neurons were cocultured for 48 h with microglia. Siglece overexpression increased whereas knockdown of Siglece decreased the relative neurite length. Representative images without sialidase treatment of three independent experiments are shown. Scale bar, 30 μm. B, Relative neurite length and neuronal cell body density were quantified. Although the neuronal cell body density was unchanged (right graph), the relative neurite length was dependent on the expression level of Siglec-E on the microglial surface (left graph). After sialidase treatment, the relative neurite length was significantly reduced. **p ≤ 0.01, ***p ≤ 0.001. C, Neurons were cocultured with microglia in the presence of Trolox. Reduced neurite length after knockdown of Siglec-E was restored after treatment with 40 nm Trolox. ***p ≤ 0.001.
Neuroprotective effect of Siglec-E is mediated via attenuation of ROS species release
To better understand the protective effect of Siglec-E, we added Trolox in the cocultures. After coculture of microglia with knockdown of Siglec-E and neurons in the presence of Trolox, the relative neurite length was unaffected and still 97 ± 2.91% for knockdown with shRNASigE1 (NTshRNA, 103 ± 1.53%; p = 1.000) and 94 ± 2.85% for shRNASigE2 (p = 0.111; Fig. 4C). Trolox treatment had no influence on the relative neurite length of neurons that were cocultured with microglia overexpressing Siglec-E (Fig. 4C). Again, the relative number of neuronal cell bodies was unaffected by the Trolox treatment (Fig. 4C). Thus, the decreasing effect of Siglece knockdown microglia on neurite density was neutralized by Trolox, indicating an involvement of ROS in the microglial effect on neurite reduction after knockdown of Siglec-E.
Discussion
The mouse Siglec family consists of five members (Siglec-2, Siglec-3, Siglec-E, Siglec-F, and Siglec-G) having an inhibitory ITIM- or ITIM-like motif (Crocker et al., 2007). Although the expression of Siglec-2/CD22 and Siglec-G is restricted to B-cells and that of Siglec-F to eosinophils, Siglec-3/CD33 and Siglec-E are expressed on several myeloid cell types (Crocker et al., 2007). Our data now demonstrate that Siglec-E is expressed on the RNA and protein level on mouse microglia. In contrast to the mouse, humans have 10 Siglecs (Siglec-2, Siglec-3, and Siglec-5 to Siglec-12) with inhibitory ITIMs (Crocker et al., 2007). Siglec-11 has been shown to be expressed on human microglia and to inhibit neurotoxicity mediated by activated microglia (Wang and Neumann, 2010). However, Siglec-11 is a primate lineage-specific molecule and has no homolog in the mouse (Crocker et al., 2007). In turn, although Siglec-E is related to human Siglec-7, Siglec-8, and Siglec-9, no direct homolog of Siglec-E can be defined in the human system (Redelinghuys et al., 2011). However, both molecules either recognize (Siglec-11) or have a preference (Siglec-E) for α2–8-linked sialic acids, which are enriched in the mammalian brain (Varki, 2011). Siglec-E and Siglec-11 are not the only inhibitory Siglecs expressed on microglia. Microarray data from Gautier et al. (2012) and our own unpublished data suggest that Siglec-3 (CD33) is also expressed on mouse and human microglia. Our data now show that Siglec-E recognized the cell surface of living neurons displaying a sialylated intact glycocalyx. This is in agreement with its natural ligand, namely sialyloligosaccharides that are broadly found in glycoproteins and gangliosides on the cell surface of primary neurons (Varki and Schauer, 2009). Thus, microglial Siglec-E is functionally related to the human inhibitory microglial Siglec-11 that also recognized intact cultured neurons (Wang and Neumann, 2010). Interestingly, recent data show that Siglec-E has a preference for the sialic acid form N-acetyl-neuraminic acid instead of N-glycolyl-neuraminic acid-terminated sequences (Redelinghuys et al., 2011). Thus, microglial Siglec-E is well fitting to the microenvironment of the brain because the mammalian brain only expresses N-acetyl-neuraminic acid and is the organ with highest sialic acid expression levels (Varki and Schauer, 2009).
Our data show that microglial Siglec-E reduced the gene transcription of the proinflammatory cytokines IL-1β and TNF-α after stimulation with neural debris. In a previous study, Siglec-E has been shown to act anti-inflammatory as well (Boyd et al., 2009). Siglec-E expression has been demonstrated to inhibit the LPS-induced activation of the transcriptional factor nuclear factor-κB and production of TNF-α (Boyd et al., 2009). Recently, it was shown that Siglec-E is an important negative regulator of the integrin CD11b-mediated activation of Syk in neutrophils and thereby limits the migration of neutrophils to inflammatory loci (McMillan et al., 2013). Interestingly, Syk activation by CD11b in neutrophils is mediated via a signaling complex involving the ITAM of DAP12 (Mócsai et al., 2006), suggesting that Siglec-E via its ITIM might be a negative regulator of the integrin–ITAM signaling cascade as shown for other Siglecs. Moreover, our data show that Siglec-E inhibits phagocytosis and the phagocytosis-associated production of superoxide, also called respiratory burst. The respiratory burst induced by the CD11b/CD18 complex in neutrophils was mediated via the ITAM–Syk signaling of DAP12 and the Fcγ chain (Mócsai et al., 2006), and the ITAM–Syk signals could result in the activation of the NOX2 complex and the production of ROS (Graham et al., 2007).
Although our data show that absence of microglial Siglec-E increased phagocytosis and excess production of superoxide contribute to reduced neurite density, our coculture system does not allow to discriminate between microglial cytotoxicity and removal of intact neurites by phagocytosis. Recent data suggest that microglial cells are involved in pruning of axons (Schafer et al., 2012), a process that might implicate removal of living structures without previous apoptosis (Brown and Neher, 2012).
In summary, our data suggest that the ITIM-containing Siglec-E receptors of microglia bind to sialic acid residues of the neuronal glycocalyx and act as negative regulators of phagocytosis and the associated superoxide release. Because radicals and excessive phagocytosis have been postulated as driving forces of neurodegeneration, the inhibitory Siglec pathway might be an interesting target for therapy.
Footnotes
This project was supported by Deutsche Forschungsgemeinschaft Grants KFO177, SFB704, and FOR1336 and the Hertie Foundation. H.N. is member of the Deutsche Forschungsgemeinschaft-funded Excellence Cluster ImmunoSensation. We thank Dr. Ajit Varki for helpful discussions. We thank Dr. Veit Hornung for the knockdown plasmids. We thank Jessica Schumacher and Rita Hass for excellent technical support of cultures and molecular biology.
The authors declare no competing financial interests.
References
- Beutner C, Roy K, Linnartz B, Napoli I, Neumann H. Generation of microglial cells from mouse embryonic stem cells. Nat Protoc. 2010;5:1481–1494. doi: 10.1038/nprot.2010.90. [DOI] [PubMed] [Google Scholar]
- Boyd CR, Orr SJ, Spence S, Burrows JF, Elliott J, Carroll HP, Brennan K, Ní Gabhann J, Coulter WA, Jones C, Crocker PR, Johnston JA, Jefferies CA. Siglec-E is up-regulated and phosphorylated following lipopolysaccharide stimulation in order to limit TLR-driven cytokine production. J Immunol. 2009;183:7703–7709. doi: 10.4049/jimmunol.0902780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Neher JJ. Eaten alive! Cell death by primary phagocytosis: “phagoptosis.”. Trends Biochem Sci. 2012;37:325–332. doi: 10.1016/j.tibs.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma'ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ Immunological Genome Consortium. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlovoy P, Larionov S, Pham TT, Neumann H. Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J. 2009;23:2502–2513. doi: 10.1096/fj.08-123877. [DOI] [PubMed] [Google Scholar]
- Graham DB, Stephenson LM, Lam SK, Brim K, Lee HM, Bautista J, Gilfillan S, Akilesh S, Fujikawa K, Swat W. An ITAM-signaling pathway controls cross-presentation of particulate but not soluble antigens in dendritic cells. J Exp Med. 2007;204:2889–2897. doi: 10.1084/jem.20071283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnartz B, Neumann H. Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia. 2013;61:37–46. doi: 10.1002/glia.22359. [DOI] [PubMed] [Google Scholar]
- Malinska D, Kudin AP, Debska-Vielhaber G, Vielhaber S, Kunz WS. Chapter 23 Quantification of superoxide production by mouse brain and skeletal muscle mitochondria. Methods Enzymol. 2009;456:419–437. doi: 10.1016/S0076-6879(08)04423-6. [DOI] [PubMed] [Google Scholar]
- McMillan SJ, Sharma RS, McKenzie EJ, Richards HE, Zhang J, Prescott A, Crocker PR. Siglec-E is a negative regulator of acute pulmonary neutrophil inflammation and suppresses CD11b beta2-integrin-dependent signalling. Blood. 2013;121:2084–2094. doi: 10.1182/blood-2012-08-449983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mócsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redelinghuys P, Antonopoulos A, Liu Y, Campanero-Rhodes MA, McKenzie E, Haslam SM, Dell A, Feizi T, Crocker PR. Early murine T-lymphocyte activation is accompanied by a switch from N-Glycolyl- to N-acetyl-neuraminic acid and generation of ligands for siglec-E. J Biol Chem. 2011;286:34522–34532. doi: 10.1074/jbc.M111.243410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Since there are PAMPs and DAMPs, there must be SAMPs? Glycan “self-associated molecular patterns” dampen innate immunity, but pathogens can mimic them. Glycobiology. 2011;21:1121–1124. doi: 10.1093/glycob/cwr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Schauer R. Essentials of glycobiology. Ed 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2009. Oligosialic and poysialic acids, Chap 14. [Google Scholar]
- Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. 2008;28:8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Neumann H. Alleviation of neurotoxicity by microglial human Siglec-11. J Neurosci. 2010;30:3482–3488. doi: 10.1523/JNEUROSCI.3940-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JQ, Biedermann B, Nitschke L, Crocker PR. The murine inhibitory receptor mSiglec-E is expressed broadly on cells of the innate immune system whereas mSiglec-F is restricted to eosinophils. Eur J Immunol. 2004;34:1175–1184. doi: 10.1002/eji.200324723. [DOI] [PubMed] [Google Scholar]